
Abstract
Multi-label classification in deep learning is a practical yet challenging task, because class overlaps in the feature space means that each instance is associated with multiple class labels. This requires a prediction of more than one class category for each input instance. To the best of our knowledge, this is the first deep learning study which quantifies uncertainty and model interpretability in multi-label classification; as well as applying it to the problem of recognising proteins expressed in cell types in testes based on immunohistochemically stained images. Multi-label classification is achieved by thresholding the class probabilities, with the optimal thresholds adaptively determined by a grid search scheme based on Matthews correlation coefficients. We adopt MC-Dropweights to approximate Bayesian Inference in multi-label classification to evaluate the usefulness of estimating uncertainty with predictive score to avoid overconfident, incorrect predictions in decision making. Our experimental results show that the MC-Dropweights visibly improve the performance to estimate uncertainty compared to state of the art approaches.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Proteins are the essential building blocks of life, and resolving the spatial distribution of all human proteins at an organ, tissue, cellular, and subcellular level greatly improves our understanding of human biology in health and disease. The testes is one of the most complex organs in the human body [15]. The spermatogenesis process results in the testes containing the most tissue-specific genes than elsewhere in the human body. Based on an integrated ‘omics’ approach using transcriptomics and antibody-based proteomics, more than 500 proteins with distinct testicular protein expression patterns have previously been identified [10], and transcriptomics data suggests that over 2,000 genes are elevated in testes compared to other organs. The function of a large proportion of these proteins are however largely unknown, and all genes involved in the complex process of spermatogenesis are yet to be characterized. Manual annotation provides the standard for scoring immunohistochemical staining pattern in different cell types. However, it is tedious, time-consuming and expensive as well as subject to human error as it is sometimes challenging to separate cell types by the human eye. It would be extremely valuable to develop an automated algorithm that can recognise the various cell types in testes based on antibody-based proteomics images while providing information on which proteins are expressed by that cell type [10]. This is, therefore, a multi-label image classification problem.

Schematic overview: cell type-specific expression of testis elevated genes [10]
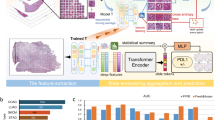
Exact Bayesian inference with deep neural networks is computationally intractable. There are many methods proposed for quantifying uncertainty or confidence estimates. Recently Gal [5] proved that a dropout neural network, a well-known regularisation technique [13], is equivalent to a specific variational approximation in Bayesian neural networks. Uncertainty estimates can be obtained by training a network with dropout and then taking Monte Carlo (MC) samples of the prediction using dropout during test time. Following Gal [5], Ghoshal et al. [7] also showed similar results for neural networks with Dropweights and Teye [14] with batch normalisation layers in training (Fig. 1).
In this paper, we aim to:
-
1.
Present the first approach in multi-label pattern recognition that can recognise various cell types-specific protein expression patterns in testes based on antibody-based proteomics images and provide information on which cell types express the protein with estimated uncertainty.
-
2.
Show Multi-Label Classification (MLC) is achieved by thresholding the class probabilities, with the Optimal Thresholds adaptively determined by a grid search scheme based on Matthews correlation coefficient.
-
3.
Demonstrate through extensive experimental results that a Deep Learning Model with MC-Dropweights [7] is significantly better than a wide spectrum of MLC algorithms such as Binary Relevance (BR), Classifier Chain (CC), Probabilistic Classifier Chain (PCC) and Condensed Filter Tree (CFT), Cost-sensitive Label Embedding with Multidimensional Scaling (CLEMS) and state-of-the-art MC-Dropout [5] algorithms across various cell types.
-
4.
Develop Saliency Maps in order to increase model interpretability visualizing descriptive regions and highlighting pixels from different areas in the input image. Deep learning models are often accused of being “black boxes”, so they need to be precise, interpretable, and uncertainty in predictions must be well understood.
