
Abstract
Fluorescence lifetime imaging ophthalmoscopy (FLIO) is an emerging technology, which enables the time-resolved in-vivo measurement of fluorescence emitted by endogenous fluorophores within the retina. The decay time of the fluorescence is a characteristic parameter for fluorescent molecules and their environment, and therefore FLIO is a promising tool to detect and assess varying metabolic states of different areas in the retina. The system is based on a confocal scanning laser ophthalmoscope with an implemented real-time eye tracking system. The fluorophores are excited by picosecond laser pulses and the fluorescence emission is detected using time correlated single photon counting (TCSPC) technology.
In this chapter the historical and technical background of FLIO technology is described first, followed by the description how the technique was integrated into a modified Spectralis system. In the subsequent sections, selected results obtained in clinical studies conducted in Bern, Salt Lake City and Jena are presented. FLIO data acquired on patients with age-related macular degeneration (AMD), diabetic retinopathy, macular dystrophies, and other diseases are discussed in regard to the additional contrast and information provided in comparison to standard intensity-based autofluorescence images.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Fluorescence lifetime imaging ophthalmoscopy (FLIO)
- Time correlated single photon counting (TCSPC)
- Autofluorescence (AF)
- Macular pigment
- Age related macular disease (AMD)
- Retinal dystrophy
- Stargardt disease
- Macula telangiectasia
- Diabetic retinopathy
1 Introduction
Whereas sodium fluorescein, a fluorescent tracer to image and assess retinal vasculature and its integrity, is used since the 1960s [1], the intrinsic fluorescence of the human retina was first reported in the 1980s [2]. Delori were the first to record fundus autofluorescence (FAF) spectra from single retinal locations [3] and first images of FAF were recorded by von Rückmann et al. in the 1990s [4]. As lipofuscin, which accumulates in the retinal pigment epithelium (RPE) and is involved in the pathogenesis of age-related macular degeneration (AMD) , was found to be a major retinal fluorophore, subsequent FAF studies addressed this disease. FAF was used to describe the progression of geographic atrophy of the RPE [5, 6], and different patterns of FAF distribution were found [7,8,9]. Specific fluorescence patterns were assigned to sub-types of AMD and to their progression rate, but these characteristic patterns did not reveal any information about the present fluorophores which might be of pathogenetic relevance. In order to distinguish fluorophores, Schweitzer et al. developed fluorescence lifetime ophthalmoscopy (FLIO) , a method to measure the fluorescence decay time. FLIO is specific for fluorophores as well as their embedding matrix [10,11,12,13,14,15,16] and offers high temporal resolution. The lifetime of isolated RPE was determined as τm = 273.6 ps (τ1 = 210 ps, α1 = 96%, τ2 = 1800 ps, α2 = 4%) [16].
Although fluorescence lifetime measurement is considered a relatively new technique in biomedical imaging (see Berezin and Achilefu for review [17]), it has been discovered in the nineteenth century already. In 1859 Edmond Bequerel developed the so called phosphoroscope with a time resolution of 10−4 s. In the 1920s, time resolution was improved to 10−8 s which enabled the first fluorescence lifetime measurements [18, 19]. However, only the availability of short pulse lasers and the introduction of time correlated single photon counting (TCSPC) [20, 21] made fluorescence lifetime measurements sufficiently sensitive for the detection of intrinsic fluorophores in living tissue. Fluorescence lifetime imaging microscopy (FLIM) evolved based on two different techniques: (1) full field illumination and the use of gated or streak cameras, an approach pursued in frequency domain technique; and (2) the time-domain method in combination with confocal scanning laser microscopy. Specifically, two-photon excitation microscopy [22], using an inherently pulsed fluorescence excitation source, was used for FLIM investigations [23]. Whereas FLIM of intrinsic fluorophores gives detailed information on cell metabolism [24] and may detect malignant changes [25,26,27], the development of genetically expressed fluorescent proteins resulted in further progress in structural as well as functional imaging [28]. Another milestone in fluorescence microscopy was the introduction of Förster resonance energy transfer (FRET) enabling the detection of interaction between labeled molecules [29].
As ocular fundus autofluorescence was reported to be a possible indicator of retinal diseases [3, 30,31,32], Schweitzer et al. first applied fluorescence lifetime imaging to the human retina in vivo [12]. They fiber-coupled a mode-locked argon-ion laser to a scanning ophthalmoscope (cLSO, Carl Zeiss, Jena, Germany) and used TCSPC for fluorescence detection. However, the lack of an image registration algorithm limited the time available for recording an image without motion artifacts to a few seconds. This resulted in images with some hundred photons per pixel only. Despite the resulting low signal to noise ratio, first fluorescence lifetime images were recorded in 2001 [11]. An offline registration of recorded images was introduced in 2002 [13], and first clinical experiments in patients with AMD were published in 2003 using a picosecond diode laser as light source [14]. Although the resolution was still low due to limited memory of the TCSPC electronics (64 × 64 pixels with a size of 80 × 80 μm2), the images clearly revealed a prolongation of lifetimes in AMD [15]. Extensive in vitro and histological studies were performed to identify the fluorophores seen in FAF and to measure their fluorescence emission spectra as well as lifetimes [16, 33]. Considerable progress was made with the use of the Heidelberg Retina Angiograph scanner (Heidelberg Engineering, Heidelberg, Germany), enabling an online image registration [34]. An industrially designed prototype device based on the Heidelberg Engineering Spectralis platform was first used by Dysli et al. [35]. During the last 6 years, the diagnostic potential of this device referred as “Spectralis FLIO” was systematically explored in clinical studies in Bern, Jena and Salt Lake City focusing on different pathologies. The results can be found in a series of interesting and seminal publications [35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. Some selected highlights of the clinical results are summarized in the Sects. 10.3–10.6 of this chapter.
Within the living human retina , many different fluorophores can be found. A previous review article about FLIO describes a variety of retinal fluorophores in detail, with a focus on natural endogenous retinal fluorophores measured with FLIM [53]. A second review article also highlights retinal fluorophores important in FLIO [48]. Additionally, a broad compilation of lifetimes of endogenous fluorophores from the literature was given by D. Schweitzer previously [54]. Therefore, we keep the presentation of these substances in this article short and focus on a compact description of the most important retinal fluorophores.
In the context of retinal fluorescence, lipofuscin is a well-known assembly of fluorophores, which accumulate within RPE cells. It is uniquely contributing to the retinal fluorescence and is very well characterized. As the dominant fluorophore at the posterior pole, it emits fluorescence with high intensity [55]. It appears in almost all phagocytes. In the RPE of human eyes it is developed through oxidative processes in the degradation of photoreceptor outer segments [56]. Lipofuscin accumulation is a general sign of cell ageing. Therefore, the amount of lipofuscin increases with increasing age [57]. This mechanism possibly also causes a prolongation of AF lifetimes [35]. Another reason for prolonged FAF lifetimes with age could also be the ageing of the lens [35]. Lipofuscin was investigated by Eldred und Katz in 1988, and further studies were conducted to better understand the constituent parts of RPE granules [58, 59]. Sparrow et al. were able to identify at least 25 different bisretinoids within these granules [60]. Lipofuscin , with an excitation maximum around 340–395 nm, shows two emission maxima (430–460 nm and 540–640 nm) [61]. The main component of the fluorescence is emitted by the hydrophobic A2E. Maximal fluorescence is emitted at an excitation wavelength of 446 nm, and the emission maximum can be found at roughly 600 nm. It shows a mean autofluorescence lifetime of approximately 0.19 ns (τ1 = 0.17 ns, α1 = 98%; τ2 = 1.12 ns, α2 = 2%) [16]. It is assumed that A2E may damage cell membranes by releasing radicals in a photo-chemical reaction, and a relation of the molecule to the development of a variety of retinal diseases such as AMD was reported [62, 63]. However, the dominance of A2E on the development of retinal diseases has recently been discussed controversially, as it is possible that A2E iso-forms rather than A2E, or even other lipofuscin components could be involved in damaging the retina [64]. Additionally, it was shown that there is no relation between A2E and an increasing FAF intensity with increasing age [65]. Therefore, phototoxic theories regarding A2E are still under discussion [66].
Although the macula appears dark in standard intensity autofluorescence images due to the strong absorption of the blue light by the macular pigment (MP) , it has been shown with FLIO [37, 50] that the MP emits a weak but measurable fluorescence with very short lifetimes. The autofluorescence lifetimes of the carotenoids lutein and zeaxanthin were determined using ex vivo FLIO imaging. Lifetimes of around 50 ps were found for the fluorescence decay of these substances (see Sect. 10.3 for details).
