
Abstract
We present some augmentations to literature Message Passing Neural Network (MPNN) architectures and benchmark their performances against a wide range of chemically and pharmaceutically relevant datasets. We analyse the effects of activation function for regularisation, we propose a new graph attention mechanism, and we implement a new edge-based memory system that should maximise the effectiveness of hidden state usage by directing and isolating information flow around the graph. We compare our results to the MolNet [14] benchmarking paper results on graph-based techniques, and also investigate the effect of method performance as a function of dataset preprocessing.
Funding from the EU H2020 MSC Grant 676434 “BigChem”.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Many fields and research areas over the past decade have benefit greatly from the rise of deep learning [3]. AI has risen in popularity notably in the pharmaceutical industry, for activities such as bioactivity and physical-chemical property prediction, de novo design, synthesis prediction and image analysis, to name a few. The rapid growth of accessible computing power thanks to graphically-accelerated computing, and the ever increasing quantity of available chemical and biochemical data, have lead to a natural desire for data-hungry machine learning techniques such as deep learning to attempt to exploit this information to the greatest possible extent.
1.1 Graph Convolution
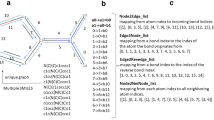
In Graph Convolutional Networks (GCNs), information propagates through a given graph much like how convolutional neural networks (CNNs) treat grid data (e.g. image data, text strings etc.). In contrast to image-data, however, graphs have irregular local connectivity, are not necessarily shift-invariant, and are not from a Euclidean domain (Fig. 1). CNNs exploit these properties for their powerful performance, and to match this on graphs these problems must be surmounted, in a manner that is invariant to the ordered representation of the graph.

Convolutional Neural Networks operating on e.g. image data (left) have a regular Euclidean representation, with a fixed dimensionality of neighbours to each data point (vertical, horizontal, and channel-depth). With graph based data, however, each node can have a variable number of neighbours (irregular), and the graph can be traversed in any order (isomorphic representation) without an easily-describable canonical representation
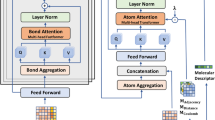
Message-Passing Neural Networks. Graph Neural Networks began in 2005 by Gori et al. [9], and in 2013 the first Graph Convolutional Network schema based on spectral graph theory was published by Bruna et al. [2]. Our work is focussed on the framework presented by Google – the Message Passing Neural Network [7], which was developed to generalize and be able to represent a selection of previously-published graph-based techniques [2, 4, 5, 10,11,12,13]. We analyse the performance effects of activation function (SELU)-based normalisation, and chemically-based dataset preprocessing, and propose two new novel architectures as extensions to the MPNN framework:
Attention MPNN (AMPNN) in which attention is performed over hidden state vector elements, dependent on edge type, allowing weighted summation in the message-passing function.
An Edge-Memory network, in which hidden states belong to directed edges and can only propagate in a single direction, designed to naturally allow for asymmetric bias and to maximise useful hidden memory information when propagating a node’s neighbourhood.
Low-Level Features from Graph Structure. Unlike traditional cheminformatic approaches to Machine Learning tasks, which use feature engineering, graphs are one of the lowest-level representations of chemical structures from which many features can be calculated directly. By directly using a chemical structure as the starting point for deep learning, feature-engineering can be avoided, and prior assumptions about task-specific knowledge don’t need to be made. Instead, task-specific features are learned within the network, and derived from the chemical structure directly. This allows for a potentially very powerful general-purpose approach to chemical task modelling, and also presents an interesting approach to the secure sharing of chemical data – the dissemination of trained models for activity prediction in lieu of chemical data itself, without the risk of reverse-engineering IP-sensitive structural information from e.g. chemical fingerprints [1, 6], and the ability to jointly-train models without the need to pre-negotiate engineered features relevant to the task.
2 Method
We evaluate our networks on a selection of benchmarking datasets, referred to as: HIV (42k compounds, classification, single-task); MUV (93k compounds, classification, 17 tasks); Tox21 (8k compounds, classification, 12 tasks); ESOL (1k compounds, regression, single-task); QM8 (22k compounds, regression, 12 tasks); SIDER (1.4k compounds, classification, 27-task), LIPO (4k compounds, regression, single-task) and BBBP (2k compounds, classification, single-task). Datasets were split and tested according to previous MolNet benchmarking [14] and hyperparameter optimisation was performed using Bayesian Optimisation in parallel with Local Penalisation [8].
3 Results
We present results for models trained on benchmarking datasets both as presented verbatim, referred to as Original Dataset, and with custom preprocessing (Charge-Parent Missing-Data – CPMD). Results are in general on-par with state-of-the-art, beating classification performance on the MUV dataset, and obtaining lower error on the smaller LIPO regression set. The charge-parent aspect of database preprocessing was found to be negligible (no performance difference between e.g. SIDER models or single-task models, with no missing data values), but the introduction of missing data values and a suitable masking loss function was found to have a strong positive effect on performance on highly sparse sets (MUV), over tripling performance relative to MolNet for the Attention, Edge and SELU networks, bringing them on-par with SVM and beating SVM with the Edge-based approach. The charge-parent aspect of the preprocessing was done to investigate how robust the model is to ionic complexes, such as those shown in Fig. 4. As the network does not model ionic bonds, it was unknown whether disjoint graphs would interfere with the message propagation, and ions such as sodium would act as noise in the training. However, due to the lack of performance difference between the two sets when all data is present, it can be assumed that the readout function safely bridges these gaps and does not interfere with models’ performance (Figs. 2 and 3).