Our objective is not to achieve state-of-the-art performance on these problems, but rather to evaluate the usefulness of estimating uncertainty leveraging MC-Dropweights with predictive score in multi-label classification to avoid overconfident, incorrect predictions for decision making.
2 Multi-label Cell-Type Recognition and Localization with Estimated Uncertainty
2.1 Problem Definition
Given a set of training data \(D\), where \(X=\left\{ x_{1}, x_{2} \ldots x_{N}\right\} \) is the set of N images and the corresponding labels \(Y=\left\{ y_{1}, y_{2} \ldots y_{N}\right\} \) is the cell-type information. The vector \(y_i=\left\{ y_{i,1}, y_{i,2} \ldots y_{i,M}\right\} \) is a binary vector, where \(y_{i,j} = 1\) indicates that the \(i^{th}\) image belongs to the \( j^{th}\) cell-type. Note that an image may belong to multiple cell-types, i.e., \(1<= \sum _{j} y_{i,j} <=M\). Based on \(D (X, Y)\), we constructed a Bayesian Deep Learning model giving an output of the predictive probability with estimated uncertainty of a given image \(x_i\) belonging to each cell category. That is, the constructed model acts as a function such that \(f : X \rightarrow Y\) using weights of neural net parameters \(\omega \) where \((0<= \hat{y}_{x,j} <= 1)\) as close as possible to the original function that has generated the outputs \(\mathrm {Y}\), output the estimated value \((\hat{y}_{i,1}, \hat{y}_{i,2},\dots , \hat{y}_{i,M})\) as close to the actual value \(({y}_{i,1}, {y}_{i,2},\dots , {y}_{i,M})\).
2.2 Solution Approach
We tailored Deep Convolutional Neural Network (DCNN) architectures for cell type detection and localisation by considering a large image capacity, binary-cross entropy loss, sigmoid activation, along with Dropweights in the fully connected layer and Batch Normalization formulation of propagating uncertainty in deep learning to estimate meaningful model uncertainty.
Multi-label Setup: There are multiple approaches to transform the multi-label classification into multiple single-label problems with the associated loss function [8]. In this study, we used immunohistochemically stained testes tissue consisting of 8 cell types corresponding to 512 testis elevated genes.
Therefore, we define a 8-dimensional class label vector \(Y=\left\{ y_{1}, y_{2} \ldots y_{N}\right\} \) ; \(Y \in \{0, 1 \}\), given 8 cell types. \(y_{c}\) indicates the presence with respect to according cell type expressing the protein in the image while an all-zero vector [0; 0; 0; 0; 0; 0; 0; 0] represents the “Absence” (no cell type expresses the protein in the scope of any of 8 categories).
Multi-label Classification Cost Function: The cost function for Multi-label Classification has to be different considering the fact that a prediction for a class is not mutually exclusive. So we selected the sigmoid function with the addition of binary cross-entropy.
Data Augmentation: We used Keras’ image pre-processing package to apply affine transformations to the images, such as rotation, scaling, shearing, and translation during training and inference. This reduces the epistemic uncertainty during training, captures heteroscedastic aleatoric uncertainty during inference and overall improves the performance of models.
Multi-label Classification Algorithm: In Bayesian classification, the mean of the predictive posterior corresponds to the parameter point estimates, and the width of the posterior reflects the confidence of the predictions. The output of the network is an M-dimensional probability vector, where each dimension indicates how likely each cell type in a given image expresses the protein. The number of cell types that simultaneously express the protein in an image varies. One method to solve this multi-label classification problem is placing thresholds on each dimension. However different dimensions may be associated with different thresholds. If the value of the \(i^{th}\) dimension of \(\hat{y}\) is greater than a threshold, we can say that the i-th cell-type is expressed in the given tissue. The main problem is defining the threshold for each class label.
A threshold based on Matthews Correlation Coefficient (MCC) is used on the model outcome to determine the predicted class to improve the accuracy of the models.