Different redox equivalents such as nicotinamide-adenine-dinucleotide (NADH) , flavin-adenine-dinucleotide (FAD) und flavin-mononucleotide (FMN) may impact FAF lifetimes, as their fluorescence properties may depend on the redox state of tissues. NADH mostly shows fluorescence in the reduced form, and NAD+ (oxidized form) shows only a very weak fluorescence [67,68,69]. While free NADH in vitro shows autofluorescence lifetimes around 0.4 ns, protein-bound NADH can show decay times of 1.2 up to 5 ns [68]. Time-resolved fluorescence lifetime imaging almost always aims to detect NADH, which is believed to be a sensitive method to investigate the redox state of tissue [70,71,72,73,74]. However, it has been discussed that a contribution of NADH to the in vivo FLIO signal is unlikely, since the short-wavelength range required to match the fluorescence excitation maximum (350 nm) cannot be used for retinal imaging due to absorption by the lens and cornea [53, 75]. Nevertheless, other studies describe autofluorescence lifetimes of approximately 1.27 ns (τ1 = 0.39 ps, α1 = 0.73, τ2 = 3.65 ns, α2 = 0.27) upon an excitation at 446 nm [16]. Therefore, the effect of NADH on in vivo FLIO measurements is still under discussion and needs further investigation.
The fluorescence of oxidized flavin FAD, located in mitochondria, is also of interest. Both, FAD as well as FMN absorb light at 450 nm wavelength and show their maximal fluorescence emission at around 530 nm; reduced forms do not fluoresce under physiological conditions [67, 68]. Typical autofluorescence lifetimes of these substances are 2.3 ns (FAD) and 4.7 ns (FMN), while protein-bound flavins show intermediate autofluorescence lifetimes (0.3–1 ns) with a weak fluorescence intensity due to quenching [68]. Skala et al. determined an autofluorescence lifetime of protein-bound FAD of 100 ps [24]. In vitro FAF lifetime investigations show flavin decay times around 2.4 ns [16].
Different components of the extracellular matrix are found in the eye. Four different types of collagen as well as elastin were previously described and are likely fluorescent [16, 69, 76,77,78,79]. The collagens (I, II, III and IV) emit fluorescence at approximately 510 nm (excitation 446 nm) [80]. Time-resolved autofluorescence investigations show different lifetimes for each type of collagen (I: 1.75 ns; II: 1.44 ns; III: 1.11 ns; IV: 1.62 ns) [16]. The autofluorescence lifetime of elastin was described to be 1.28 ns [16].
The skin pigments melanin and bilirubin emit fluorescence at a maximum of 436 nm (melanin) and 520–540 nm (bilirubin) [16, 81]. As melanin occurs not only in the iris but also in the choroid and the RPE layer, it is of special interest when investigating FAF lifetimes. The excitation maximum of melanin, however, is at 360 nm, but also excitation at longer wavelengths can yield lower but measurable fluorescence intensities [82]. Time-resolved in vitro measurements at 446 nm excitation showed mean autofluorescence lifetimes of melanin powder at 916 ps (τ1 = 280 ps, α1 = 70%; τ2 = 2.4 ns, α2 = 30%) [16]. Considerably shorter lifetimes were determined for melanin dissolved in PBS (τ1 = 0.03 ns, τ2 = 0.62 ns, τ3 = 3.24 ns) [83].
The aromatic amino acids tyrosine , phenylalanine and tryptophan also fluoresce, but the excitation lies within the ultra-violet range (260–295 nm), and the emission maxima can be found between 280 and 350 nm [68]. Therefore, a fluorescence detection of these substances is unlikely with FLIO imaging.
Protoporphyrin IX , occurring within the cytochrome-c complex in mitochondria and as a by-product in the synthesis of hemoglobin, shows fluorescence at an emission maximum of 635 nm, with an autofluorescence lifetime in the nanosecond range [67]. Its detection was discussed in relation with tumorigenesis, as the amount of protoporphyrin may increase in proliferative tissue [23, 84, 85].
Pathological fluorophores, such as advanced glycation end products (AGEs) , also show fluorescence at the retina [16, 86]. They are discussed within the disease-related sections of this manuscript.
2 Technical Realization Based on the Spectralis Platform
While fluorescence lifetime imaging microscopy is already a well-established imaging technology, the transfer of this modality to in vivo imaging of the retina in ophthalmology involves several challenges. Examples are the low fluorescence yield at limited excitation power as well as unpredictable eye movements. However, with the Spectralis FLIO , a Spectralis variant which is released for clinical studies, these challenges have been solved. Within an acquisition time of typically 120–180 s reliable and reproducible in vivo FLIO data can be acquired on a patient eye with a dilated pupil.
The FLIO systems used in these clinical studies were developed and assembled on the basis of the Spectralis platform, a well-established imaging platform, which is widely used for multicolor, fluorescence (angiography and autofluorescence ), and OCT imaging on a daily basis in clinical routine. Recently, the Spectralis modalities were extended with a high resolution OCTA-mode (OCT angiography ), which allows to represent the vascular plexus in three dimensions without the invasive injection of a fluorescence dye (see Chap. 7 and references for further details).
In the Spectralis OCT a second completely independent scanning system is implemented for the OCT path in order to actively track and compensate for eye movements, which were detected by online processing of the continuously acquired SLO images and fed back to the OCT scanner control electronics. In contrast, in the Spectralis FLIO system both lasers, the infrared laser for the reference image and the picosecond laser for fluorescence excitation, are delivered to the retina by the same SLO scanning system . The eye movements are detected by online image processing of continuously acquired near infrared reference images, and the spatial assignment of each detected fluorescence photon is based on the preceding detected eye position. If an eye movement is detected within two consecutive reference images, the photons which were detected within this period and thus cannot unambiguously be assigned to a spatial position, are rejected.
The FLIO set-up is schematically represented in Fig. 10.1. The blue (470 nm) picosecond laser pulses are superimposed by means of beam splitter to the continuous, near infrared (815 nm) reflection laser and both beams are simultaneously deflected in X and Y directions in the SLO scanning unit, in a way that the retina is scanned line by line. The field of view for FLIO images is usually 30° × 30°, corresponding to an area of approximately 8.9 × 8.9 mm2 for emmetropic eyes. The repetition rate of the FLIO laser pulses is 80 MHz, i.e. the pulse separation of two consecutive pulses is 12.5 ns. The pixel clock for standard Spectralis SLO images is 10 MHz (100 ns pixel separation) in the high speed mode (HS-mode), which is normally used for the FLIO application. This means, that for each HS pixel of the infrared SLO image about 8 laser pulses are applied; the pulse width of each single pulse is in the order of 70–100 ps.

Left part: schematics of the optical set-up of the FLIO camera head and laser & detection unit. See text for detailed explanation. Right part: scan algorithm of IR tracking laser with superimposed picosecond laser pulses. Green resp. yellow circles represent photons detected in the short wave detection channel (SSC) resp. long wave detection channel (LSC)
The fluorescence light as well as the back scattered infrared light originating from the focus volume is then travelling the same optical path backwards, is “descanned” to a stationary beam, and deflected by another beam splitter towards the detection arm. A multimode fiber with core diameter of 100 μm serves as spatial aperture, resulting in a z-transfer point spread function of about 2 mm (FWHM). This confocal set-up provides an efficient suppression of out of focus light; in particular it efficiently blocks the intrinsic fluorescence of the lens tissue. If there was no efficient blocking of the strong fluorescence of the lens tissue with a mean lifetime of >2 ns, it would, for example, not be possible to measure the faster (≈50 ps) picosecond decay time of the weaker fluorescence of macula pigment. It cannot be completely excluded that for patients with very dense cataract, where on one hand the lens fluorescence signal is strong and on the other hand the retina signal is reduced due to the double-pass through the strongly scattering lens tissue, the result of the lifetime measurement of the retinal fluorophore s may be biased due to the contribution of the lens fluorescence. Further research is required to quantitatively assess this influence, e.g. by systematically measuring patients before and after intraocular lens (IOL) implantation.