Relative Performance of Classification (left) and Relative Error of Regression (right) models against the best presented MolNet model, on the original datasets. Unless otherwise stated, classification sets were evaluated using the ROC-AUC metric.

Relative Performance of Classification (left) and Relative Error of Regression (right) models against the best presented MolNet model, on the CPMD datasets. Unless otherwise stated, classification sets were evaluated using the ROC-AUC metric.

Examples of ionic complexes in the Original Dataset
References
Bologa, C., Allu, T.K., Olah, M., Kappler, M.A., Oprea, T.I.: Descriptor collision and confusion: toward the design of descriptors to mask chemical structures. J. Comput. Aided Mol. Des. 19(9–10), 625–635 (2005). https://doi.org/10.1007/s10822-005-9020-4
Bruna, J., Zaremba, W., Szlam, A., LeCun, Y.: Spectral networks and locally connected networks on graphs. arXiv:1312.6203 [cs], December 2013. http://arxiv.org/abs/1312.6203
Chen, H., Engkvist, O., Wang, Y., Olivecrona, M., Blaschke, T.: The rise of deep learning in drug discovery. Drug Discov. Today 23(6), 1241–1250 (2018). https://doi.org/10.1016/j.drudis.2018.01.039, http://www.sciencedirect.com/science/article/pii/S1359644617303598
Defferrard, M., Bresson, X., Vandergheynst, P.: Convolutional neural networks on graphs with fast localized spectral filtering. In: Lee, D.D., Sugiyama, M., Luxburg, U.V., Guyon, I., Garnett, R. (eds.) Advances in Neural Information Processing Systems, vol. 29, pp. 3844–3852. Curran Associates Inc. (2016). http://papers.nips.cc/paper/6081-convolutional-neural-networks-on-graphs-with-fast-localized-spectral-filtering.pdf
Duvenaud, D.K., et al.: Convolutional networks on graphs for learning molecular fingerprints. In: Cortes, C., Lawrence, N.D., Lee, D.D., Sugiyama, M., Garnett, R. (eds.) Advances in Neural Information Processing Systems, vol. 28, pp. 2224–2232. Curran Associates Inc. (2015). http://papers.nips.cc/paper/5954-convolutional-networks-on-graphs-for-learning-molecular-fingerprints.pdf
Filimonov, D., Poroikov, V.: Why relevant chemical information cannot be exchanged without disclosing structures. J. Comput. Aided Mol. Des. 19(9–10), 705–713 (2005). https://doi.org/10.1007/s10822-005-9014-2
Gilmer, J., Schoenholz, S.S., Riley, P.F., Vinyals, O., Dahl, G.E.: Neural message passing for quantum chemistry. arXiv:1704.01212 [cs], April 2017. http://arxiv.org/abs/1704.01212
González, J., Dai, Z., Hennig, P., Lawrence, N.D.: Batch Bayesian Optimization via Local Penalization. arXiv:1505.08052 [stat], May 2015. http://arxiv.org/abs/1505.08052
Gori, M., Monfardini, G., Scarselli, F.: A new model for learning in graph domains. In: Proceedings of the 2005 IEEE International Joint Conference on Neural Networks 2005, vol. 2, pp. 729–734, July 2005. https://doi.org/10.1109/IJCNN.2005.1555942
Kearnes, S., McCloskey, K., Berndl, M., Pande, V., Riley, P.: Molecular graph convolutions: moving beyond fingerprints. J. Comput. Aided Mol. Des. 30(8), 595–608 (2016). https://doi.org/10.1007/s10822-016-9938-8
Kipf, T.N., Welling, M.: Semi-supervised classification with graph convolutional networks. arXiv:1609.02907 [cs, stat], September 2016. http://arxiv.org/abs/1609.02907
Li, Y., Tarlow, D., Brockschmidt, M., Zemel, R.: Gated Graph Sequence Neural Networks. arXiv:1511.05493 [cs, stat], November 2015. http://arxiv.org/abs/1511.05493
Schütt, K.T., Arbabzadah, F., Chmiela, S., Müller, K.R., Tkatchenko, A.: Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 8, 13890 (2017). https://doi.org/10.1038/ncomms13890
Wu, Z., et al.: MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 9(2), 513–530 (2018). https://doi.org/10.1039/C7SC02664A, https://pubs.rsc.org/en/content/articlelanding/2018/sc/c7sc02664a
Acknowledgements
The project leading to this article received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 676434, “Big Data in Chemistry” (“BIGCHEM”, http://bigchem.eu). The article reflects only the authors’ view, and neither the European Commission nor the Research Executive Agency are responsible for any use that may be made of the information it contains.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this paper

Cite this paper
Withnall, M., Lindelöf, E., Engkvist, O., Chen, H. (2019). Attention and Edge Memory Convolution for Bioactivity Prediction. In: Tetko, I., Kůrková, V., Karpov, P., Theis, F. (eds) Artificial Neural Networks and Machine Learning – ICANN 2019: Workshop and Special Sessions. ICANN 2019. Lecture Notes in Computer Science(), vol 11731. Springer, Cham. https://doi.org/10.1007/978-3-030-30493-5_69
Download citation
DOI: https://doi.org/10.1007/978-3-030-30493-5_69
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-30492-8
Online ISBN: 978-3-030-30493-5
eBook Packages: Computer ScienceComputer Science (R0)