We adopted a grid search scheme based on Matthews Correlation Coefficients (MCC) to estimate the optimal thresholds for each cell type-specific protein expression [2]. Details of the optimal threshold finding algorithm is shown in Algorithm 1.

The idea is to estimate the threshold for each cell category in an image separately. We convert the predicted probability vector with the estimated threshold into binary and calculate the Matthews correlation coefficient (MCC) between the threshold value and the actual value. The Matthews correlation coefficient for all thresholds are stored in the vector \(\omega \), from which we find the index of threshold that causes the largest correlation. The Optimal Threshold for the \(i^{th}\) dimension is then determined by the corresponding value. We then leveraged Bias-Corrected Uncertainty quantification method [6] using Deep Convolutional Neural Network (DCNN) architectures with Dropweights [7].
Network Architecture: Our models are trained and evaluated using Keras with Tensorflow backend. For the DNN architecture, we used a generic building block containing the following model structure: Conv-Relu-BatchNorm-MaxPool-Conv-Relu-BatchNorm-MaxPool-Dense-Relu-Dropweights and Dense-Relu-Dropweights-Dense-Sigmoid, with 32 convolution kernels, 3 \(\times \) 3 kernel size, 2 \(\times \) 2 pooling, dense layer with 512 units, 128 units, and 8 feed-forward Dropweights probabilities 0.3. We optimised the model using Adam optimizer with the default learning rate of 0.001. The training process was conducted in 1000 epochs, with mini-batch size 32. We repeated our experiments three times for an algorithm and calculated a mean of the results.
3 Estimating Bias-Corrected Uncertainty Using Jackknife Resampling Method
3.1 Bayesian Deep Learning and Estimating Uncertainty
There are many measures to estimate uncertainty such as softmax variance, expected entropy, mutual information, predictive entropy and averaging predictions over multiple models. In supervised learning, information gain, i.e. mutual information between the input data and the model parameters is considered as the most relevant measure of the epistemic uncertainty [4, 12]. Estimation of entropy from the finite set of data suffers from a severe downward bias when the data is under-sampled. Even small biases can result in significant inaccuracies when estimating entropy [9]. We leveraged Jackknife resampling method to calculate bias-corrected entropy [11].
Given a set of training data \(D\), where \(\mathbf {X}=\left\{ x_{1}, x_{2} \ldots x_{N}\right\} \) is the set of N images and the corresponding labels \(\mathbf {Y}=\left\{ y_{1}, y_{2} \ldots y_{N}\right\} \), a BNN is defined in terms of a prior \(p(\omega )\) on the weights, as well as the likelihood \(p(D | \omega )\). Consider class probabilities \(p(y_{x_i}=c \mid x_i, \omega _t, D)\) with \(\omega _t \sim q(\omega \mid D)\) with \(\mathcal {W} = (\omega _t)_{t=1}^T\), a set of independent and identically distributed (i.i.d.) samples draws from \(q(\omega \mid , D)\). The below procedure computes the Monte Carlo (MC) estimate of the posterior predictive distribution, its Entropy and Mutual Information(MI):
where
The stochastic predictive entropy is \({H}[y\mid x,\omega ] = \mathbb {H}(\hat{p}) = -\sum _{c}\hat{p}_c\log (\hat{p}_c)\), where \(\hat{p}_c = \tfrac{1}{T} \sum _{t} p_{tc}\) is the entire sample maximum likelihood estimator of probabilities.
The first term in the MC estimate of the mutual information is called the plug-in estimator of the entropy. It has long been known that the plug-in estimator underestimates the true entropy and plug-in estimate is biased [11, 17].