The signal light is then guided via the multimode fiber out of the camera head and launched to the external detection unit. The fiber output is collimated and the beam is filtered by a blocking filter (F1) to remove back scattered light of the excitation laser at 470 nm. The back scattered infrared (IR) light is separated from the fluorescence photons by means of beam splitter plate (BSP 1) and then focused on an avalanche photodiode (APD) with high quantum yield in the near IR range. The APD-signal is digitized to 8 bits and then transferred to the computer. The real-time image processing algorithm calculates the correction parameters in form of an affine transformation, describing the eye movements with respect to the first frame during the measurement.
The fluorescence light path is split (BSP 2) into two branches: the short spectral channel (SSC: ≈498–560 nm) and the long spectral channel (LSC: ≈560–720 nm). Finally, the fluorescence photons are detected by two time-correlated single photon counting (TCSPC) detection units, each consisting of a hybrid detector, which combines a highly sensitive GaAsP photocathode with an avalanche-like multiplication of the photo-electrons as used in standard APD detectors and ultrafast read-out electronics. The time resolved measurement works as follows: each picosecond laser pulse triggers the start of two electronic time clocks, one for the short wave channel and one for the long wave channel. The time clock is stopped either by the detection of a fluorescence photon in the corresponding detection channel, or by the consecutive picosecond laser pulse triggering a new measuring period. Each detected photon is then assigned to a spatial location (XY-pixel) by considering the preceding result of the eye movement detection algorithm and is provided with a time stamp, indicating the elapsed time between laser trigger and detection of the photon.
The maximum laser power is 300 μW, which corresponds at 80 MHz laser repetition to a pulse energy of 3.75 pJ. Since the laser is modulated off during the resetting periods of the horizontal and vertical scanners, only a mean average power of 200 μW is measured in front of the objective and applied to the patients retina. For a rigorous laser safety classification corresponding to the IEC standard 60825-1:2014 the action of a single pulse (class 1 limit in the standard: 77 nJ) as well as the accumulative action of consecutive pulses within a defined angular subtense of (1.5 mrad)2 must be assessed for different time regimes according to the three rules described in Sect. 4.3.f of the IEC standard. For all considered cases, the accessible emission for the FLIO system (AEFLIO) is below 1% of the corresponding accessible emission limit for class 1 systems (AELclass 1). Thus, the Spectralis FLIO is classified as class 1 laser product and is safe for examinations on humans. See also the published laser safety assessment in Ref. [37] (still referring to the IEC standard 60825-1:2007 with slightly different limits) and in the supplement of Ref. [39].
It might be of interest to compare the FLIO laser exposure with the exposure during fluorescence angiography (FA) with the standard Spectralis. The laser power of the Spectralis FA-mode is ≈1.4× higher compared to the FLIO. However, since the laser wavelength 470 nm is ≈2.1× more critical as the wavelength 486 nm used in the Spectralis FA mode (IEC Standard: factor C3 is 2.51 for 470 nm and 5.25 for 486 nm), the accessible emission of the FLIO mode is according to the Standard slightly higher (≈1.5×) but comparable to the exposure of the Spectralis FA-mode.
Since the maximum count rate of the detection units is 10 MHz, the laser power applied to the patient’s eye often has to be reduced by adding neutral density filters just in front of the single-mode fiber coupler, in order to avoid an overload of the detector.
During the examination for each XY pixel position a histogram is build up, and the number of photon counts is sorted against their arrival time with respect to the precedent laser pulse. The Spectralis FLIO acquisition software bins 3 × 3 high speed pixels (in total 768 × 768 pixels acquired at 10 MHz pixel rate for IR reference image) to one FLIO super pixel , so that the FLIO images consists of 256 × 256 pixels. Thus, a mean detection rate of typically 2 MHz results in the collection of about 3 × 108 Photons during 150 s (typical examination time is 2–3 min), resulting in a mean number of about 4500 photons per FLIO pixel. The measurement usually is stopped after about 1000 photons/histogram have been collected in the darkest pixels within the area of interest (typically within the macula), since this minimum number of counts still allows for evaluating the mean lifetime with sufficient accuracy. Finally, the acquired pixel histograms are saved to the hard drive.
Different software can be used to approximate the autofluorescence decay, such as FLIMX [87] or SPC Image (Becker & Hickl GmbH) [88], the latter being the most commonly used software in FLIO investigations. The photon arrival decay in each of the 65,536 pixels is hereby approximated according to:
Here ⨂ indicates the convolution integral with the instrument response function (IRF). Bi- and tri-exponential decays were previously used to investigate the retinal fluorescence in vivo. A tri-exponential approach leads to three different lifetimes (τ1, τ2, and τ3) as well as three corresponding amplitudes, a bi-exponential approach leads to two different lifetimes (τ1 and τ2) as well as two corresponding amplitudes. The amplitudes represent the contribution of each component to the total fluorescence decay.
As the FAF in vivo typically does not completely decay within 12.5 ns (time between two excitation pulses upon 80 MHz laser repetition rate), the application of an incomplete multi-exponential decay mode is standard. The digital time resolution is given by the temporal width of the histogram binning and is 12.2 ps (interval between two laser pulses 12.5 ns divided by number of channels 1024). However, since at least three supporting points are necessary for the reconstruction of a decay, the shortest detectable lifetimes are about 30 ps [37]. For further data analysis, image processing, and especially to average FAF lifetimes over certain regions of interest, different software packages have been used. Common is the FLIO-reader (ARTORG Center for Biomedical Engineering Research, University of Bern, Bern, Switzerland) and the software FLIMX (Institute of Biomedical Engineering and Informatics, Technische Universität Ilmenau, Ilmenau, Germany) [87].
3 Clinical Applications I: The Healthy Eye
Retinal autofluorescence imaging is a commonly used tool in ophthalmology for clinical investigation and research. In addition to fundus autofluorescence intensity imaging, which derives contrast mainly from lipofuscin and its derivatives, fluorescence lifetime imaging has the potential to provide additional contrast, as fluorescence lifetimes are largely independent of the fluorophores concentration and intensity. In the last decade, fluorescence lifetime imaging has gathered momentum through the research efforts and findings of several groups, and has contributed to the understanding of the pathophysiology of the healthy eye as well as retinal disorders ranging from degenerative diseases to retinal dystrophies and other diseases that affect the retina.
The FLIO signal in healthy eyes is very well established and has been shown to be highly reproducible between different groups. In the 30° FLIO field centered at the macula, shortest lifetimes can be found right at the center of the fovea. Intermediate lifetimes can be detected all across the retina and longest decay times can be found at the optic disc. Schweitzer et al. first published this typical pattern using an experimental FLIO device before it was coupled to a Spectralis [15]. The group of Dysli et al. in Bern [35] were the first to show this pattern in healthy eyes using the Spectralis FLIO, followed by Sauer et al. in Jena as well as Klemm et al. in Ilmenau. In the process of investigating the FLIO signal in healthy eyes, it was possible to show an individual fluorescence of the macular pigment. Here this finding is highlighted.
3.1 Macular Pigment
In the healthy eye, the anti-oxidative carotenoids lutein, zeaxanthin and meso-zeaxanthin accumulate in the center of the macula [89, 90]. Thus, they are called macular pigment (MP) . Whereas the amount lutein and zeaxanthin in the MP depends in part on dietary intake, meso-zeaxanthin is formed inside the retinal pigment epithelium from lutein by the RPE [91]. Based on the existence of highly specific binding proteins at the area of the fovea, MP accumulates at an area of 0.5 mm in diameter at the posterior pole of the eye [92,93,94,95,96,97,98]. These carotenoids are most concentrated in the foveal Müller cells and the Henle fiber layer, but also in other inner retinal layers [99, 100].
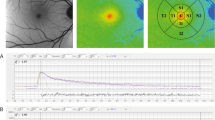
MP is believed to protect the eye from light damage, especially within the blue-light range around 460 nm [92, 101, 102]. MP may absorb blue light, which is potentially photo-toxic, before it reaches the photoreceptor layer; it may also quench free radicals [100, 103,104,105]. Levels and distribution can be altered in different retinal diseases such as macular telangiectasia type 2 (MacTel) as well as age-related macular degeneration (AMD) . In healthy eyes, different distribution patterns have been described. MP can show a slim cone-like distribution, a broader plateau or even ring-like distributions within the 1 mm ring (Fig. 10.2).