A classic method for correcting the bias is the Jackknife resampling method [3]. In order to solve the bias problem, we propose a Jackknife estimator to estimate the epistemic uncertainty to improve an entropy-based estimation model. Unlike MC-Dropout, it does not assume constant variance. If \(\mathcal D(X, Y)\) is the observed random sample, the \(i^{th}\) Jackknife sample, \(x_{i}\), is the subset of the sample that leaves-one-out observation \(x_{i}: x_{(i)} = ({x}_1,\dots {x}_{i-1},{x}_{i+1} \dots {x}_n)\). For sample size \(N\), the Jackknife standard error \(\hat{\sigma }\) is defined as: \(\sqrt{\frac{(N-1)}{N} {\sum _{{i}=1}^{{N}} (\hat{\sigma }_{i} - \hat{\sigma }_{(\odot )}})^{2}} \) , where \(\hat{\sigma }_{(\odot )}\) is the empirical average of the Jackknife replicates: \( \frac{1}{N} \sum _{{i}=1}^{{N}} \hat{\sigma }_{(i)}\). Here, the Jackknife estimator is an unbiased estimator of the variance of the sample mean. The Jackknife correction of a plug-in estimator \(\mathbb {H}(\cdot )\) is computed according to the method below [3]:
Given a sample \((p_t)_{t=1}^T\) with \(p_t\) discrete distribution on 1...C classes, T corresponds to the total number of MC-Dropweights forward passes during the test.
-
1.
for each \(t=1... T\)
-
calculate the leave-one-out estimator: \(\hat{p}_c^{-t} = \tfrac{1}{T-1} \sum _{j\ne i} p_{jc}\)
-
calculate the plug-in entropy estimate: \(\hat{H}_{-t} = \mathbb {H}(\hat{p}^{-t})\)
-
-
2.
calculate the bias-corrected entropy \(\hat{H}_{J} = T\hat{H} + \frac{(T-1)}{T} \sum _{t=1}^T {\hat{H}_{(-i)}}\), where \(\hat{H}_{(-i)}\) is the observed entropy based on a sub-sample in which the \(i\)th individual is removed.
We leveraged the following relation:
while resolving the i-th data point out of the sample mean \(\mu = \tfrac{1}{T} \sum _i x_i\) and recompute the mean \(\mu _{-i}\). This makes it possible to quickly calculate leave-one-out estimators of a discrete probability distribution.
The epistemic uncertainty can be obtained as the difference between the approximate predictive posterior entropy (or total entropy) and the average uncertainty in predictions (i.e: aleatoric entropy):
Therefore, the mutual information \(I(\mathbf {y}:\mathbf {\omega })\) i.e. as a measure of bias-corrected epistemic uncertainty, represents the variability in the predictions made by the neural network weight configurations drawn from approximate posteriors. It derives an estimate of the finite sample bias from the leave-one-out estimators of the entropy and reduces bias considerably down to \(O({n}^{-2})\) [3].
The bias-corrected uncertainty estimation model explains regions of ambiguous data space or difficult to classify, as data distribution with noise in the inputs or model, which was trained with different domain data. Consequently, these inputs should be assigned a higher aleatoric uncertainty. As a result, we can expect high model uncertainty in these regions.
Following Gal [5], we define the stochastic versions of Bayesian uncertainty using MC-Dropweights, where the class probabilities \(p(y_{x_i}=c \mid x_i, \omega _t, D)\) with \(\omega _t \sim q(\omega \mid D)\) and \(\mathcal {W} = (\omega _t)_{t=1}^T\) along with a set of independent and identically distributed (i.i.d.) samples drawn from \(q(\omega \mid , D)\), can be approximated by the average over the MC-Dropweights forward pass.
We trained the multi-label classification network with all eight classes. We dichotomised the network outputs using optimal threshold with Algorithm 1 for each cell type, with a 1000 MC-Dropweights forward passes at test time. In these detection tasks, \(p(y_{x_i} >= 0; OptimalThreshold_i \mid x_i, \omega _t, D)\), where 1 marks the presence of cell type, is sufficient to indicate the most likely decision along with estimated uncertainty.
3.2 Dataset
Our main dataset is taken from The Human Protein Atlas project, that maps the distribution of all human proteins in human tissues and organs [15]. Here, we used high-resolution digital images of immunohistochemically stained testes tissue consisting of 8 cell types: spermatogonia, preleptotene spermatocytes, pachytene spermatocytes, round/early spermatids, elongated/late spermatids, sertoli cells, leydig cells, and peritubular cells, publicly available on the Human Protein Atlas version 18 (v18.proteinatlas.org), as shown in Fig. 2:

Examples of proteins expressed only in one cell-type [10]
A relationship was observed between spermatogonia and preleptotene spermatocytes cell types and between round/early spermatids and elongated/late spermatids cell types along with Pachytene spermatocytes cells. Figure 3 illustrates the correlation coefficients between cell types. The observable pattern is that very few cell types are strongly correlated with each other.

Annotated heatmap of a correlation matrix between cell types
3.3 Results and Discussions
We conducted the experiments on Human Protein Atlas datasets to validate the proposed algorithm, MC-Dropweights in Multi-Label Classification.
Multi-label Classification Model Performance: Model evaluation metrics for multi-label classification are different from those used in multi-class (or binary) classification. The performance metrics of multi-label classifiers can be classified as label-based (i.e.: it is assumed that labels are mutually exclusive) and example-based [16]. In this work, example-based measures (Accuracy score, Hamming-loss, F1-Score) and Rank-Loss are used to evaluate the performance of the classifiers.
In the first experiment, we compared the MC-Dropweights neural network-based method with five machine learning MLC algorithms introduced in Sect. 1: binary relevance (BR), Classifier Chain (CC), Probabilistic Classifier Chain (PCC) and Condensed Filter Tree (CFT), Cost-Sensitive Label Embedding with Multi-dimensional Scaling (CLEMS) and the MC-Dropout neural network model. Table 1 shows that MC-Dropweights exhibits considerably better performance overall the algorithms, which demonstrates the importance of considering the Dropweights in the neural network.
Cell Type-Specific Predictive Uncertainty: The relationship between uncertainty and predictive accuracy grouped by correct and incorrect predictions is shown in Fig. 4. It is interesting to note that, on average, the highest uncertainty is associated with Elongated/late Spermatids and Round/early Spermatids. This indicates that there is some feature which contributes greater uncertainty to the Spermatids class types than to the other cell types.

Distribution of uncertainty values for all protein images, grouped by correct and incorrect predictions. Label assignment was based on optimal thresholding (Algorithm 1). For an incorrect prediction, there is a strong likelihood that the predictive uncertainty is also high in all cases except for Spermatids.
Cell Type Localization: Estimated uncertainty with Saliency Mapping is a simple technique to uncover discriminative image regions that strongly influence the network prediction in identifying a specific class label in the image. It highlights the most influential features in the image space that affect the predictions of the model [1] and visualises the contributions of individual pixels to epistemic and aleatoric uncertainties separately. We calculated the class activation maps (CAM) [18] using the activations of the fully connected layer and the weights from the prediction layer as shown in Fig. 5.

Saliency maps for some common methods towards model explanation
4 Conclusion and Discussion
In this study, a multi-label classification method was developed using deep learning architecture with Dropweights for the purposes of predicting cell types-specific protein expression with estimated uncertainty, which can increase the ability to interpret, with confidence and make models based on deep learning more applicable in practice. The results show that a Deep Learning Model with MC-Dropweights yields the best performance among all popular classifiers.
Building truly large-scale, fully-automated, high precision, very high dimensional, image analysis system that can recognise various cell type-specific protein expression, specifically for Elongated/Late Spermatids and Round/early Spermatids remains a strenuous task. The properties in the dataset such as label correlations, label cardinality can strongly affect the uncertainty quantification in predictive probability performance of a Bayesian Deep learning algorithm in multi-label settings. There is no systematic study on how and why the performance varies over different data properties; any such study would be of great benefit in progressing multi-label algorithms.