Healthy eyes with different characteristic MP distribution patterns
On fundus autofluorescence intensity images, MP appears as a dark spot in the center of the fovea because of its absorption of the blue excitation light [106]. Therefore, it was believed to only absorb blue wavelength light and not show any fluorescence characteristics. Based on this assumption, dual wavelength autofluorescence at blue (absorption) and green (non-absorption) wavelength light are used to calculate individual amounts of MP . However, a resonance-Raman-based study demonstrated that MP shows fluorescence characteristics, albeit with low quantum efficiency [107, 108]. As fluorescence lifetimes are independent of the fluorescence intensity, FLIO can also detect the fluorescence of carotenoids [37]. This was first described by Sauer et al., who found a strong correlation between mean fundus autofluorescence lifetimes and the amount of MP [37]. The short autofluorescence lifetimes at the fovea, depicted in red color in FLIO images, were therefore attributed to retinal carotenoids (Fig. 10.2).
A further study investigated autofluorescence lifetimes in patients with macular holes [44]. Interestingly, the distribution of MP and short autofluorescence lifetimes were identical. In the center of macular holes, where no MP could be localized, the short autofluorescence lifetimes were absent. These were found adjacent to the macular hole, corresponding to the MP distribution. This study also describes a follow-up of patients before and after successful vitreoretinal surgery with closure of the macular hole [44]. It was reported that short autofluorescence lifetimes corresponding to the macular pigment, migrate back to the fovea as the macular hole was closed. Furthermore, the increase of short autofluorescence lifetimes in the fovea after surgery was described to result in a better visual outcome for the patients.
A different FLIO study investigated fundus autofluorescence lifetimes in patients with albinism [50]. These patients usually do not have a foveal depression, which is often described as foveal hypoplasia. Interestingly, these patients do not have macular pigment accumulation at the posterior pole. Two patients were examined, and the albinism was electro-physiologically confirmed based on reversed pattern onset of visual evoked potentials (VEP) across the occipital scalp, which is indicative of optic misrouting associated with albinism. Only one of the four investigated eyes showed small amounts of MP , consistent with a small area of short autofluorescence lifetimes. The other three eyes did not show any macular pigment, and short autofluorescence lifetimes were absent from the fovea.
Finally, ex vivo measurements were performed on the carotenoids lutein and zeaxanthin [50]. These measurements confirm the weak fluorescence at short lifetimes of carotenoids with FAF lifetimes of 50 ps for lutein and 60 ps for zeaxanthin in the SSC. Furthermore, it was reported that in combination with binding proteins, autofluorescence lifetimes showed prolonged means.
Overall, FLIO will probably not replace existing methods for MP measurement, but it may give additional interesting insights about it. Furthermore, to investigate MP with FLIO, there is no need for a reference region to calculate the amounts of MP . This may be especially helpful in diseases such as retinitis pigmentosa and choroideremia, as well as in geographic atrophy in end-stage AMD. Here, retinal degenerations affect the reference areas of the MP measurement and calculation of the MP amount is therefore not possible. FLIO may fill the gap to assess MP in these patients, where other methods fail to give reliable measures.
4 Clinical Applications II: AMD and Retinal Dystrophies
4.1 Age-Related Macular Degeneration
Age-related macular degeneration (AMD) is one of the major causes of vision loss in the elderly population [109]. Besides genetic predisposition, several other factors such as diet, hypertension, arteriosclerosis, elevated serum lipids, smoking, alcohol abuse, and exposure to ultraviolet light have been identified [110,111,112,113,114].
Several stages of AMD can be identified: early, intermediate and advanced AMD [115, 116]. Hallmarks of AMD are retinal drusen and retinal pigment epithelium (RPE) abnormalities like hypo- or hyperpigmentation. Several subgroups of drusen exist such as soft drusen, hard drusen, cuticular drusen, crystalline drusen, and reticular pseudodrusen [110, 117]. Retinal drusen are focal deposits of extracellular debris situated between the basal lamina of the RPE and the inner collagenous layer of Bruch membrane . Soft drusen consist of lipid rich material and other constituents such as zinc, oligosaccharides, amyloid, apolipoproteins , and complement factors [118, 119]. Reticular pseudodrusen have similar constituents as soft drusen [110, 120]. The advanced stage of AMD is characterized by either geographic atrophy of the RPE involving the foveal center or neovascular maculopathy or a combination of both.
Major changes in several fluorophores have been identified in all stages of AMD . The most important change is accumulation of lipofuscin within the RPE, and in geographic atrophy absence of lipofuscin [110]. Fundus autofluorescence (FAF) has been used to quantify lipofuscin in the RPE, and identify areas of geographic atrophy which appear as hypo-fluorescent lesions in FAF.
In the past decade, several studies have investigated fluorescence lifetime patterns in various stages of AMD using FLIO. Fluorescence lifetime measurements derive their signal not only from lipofuscin but also from many other endogenous fluorophores such as visual cycle end products like retinal derivates, and therefore offer the ability to resolve retinal abnormalities at early stages of the disease. Most of these studies employed a modified Spectralis system (Heidelberg Engineering) using a 470 nm pulsed laser (80 MHz; <100 ps pulse width) and highly sensitive hybrid detectors for time correlated single photon counting. Two separate channels are used: A short spectral channel (SSC; 498–560 nm) and a long spectral channel (LSC: 560–720 nm).
Recent studies have shown that mean retinal autofluorescence lifetimes of the macula in patients with AMD are generally significantly prolonged [15, 41, 43]. This prolongation was found to occur in a ring-shaped manner, particularly visible in the LSC with colors set from 300 to 500 ps [51]. If color ranges are set differently, the pattern may be difficult to be observed. The prolongation occurs in the area between the large arcade vessels and is most pronounced at the nasal and temporal macula. Figure 10.3 shows the pattern. The pattern may indicate the first sign of AMD , as a trace pattern was also found in patients at high risk to develop the disease.

FLIO and FAF intensity images from two healthy individuals and two patients with AMD. (Reprinted from [51])
Areas of retinal drusen, however, are heterogeneous and can have shortened, normal or prolonged autofluorescence lifetimes. This may be a result of different forms of AMD . A study that investigated drusen in non-exudative AMD only did not find shortened autofluorescence lifetimes [51]. However, drusen with very short fluorescence lifetimes may represent newly formed deposits [40]. Longitudinal studies are needed to show whether fluorescence lifetime features change within individual drusen over time and whether this may help to identify newly formed drusen. In intermediate AMD , areas of intraretinal hyper-reflective deposits (possibly melanolipofuscin) display long lifetimes whereas deposits within the photoreceptor outer band display relatively short lifetimes.
In patients with geographic atrophy due to advanced AMD , fluorescence lifetimes are significantly prolonged both in the SSC and the LSC within areas of atrophy (Fig. 10.4) [41, 49]. In the border zone of geographic atrophy, where characteristic FAF distribution of hyper-fluorescence can be observed [121], distinct patterns of only marginally prolonged fluorescence lifetimes were observed [41]. In many cases with geographic atrophy, areas with very short fluorescence lifetimes can be observed in the foveal area . These short lifetimes might originate from residual macular pigment within the outer nuclear and plexiform layers [37]. A correlation between these short lifetimes in both spectral channels in the fovea and BCVA was shown [41]. Short fluorescence lifetimes within the macular center therefore may provide useful information about the integrity of the foveal photoreceptors.

Fundus autofluorescence imaging in a patient with geographic atrophy due to age-related macular degeneration. Fundus autofluorescence image (FAF) and color coded image of fluorescence lifetime imaging ophthalmoscopy (FLIO). The lifetimes within the atrophy are prolonged; however, in the foveal center short lifetimes persist. These short lifetimes have been correlated with the presence of macular pigment
Neovascular AMD as the second form of advanced AMD is characterized by the presence of choroidal neovascularization (CNV). According to the localization of the CNV complex neovascular AMD can be classified into type 1 (below the RPE) or type 2 (above the RPE) CNV [116]. In pilot studies, there was only minimal contrast of the CNV complex in fluorescence lifetime imaging (unpublished data). Areas of CNV display only slightly prolonged fluorescence lifetimes. In the active stages of this disease, hyporeflective areas of intra- or subretinal fluid can be seen in optical coherence tomography (OCT) . These areas of intra or subretinal fluid are not directly identifiable in fluorescence lifetime maps acquired by FLIO (unpublished data).
4.2 Retinal Dystrophies
Fundus autofluorescence intensity measurement has emerged as one of the key tools for noninvasive retinal imaging in retinal dystrophies for diagnostic purposes as well as for follow-up examinations [122]. In association with upcoming genetic specification and differentiation as well as emergent trials addressing genetic modification in retinal dystrophies [123,124,125], imaging modalities to record subtle changes in retinal metabolism and structures are essential.