References
Adebayo, J., Gilmer, J., Muelly, M., Goodfellow, I., Hardt, M., Kim, B.: Sanity checks for saliency maps. In: Advances in Neural Information Processing Systems, pp. 9505–9515 (2018)
Chu, W.T., Guo, H.J.: Movie genre classification based on poster images with deep neural networks. In: Proceedings of the Workshop on Multimodal Understanding of Social, Affective and Subjective Attributes, pp. 39–45. ACM (2017)
DasGupta, A.: Asymptotic Theory of Statistics and Probability. Springer, New York (2008). https://doi.org/10.1007/978-0-387-75971-5
Depeweg, S., Hernández-Lobato, J.M., Doshi-Velez, F., Udluft, S.: Decomposition of uncertainty in Bayesian deep learning for efficient and risk-sensitive learning. arXiv preprint arXiv:1710.07283 (2017)
Gal, Y.: Uncertainty in deep learning. Ph.D. thesis, University of Cambridge (2016)
Ghoshal, B., Tucker, A., Sanghera, B., Wong, W.: Estimating uncertainty in deep learning for reporting confidence to clinicians in medical image segmentation and diseases detection. In: Computational Intelligence - Special Issue on Foundations of Biomedical (Big) Data Science, vol. 1 (2019)
Ghoshal, B., Tucker, A., Sanghera, B., Wong, W.: Estimating uncertainty in deep learning for reporting confidence to clinicians when segmenting nuclei image data. 2019 IEEE 32nd International Symposium on Computer-Based Medical Systems (CBMS), vol. 1, pp. 318–324, June 2019. https://doi.org/10.1109/CBMS.2019.00072
Huang, K.H., Lin, H.T.: Cost-sensitive label embedding for multi-label classification. Mach. Learn. 106(9–10), 1725–1746 (2017)
Macke, J., Murray, I., Latham, P.: Estimation bias in maximum entropy models. Entropy 15(8), 3109–3129 (2013)
Pineau, C., et al.: Cell type-specific expression of testis elevated genes based on transcriptomics and antibody-based proteomics. J. Proteome Res. 18, 4215–4230 (2019)
Quenouille, M.H.: Notes on bias in estimation. Biometrika 43(3/4), 353–360 (1956)
Shannon, C.E.: A mathematical theory of communication. Bell Syst. Tech. J. 27(3), 379–423 (1948)
Srivastava, N., Hinton, G., Krizhevsky, A., Sutskever, I., Salakhutdinov, R.: Dropout: a simple way to prevent neural networks from overfitting. Journal Mach. Learn. Res. 15(1), 1929–1958 (2014)
Teye, M., Azizpour, H., Smith, K.: Bayesian uncertainty estimation for batch normalized deep networks. arXiv preprint arXiv:1802.06455 (2018)
Uhlén, M., et al.: Tissue-based map of the human proteome. Science 347(6220), 1260419 (2015)
Wu, X.Z., Zhou, Z.H.: A unified view of multi-label performance measures. In: Proceedings of the 34th International Conference on Machine Learning, vol. 70, pp. 3780–3788. JMLR. org (2017)
Yeung, R.W.: A new outlook on Shannon’s information measures. IEEE Trans. Inf. Theory 37(3), 466–474 (1991)
Zhou, B., Khosla, A., Lapedriza, A., Oliva, A., Torralba, A.: Learning deep features for discriminative localization. In: CVPR (2016)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Ghoshal, B., Lindskog, C., Tucker, A. (2020). Estimating Uncertainty in Deep Learning for Reporting Confidence: An Application on Cell Type Prediction in Testes Based on Proteomics. In: Berthold, M., Feelders, A., Krempl, G. (eds) Advances in Intelligent Data Analysis XVIII. IDA 2020. Lecture Notes in Computer Science(), vol 12080. Springer, Cham. https://doi.org/10.1007/978-3-030-44584-3_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-44584-3_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-44583-6
Online ISBN: 978-3-030-44584-3
eBook Packages: Computer ScienceComputer Science (R0)