Retinitis pigmentosa summarizes a genetically heterogeneous group of degenerative retinal diseases with different inheritance patterns and penetrance. Progressive rod followed by cone dysfunction clinically leads to primary night blindness and progressive constriction of the visual field [126]. In FAF intensity measurement a hyperfluorescent ring may be identified, delineating the border between morphologically intact retinal layer structure and altered outer retinal layers [127]. Using fluorescence lifetime imaging , ring characteristic structures with specific grades of degeneration can be identified: intact retina, photoreceptor atrophy, and combined photoreceptor and RPE atrophy (Fig. 10.5a) [47, 52]. This lifetime pattern may allow for more differentiated clinical assessment of the patients and their follow-up examination over time.

Fundus autofluorescence imaging in patients with retinal dystrophies. (a) Patient with retinitis pigmentosa, (b) patient with Stargardt disease, (c) patient with choroideremia (FAF fundus autofluorescence intensity image, FLIO fluorescence lifetime imaging ophthalmoscopy)
Stargardt disease is the most common monogenetic juvenile retinal dystrophy, inherited in an autosomal recessive pattern [128]. Due to a mutation in the ABCA4 gene, coding for the ABCA4 transmembrane transporter in the photoreceptor outer segments, visual cycle byproducts accumulate, leading to progressive dysfunction and destruction of the outer retina and the RPE [129]. Clinically, progressive accumulation of yellowish retinal deposits is visible which appear as hyperfluorescent flecks using FAF intensity measurement. In advanced disease stage, RPE atrophy manifests as hypoautofluorescence in FAF intensity images. Fluorescence lifetime measurement in Stargardt disease revealed that hyperfluorescent flecks may feature shorter or longer lifetimes compared to the surrounding retina (Fig. 10.5b) [40]. In a subgroup of patients, areas and flecks with shorter fluorescence lifetimes have been identified even before they were visible in the FAF intensity measurement . Over time, they appear in FAF and the fluorescence lifetimes become gradually longer. In areas of RPE atrophy, generally prolonged fluorescence lifetimes were observed.
Choroideremia is a rare monogenetic retinal dystrophy with an x-linked inheritance pattern, thus affecting mainly young male subjects [130]. It is caused by a mutation in the CHM gene, coding for a protein responsible for membrane trafficking in the retina and the RPE . Clinically, progressive degeneration of the choroid, the RPE and the neurosensory retina is observed, leading to progressive impairment of visual function, and finally complete blindness [130]. Whereas FAF intensity sharply delineates the borders of the RPE, FLIO provides additional information within the area of RPE atrophy (Fig. 10.5c) [40]. Thereby, areas with remaining RPE feature the shortest lifetimes, followed by RPE atrophy and remaining photoreceptor layers in OCT, and complete atrophy of RPE and the outer nuclear layers with the longest fluorescence lifetimes. Follow-up examinations in patients with choroideremia have shown that FLIO is a sensitive tool to monitor subtle changes of retinal degeneration over time.
In summary, FLIO enables detection and identification of early disease associated changes on the level of the RPE, the outer retinal layers, and possibly also the choroid. In Stargardt disease, retinitis pigmentosa and choroideremia we have shown that FLIO provides supplementary information in addition to commonly used standard imaging modalities such as FAF intensity measurement, color fundus imaging, and OCT.
5 Clinical Applications III: Macula Telangiectasia
5.1 Macular Telangiectasia
Macular Telangiectasia type 2 (MacTel) is an inherited retinal disease with an onset of about 40–60 years, but cases of younger patients have also been reported [131,132,133]. The youngest affected patient was diagnosed at the age of 21. Although patients usually do not proceed to legal blindness, vision is often significantly disturbed. Patients initially report metamorphopsia and difficulties with reading, which proceeds to disturbance in the vision affecting the daily life [134]. MacTel affects an oval-shaped area of approximately 5°–6° from the foveal center. Macular pigment can be found around the MacTel area with an eccentricity of 5°–9° instead of in the central fovea [101, 135,136,137,138].
Initially, it was believed that MacTel is a rare disease, but as researchers find out more about the disease characteristics and doctors learn to distinguish MacTel from other retinal diseases such as AMD, it is believed that the disease prevalence is much higher than initially assumed [139, 140]. So far, despite intensive research, no causative gene has been found for MacTel but a dominant genetic inheritance with reduced penetrance is likely [141,142,143,144,145]. To truly distinguish it from other retinal diseases such as AMD which may show similar features to those of MacTel , retinal imaging plays a very important role. Several imaging modalities have been used to describe features of MacTel [131, 146,147,148]. Fundus photography may show retinal greying, a feature that is often difficult to truly distinguish. In OCT imaging, retinal cysts and ellipsoid-zone loss may be observed at the temporal side of the fovea [149, 150]. However, these cysts may also in some cases be found at the nasal side, or may be absent especially in early disease stages. Furthermore, these cysts may be mistaken as changes caused by neovascular AMD. Blue-light reflectance imaging was described to show changes related to MacTel , but also current results are not satisfying. Autofluorescence imaging is often relatively normal in early stages, except for the decrease of the central hypofluorescence, leading to a hyperfluorescent macular area. Macular pigment levels are often reduced in initial stages of the disease and can evolve to ring-like distributions in later stages [151]. Fluorescein angiography is usually able to show leakage indicative of MacTel, but non-invasive imaging modalities would be preferred. Recently, FLIO has emerged as a novel and non-invasive tool to detect changes in MacTel with extremely high contrast [45]. It highlights the MacTel area in affected individuals, and especially in early stages, shows a temporal, crescent shaped prolongation of FAF lifetimes (Fig. 10.6). Especially the SSC in FLIO imaging seems to highlight MacTel-related changes. A standardized grid (ETDRS grid) was used to characterize different areas of the fundus; the area corresponding to the MacTel region (T1) showed significantly prolonged FAF lifetimes as compared to the reference region (T2), whereas in healthy eyes, T2 showed slightly longer fluorescence decays. Based on this finding, a ratio was established to quantify definite MacTel from definite healthy. If T2/T1 is larger than 1.0, the person is likely healthy and a ratio below 0.9 (in combination with typical FLIO findings) likely indicates MacTel. This ratio, as well as the images obtained with FLIO, may help to identify patients with MacTel . Furthermore, FLIO may also indicate affected individuals in a stage where they clinically still show a healthy fundus exam [45]. This was already shown for clinically unaffected parents of MacTel patients and is currently being investigated for the second generation (children of MacTel patients, unpublished data). The earliest changes visible with FLIO, however, seem to be not at the temporal side of the fovea but rather superiorly, again presenting as prolonged FAF lifetimes.

FLIO and FAF intensity images for a healthy person and a MacTel patient
Over all, FLIO is a novel tool for the detection of MacTel and likely gives the best contrast of all non-invasive imaging modalities. It highlights the MacTel area and may be capable of indicating MacTel-related retinal alterations at the earliest stages. Although the availability of FLIO is currently still limited, it is likely to emerge as a very helpful tool for the detection of MacTel, especially at its earliest stages.
6 Clinical Applications IV: Diabetic Retinopathy
Diabetic retinopathy is a micro-vascular complication in diabetes [152]. Micro-vasculopathy and inflammation finally result in neuronal degeneration [153] and a breakdown of the blood–retina barrier (BRB), causing retinopathy and macular edema [154]. As hyperglycemia is a primary event in diabetes, this causes not only an impairment of the vascular endothelium but also a general protein glycation. This formation of advanced glycation end products (AGEs) in the non-enzymatic Maillard reaction of proteins with glucose and other sugar molecules is involved in BRB breakdown [154]. In addition, endothelial dysfunction and the protein-kinase C pathway may play a role [155]. As protein glycation is a process generally taking place in ageing tissue and predominantly affecting long-living proteins , it is greatly enhanced in diabetes mellitus [156]. It correlates with the level as well as the duration of hyperglycemia [157]. Protein glycation comprises several steps. First, Schiff’s bases are formed in a reaction of the Aldehyde- and Ketone groups of sugars with amino groups of the proteins. This is followed by the Amadori rearrangement finally resulting in the AGE. This is further enhanced by highly reactive Carbonyl groups of intermediates, such as α-Oxoaldehyde , Glyoxal , and Methylglyoxal [158]. These intermediates are not only generated by the Maillard reaction but also by other pathways such as auto-oxidation of sugars and glycolysis [159]. Oxidative end products , such as Pentosidin and N-Carboxymethyllysin, as well as non-oxidative AGEs (e.g. Hydroimidazolone and Pyrralin ) are distinguished [158, 160]. A well-known AGE is the glycated Hemoglobin HbA1C which is used clinically for long-term monitoring of diabetes [156, 161,162,163]. Upon hyperglycemia, AGEs accumulate in the lens, the cornea, the vitreous, and the retina of the eye. Thus, AGEs contribute to diabetic retinopathy in different ways. They may damage the vascular endothelium and, subsequently, also affect the pericytes. This leads to a disruption of the BRB and can result in diabetic macular edema, one of the most sight-threatening complications of diabetes [164, 165]. Furthermore, AGEs have procoagulant potential contributing to capillary occlusion which is typical for diabetic retinopathy [158]. However, neuronal cells are also directly affected [166]. Animal experiments showed AGE deposition in the vascular as well as in the neuronal compartment of the retina [160]. Protein crosslinking , namely the covalent binding of Lysine residues, alters the tertiary structure of proteins and, thus, impairs their function. However, the modified proteins are able to bind to receptors for advanced glycation end products (RAGE) which is expressed by various cell types such as macrophages, monocytes, endothelial cells, glial cells, and neurons. Besides inflammatory reactions [167], this results in the secretion of cytokines, adhesion molecules, and growth factors like vascular endothelial growth factor (VEGF) [164]. VEGF stimulates neovascularization, which is the diagnostic criterion for proliferative diabetic retinopathy. Finally, the activation of the RAGE may exert oxidative stress to the cells by generation of reactive oxygen species (ROS) leading to neuronal cells death [168].
Changes of FLIO lifetimes in healthy subjects with different states of glucose were previously described by Klemm and coworkers [169]. Additionally, AGEs seem to show a fluorescence, and their concentration in serum was found to increase with the severity of diabetic retinopathy [170]. As increased fundus autofluorescence (FAF) has been found in diabetic macular edema in association with decreased macular sensitivity [171], Schweitzer et al. [38] and Schmidt et al. [46] investigated fluorescence lifetimes in diabetic patients.
Schweitzer et al. [38] compared the fluorescence decay upon a 448 nm excitation for a group of 48 patients suffering from type 2 diabetes without retinopathy to 48 healthy control subjects of same age. They found a general prolongation in the fundus autofluorescence lifetimes in diabetic eyes. Using a three-exponential fit of the decay and a sophisticated statistical procedure, they revealed a good discrimination of both groups with a sensitivity of 73% and 70% as well as a specificity of 84% and 64% for the two spectral channels (490–560 nm and 560–700 nm) respectively for the mean fluorescence lifetime τm. The best discrimination, however, was achieved by the intermediate decay time component τ2 at 490–560 nm (sensitivity 84%, specificity 76%), which the authors assigned to fluorophores in the retina. They discuss this as potentially being a result of reduced protein binding of FAD, as well as protein glycation that may lead to an accumulation of AGEs. In a subgroup analysis, they found a considerably better discrimination in phakic patients and controls as in pseudo-phakic eyes. Thus, they concluded an influence of the lens fluorescence on the measurements at the fundus despite the use of a confocal scanning laser system. This is due to the extremely strong fluorescence emission from the lens. An accumulation of AGEs in the lens is well known [172], this could in part account for the prolonged lifetimes measured in diabetic retinas.
Schmidt et al. [46] extended this study to patients with diabetic retinopathy. They compared fluorescence lifetimes upon excitation at 470 nm in 34 patients suffering from non-proliferative diabetic retinopathy (NPDR) with that of 28 age-matched healthy controls. An example of a patient with diabetic retinopathy is given in Fig. 10.7. Fluorescence lifetimes were recorded in the macula and at two concentric annuli given by the standard ETDRS grid (Fig. 10.7, middle left), data from a three-exponential fit of the decays was used.

Mean funds autofluorescence (FAF) lifetime images (FLIO) from two spectral channels, as well as and FAF intensity images from the retina of a healthy control (left) and a diabetic retinopathy patient (right). Middle left panel comprises a standardized ETDRS grid
Consistent with Schweitzer et al., they showed increased lifetimes in the patient group in all investigated retinal fields (Fig. 10.8). This holds true for both spectral channels, however, was more pronounced in the SSC (498–560 nm, p ≤ 0.002) than in the LSC (560–700 nm, p < 0.05). A ROC analysis using a logistic regression model resulted in a sensitivity of 90% and a specificity of 71% for the discrimination of NPDR patients . In contrast to Schweitzer et al., Schmidt et al. found the best discrimination for the long-living fluorescence component τ3 instead of τ2. This might result from the longer excitation wavelength used. Again, the formation of AGEs in neurons, vascular, and glial cells was discussed as source of the prolongation of lifetimes. This was corroborated by FLIO measurements at the lenses of the subjects. These showed shorter lifetimes in the patients, again predominantly in the SSC. As AGE (bovine serum albumin incubated with glucose) showed a decay time of 1.7 ns and an emission maximum of 523 nm [16], its accumulation must increase the physiologically shorter fundus autofluorescence lifetime, but decrease that of the lens which is known to be longer in healthy state. The assumption of AGEs as source of an additional fluorescence from the ocular fundus is corroborated by the finding of a correlation of the abundance of the intermediate lifetime component with the HbA1C value of the patients (SSC: p = 0.009 and LSC: p = 0.016).

Mean fluorescence lifetime of the fundus of NPDR patients (green) and controls (blue) in different regions of the retina (c = central, is = inner superior, os = outer superior segment of the ETDRS-grid in the short wavelength channel (498–560 nm))
In conclusion, these investigations indicate that FLIO has the potential to show protein glycation as well as alterations in coenzymes of the cellular energy metabolism that are associated with diabetes. This might help elucidate pathways leading to diabetic retinopathy and, thus, provides opportunities for differential diagnostics with the option of individualized therapy.
7 Conclusion and Summary
FLIM (fluorescence lifetime imaging microscopy) is a well-established technology in the field of microscopy, which provides additional information on the temporal characteristics of the fluorescence decay. In the last 10 years, this technology has been transferred to be used in ophthalmology (FLIO), aiming for a better understanding of the nature of the endogenous fluorophores within the retina and their role and changes during the evolvement of retinal pathologies. The FLIO modality was evaluated in several investigational studies on patients with different retinal diseases and it has been shown, that reliable and reproducible data can be acquired in a clinical setting. In many aspects a good correlation of FLIO data with other existing imaging modalities was shown, and in some diseases the lifetime contrast could provide an earlier or more reliable diagnosis and a finer grading of the stage of the disease compared to standard autofluorescence imaging or OCT modality.
References
Novotny HR, Alvis DL. A method of photographing fluorescence in circulating blood in the human retina. Circulation. 1961;24:82–6.
Teich JM. The theory and development of a noninvasive retinal fluorescence scanner with application to early diagnosis of diabetic retinopathy. MIT: Cambridge; 1985.
Delori FC. Spectrometer for noninvasive measurement of intrinsic fluorescence and reflectance of ocular fundus. Appl Opt. 1994;33(31):7439–52.
von Ruckmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol. 1995;79(5):407–12.
Schmitz-Valckenberg S, et al. Correlation between the area of increased autofluorescence surrounding geographic atrophy and disease progression in patients with AMD. Invest Ophthalmol Vis Sci. 2006;47(6):2648–54.
Schmitz-Valckenberg S, et al. Semiautomated image processing method for identification and quantification of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(10):7640–6.
Bindewald A, et al. Classification of fundus autofluorescence patterns in early age-related macular disease. Invest Ophthalmol Vis Sci. 2005;46(9):3309–14.
Bindewald A, et al. Classification of abnormal fundus autofluorescence patterns in the junctional zone of geographic atrophy in patients with age related macular degeneration. Br J Ophthalmol. 2005;89(7):874–8.
Einbock W, et al. Changes in fundus autofluorescence in patients with age-related maculopathy. Correlation to visual function: a prospective study. Graefes Arch Clin Exp Ophthalmol. 2005;243(4):300–5.
Schweitzer D, et al. Tau-mapping of the autofluorescence of the human ocular fundus. Proc SPIE. 2000;4164:79–89.
Schweitzer D, Kolb A, Hammer M. Autofluorescence lifetime measurements in images of the human ocular fundus. Proc SPIE. 2001;4432:29–39.
Schweitzer D, et al. Basic investigations for 2-dimensional time-resolved fluorescence measurements at the fundus. Int Ophthalmol. 2001;23:399–404.
Schweitzer D, et al. Zeitaufgelöste Messung der Autofluoreszenz - ein Werkzeug zur Erfassung von Stoffwechselvorgängen am Augenhintergrund. Der Opthalmologe. 2002;99(10):774–9.
Schweitzer D, et al. Evaluation of time-resolved autofluorescence images of the ocular fundus. In: Diagnostic Optical Spectroscopy in Biomedicine II, Munich; 2003.
Schweitzer D, et al. In vivo measurement of time-resolved autofluorescence at the human fundus. J Biomed Opt. 2004;9(6):1214–22.
Schweitzer D, et al. Towards metabolic mapping of the human retina. Microsc Res Tech. 2007;70(5):410–9.
Berezin MY, Achilefu S. Fluorescence lifetime measurements and biological imaging. Chem Rev. 2010;110(5):2641–84.
Gottling PF. Determination of the time between excitation and emission for certain fluorescent solids. Phys Rev. 1923;22:566.
Gaviola E. The dacay-time of dye stuff fluorescence. Ann Phys. 1926;81:681.
Leskovar B, et al. Photon-counting system for subnanosecond fluorescence lifetime measurements. Rev Sci Instrum. 1976;47(9):1113–21.
Lewis C, et al. Measurement of short-lived fluorescence decay using single photon-counting method. Rev Sci Instrum. 1973;44(2):107–14.
Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248(4951):73–6.
Cubeddu R, et al. Fluorescence lifetime imaging: an application to the detection of skin tumors. IEEE J Sel Top Quantum Electron. 1999;5(4):923–9.
Skala MC, et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci U S A. 2007;104(49):19494–9.
Walsh AJ, et al. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res. 2013;73(20):6164–74.
Walsh AJ, et al. Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res. 2014;74(18):5184–94.
Walsh AJ, et al. Temporal binning of time-correlated single photon counting data improves exponential decay fits and imaging speed. Biomed Opt Express. 2016;7(4):1385–99.
Winkler K, et al. Ultrafast dynamics in the excited state of green fluorescent protein (wt) studied by frequency-resolved femtosecond pump-probe spectroscopy. Phys Chem Chem Phys. 2002;4(6):1072–81.
Chen Y, Periasamy A. Characterization of two-photon excitation fluorescence lifetime imaging microscopy for protein localization. Microsc Res Tech. 2004;63(1):72–80.
Delori FC, et al. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Investig Ophthalmol. 1995;36:718–29.
von Rückmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol. 1995;79:407–12.
von Rückmann A, Fitzke FW, Bird AC. Clinical application of in vivo imaging of fundus autofluorescence. Investig Ophthalmol. 1995;36(4):238.
Schweitzer D, et al. Interpretation of measurements of dynamic fluorescence of the eye. Boston, MA: SPIE; 2007.
Hammer M et al. In-vivo and in-vitro investigations of retinal fluorophores in age-related macular degeneration by fluorescence lifetime imaging. In: SPIE Photonics West. SPIE; 2009.
Dysli C, et al. Quantitative analysis of fluorescence lifetime measurements of the macula using the fluorescence lifetime imaging ophthalmoscope in healthy subjects. Invest Ophthalmol Vis Sci. 2014;55(4):2106–13.
Dysli C, Wolf S, Zinkernagel MS. Fluorescence lifetime imaging in retinal artery occlusion. Invest Ophthalmol Vis Sci. 2015;56(5):3329–36.
Sauer L, et al. Impact of macular pigment on fundus autofluorescence lifetimes. Invest Ophthalmol Vis Sci. 2015;56(8):4668–79.
Schweitzer D, et al. Fluorescence lifetime imaging ophthalmoscopy in type 2 diabetic patients who have no signs of diabetic retinopathy. J Biomed Opt. 2015;20(6):61106.
Dysli C, et al. Fluorescence lifetime imaging in Stargardt disease: potential marker for disease progression. Invest Ophthalmol Vis Sci. 2016;57(3):832–41.
Dysli C, et al. Autofluorescence lifetimes in patients with choroideremia identify photoreceptors in areas with retinal pigment epithelium atrophy. Invest Ophthalmol Vis Sci. 2016;57(15):6714–21.
Dysli C, Wolf S, Zinkernagel MS. Autofluorescence lifetimes in geographic atrophy in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2016;57(6):2479–87.
Dysli C, et al. Fundus autofluorescence lifetimes and central serous chorioretinopathy. Retina. 2017;37(11):2151–61.
Dysli C, et al. Fluorescence lifetimes of drusen in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2017;58(11):4856–62.
Sauer L, et al. Monitoring macular pigment changes in macular holes using fluorescence lifetime imaging ophthalmoscopy. Acta Ophthalmol. 2017;95(5):481–92.
Sauer L, Gensure RH, Hammer M, Bernstein PS. Fluorescence lifetime imaging ophthalmoscopy (FLIO) – a novel way to assess macular telangiectasia type 2 (MacTel). Ophthalmol Retina. 2018;2(6):587–98.
Schmidt J, et al. Fundus autofluorescence lifetimes are increased in non-proliferative diabetic retinopathy. Acta Ophthalmol. 2017;95(1):33–40.
Dysli C, et al. Fundus autofluorescence lifetime patterns in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2018;59(5):1769–78.
Sauer L, et al. Review of clinical approaches in fluorescence lifetime imaging ophthalmoscopy. J Biomed Opt. 2018;23(9):1–20.
Sauer L, et al. Monitoring foveal sparing in geographic atrophy with fluorescence lifetime imaging ophthalmoscopy - a novel approach. Acta Ophthalmol. 2018;96(3):257–66.
Sauer L, Andersen KM, Li B, Gensure RH, Hammer M, Bernstein PS. Fluorescence lifetime imaging ophthalmoscopy (FLIO) of macular pigment. Invest Ophthalmol Vis Sci. 2018;59(7):3094–103.
Sauer L, et al. Patterns of fundus autofluorescence lifetimes in eyes of individuals with nonexudative age-related macular degeneration. Invest Ophthalmol Vis Sci. 2018;59(4):AMD65–77.
Andersen KM, et al. Characterization of retinitis pigmentosa using fluorescence lifetime imaging ophthalmoscopy (FLIO). Transl Vis Sci Technol. 2018;7(3):20.
Dysli C, et al. Fluorescence lifetime imaging ophthalmoscopy. Prog Retin Eye Res. 2017;60:120–43.
Schweitzer D. Autofluorescence diagnostics of ophthalmic diseases. In: Ghukasyan VV, Heikal AA, editors. Natural biomarkers for cellular metabolism-biology, techniques, and applications. Boca Raton: Taylor&Francis Group; 2014. p. 317–44.
Delori FC, et al. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci. 1995;36(3):718–29.
Katz ML, et al. Influence of early photoreceptor degeneration on lipofuscin in the retinal pigment epithelium. Exp Eye Res. 1986;43(4):561–73.
Delori FC, Goger DG, Dorey CK. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 2001;42(8):1855–66.
Eldred GE, Katz ML. Fluorophores of the human retinal pigment epithelium: separation and spectral characterization. Exp Eye Res. 1988;47(1):71–86.
Chowdhury PK, et al. Generation of fluorescent adducts of malondialdehyde and amino acids: toward an understanding of lipofuscin. Photochem Photobiol. 2004;79(1):21–5.
Sparrow JR, et al. The bisretinoids of retinal pigment epithelium. Prog Retin Eye Res. 2012;31(2):121–35.
Sohal RS. Assay of lipofuscin/ceroid pigment in vivo during aging. Methods Enzymol. 1984;105:484–7.
Holz FG, et al. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 1999;40(3):737–43.
Holz FG, et al. Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2001;42(5):1051–6.
Murdaugh LS, et al. Compositional studies of human RPE lipofuscin. J Mass Spectrom. 2010;45(10):1139–47.
Ablonczy Z, et al. Lack of correlation between the spatial distribution of A2E and lipofuscin fluorescence in the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2013;54(8):5535–42.
Smith RT, Bernstein PS, Curcio CA. Rethinking A2E. Invest Ophthalmol Vis Sci. 2013;54(8):5543.
Koenig K, Schneckenburger H. Laser-induced autofluorescence for medical diagnosis. J Fluoresc. 1994;4(1):17–40.
Lakowicz JR. Principles of fluorescence spectroscopy. New York: Springer; 2007.
Chorvat D, Chorvatova A. Multi-wavelength fluorescence lifetime spectroscopy: a new approach to the study of endogenous fluorescence in living cells and tissues. Laser Phys Lett. 2009;6(3):175–93.
Chance B. Pyridine nucleotide as an indicator of the oxygen requirements for energy-linked functions of mitochondria. Circ Res. 1976;38(5 Suppl 1):I31–8.
Schweitzer D, et al. Time-correlated measurement of autofluorescence. A method to detect metabolic changes in the fundus. Ophthalmologe. 2002;99(10):774–9.
Wakita M, Nishimura G, Tamura M. Some characteristics of the fluorescence lifetime of reduced pyridine nucleotides in isolated mitochondria, isolated hepatocytes, and perfused rat liver in situ. J Biochem. 1995;118(6):1151–60.
Niesner R, et al. Noniterative biexponential fluorescence lifetime imaging in the investigation of cellular metabolism by means of NAD(P)H autofluorescence. ChemPhysChem. 2004;5(8):1141–9.
Schneckenburger H, et al. Autofluorescence lifetime imaging of cultivated cells using a UV picosecond laser diode. J Fluoresc. 2004;14(5):649–54.
Kierdaszuk B, et al. Fluorescence of reduced nicotinamides using one- and two-photon excitation. Biophys Chem. 1996;62(1–3):1–13.
Ihanamaki T, Pelliniemi LJ, Vuorio E. Collagens and collagen-related matrix components in the human and mouse eye. Prog Retin Eye Res. 2004;23(4):403–34.
Blomfield J, Farrar JF. The fluorescent properties of maturing arterial elastin. Cardiovasc Res. 1969;3(2):161–70.
Fujimoto D, Moriguchi T. Pyridinoline, a non-reducible crosslink of collagen. Quantitative determination, distribution, and isolation of a crosslinked peptide. J Biochem. 1978;83(3):863–7.
Richards-Kortum R, Sevick-Muraca E. Quantitative optical spectroscopy for tissue diagnosis. Annu Rev Phys Chem. 1996;47:555–606.
Schweitzer D, et al. Spectral and time-resolved studies on ocular structures. In: SPIE Proceedings; 2007.
Aiken JH, Huie CW. Detection of bilirubin using surfactant fluorescence enhancement and visible laser fluorometry. Anal Lett. 1991;24(1):167–80.
Keilhauer CN, Delori FC. Near-infrared autofluorescence imaging of the fundus: visualization of ocular melanin. Invest Ophthalmol Vis Sci. 2006;47(8):3556–64.
Colbert A, Heikal AA. Towards probing skin cancer using endogenous melanin fluorescence. Penn State McNair J. 2005:8.
Ghadially FN, Neish WJ, Dawkins HC. Mechanisms involved in the production of red fluorescence of human and experimental tumours. J Pathol Bacteriol. 1963;85:77–92.
Marcu L. Fluorescence lifetime techniques in medical applications. Ann Biomed Eng. 2012;40(2):304–31.
John WG, Lamb EJ. The Maillard or browning reaction in diabetes. Eye (Lond). 1993;7(Pt 2):230–7.
Klemm M, et al. FLIMX: a software package to determine and analyze the fluorescence lifetime in time-resolved fluorescence data from the human eye. PLoS One. 2015;10(7):e0131640.
Becker W. Fluorescence lifetime imaging–techniques and applications. J Microsc. 2012;247(2):119–36.
Bone RA, Landrum JT, Tarsis SL. Preliminary identification of the human macular pigment. Vis Res. 1985;25(11):1531–5.
Bone RA, et al. Stereochemistry of the human macular carotenoids. Invest Ophthalmol Vis Sci. 1993;34(6):2033–40.
Shyam R, et al. RPE65 has an additional function as the lutein to meso-zeaxanthin isomerase in the vertebrate eye. Proc Natl Acad Sci U S A. 2017;114(41):10882–7.
Sharifzadeh M, Bernstein PS, Gellermann W. Nonmydriatic fluorescence-based quantitative imaging of human macular pigment distributions. J Opt Soc Am A Opt Image Sci Vis. 2006;23(10):2373–87.
Bhosale P, Bernstein PS. Vertebrate and invertebrate carotenoid-binding proteins. Arch Biochem Biophys. 2007;458(2):121–7.
Loane E, et al. Transport and retinal capture of lutein and zeaxanthin with reference to age-related macular degeneration. Surv Ophthalmol. 2008;53(1):68–81.
Bhosale P, et al. Identification and characterization of a Pi isoform of glutathione S-transferase (GSTP1) as a zeaxanthin-binding protein in the macula of the human eye. J Biol Chem. 2004;279(47):49447–54.
Bhosale P, Bernstein PS. Synergistic effects of zeaxanthin and its binding protein in the prevention of lipid membrane oxidation. Biochim Biophys Acta. 2005;1740(2):116–21.
Bhosale P, et al. Purification and partial characterization of a lutein-binding protein from human retina. Biochemistry. 2009;48(22):4798–807.
Bernstein PS, et al. Retinal tubulin binds macular carotenoids. Invest Ophthalmol Vis Sci. 1997;38(1):167–75.
Snodderly DM, Auran JD, Delori FC. The macular pigment. II. Spatial distribution in primate retinas. Invest Ophthalmol Vis Sci. 1984;25(6):674–85.
Kijlstra A, et al. Lutein: more than just a filter for blue light. Prog Retin Eye Res. 2012;31(4):303–15.
Bernstein PS, et al. Lutein, zeaxanthin, and meso-zeaxanthin: the basic and clinical science underlying carotenoid-based nutritional interventions against ocular disease. Prog Retin Eye Res. 2016;50:34–66.
Woodall AA, Britton G, Jackson MJ. Carotenoids and protection of phospholipids in solution or in liposomes against oxidation by peroxyl radicals: relationship between carotenoid structure and protective ability. Biochim Biophys Acta. 1997;1336(3):575–86.
Ham WT Jr, et al. Histologic analysis of photochemical lesions produced in rhesus retina by short-wave-length light. Invest Ophthalmol Vis Sci. 1978;17(10):1029–35.
Krinsky NI. Antioxidant functions of carotenoids. Free Radic Biol Med. 1989;7(6):617–35.
Bone RA, et al. Distribution of lutein and zeaxanthin stereoisomers in the human retina. Exp Eye Res. 1997;64(2):211–8.
Holz FG, Spaide RF. Medical retina: focus on retinal imaging. New York: Springer; 2010.
Ermakov IV, et al. Resonant Raman detection of macular pigment levels in the living human retina. Opt Lett. 2001;26(4):202–4.
Ermakov IV, Ermakova MR, Gellermann W. Simple Raman instrument for in vivo detection of macular pigments. Appl Spectrosc. 2005;59(7):861–7.
Wilde C, et al. Prevalence of age-related macular degeneration in an elderly UK Caucasian population-the Bridlington Eye Assessment Project: a cross-sectional study. Eye (Lond). 2017;31(7):1042–50.
Khan KN, et al. Differentiating drusen: drusen and drusen-like appearances associated with ageing, age-related macular degeneration, inherited eye disease and other pathological processes. Prog Retin Eye Res. 2016;53:70–106.
Korner-Stiefbold U. Age-related macular degeneration (AMD)--therapeutic possibilities and new approaches. Ther Umsch. 2001;58(1):28–35.
Lambert NG, et al. Risk factors and biomarkers of age-related macular degeneration. Prog Retin Eye Res. 2016;54:64–102.
Tomany SC, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111(7):1280–7.
Zinkernagel MS, et al. Association of the intestinal microbiome with the development of neovascular age-related macular degeneration. Sci Rep. 2017;7:40826.
Holz FG, et al. Geographic atrophy: clinical features and potential therapeutic approaches. Ophthalmology. 2014;121(5):1079–91.
Yung M, Klufas MA, Sarraf D. Clinical applications of fundus autofluorescence in retinal disease. Int J Retina Vitreous. 2016;2:12.
Reim M, et al. Diagnosen am Augenhintergrund. Georg Thieme: Stuttgart; 2004.
Russell SR, et al. Location, substructure, and composition of basal laminar drusen compared with drusen associated with aging and age-related macular degeneration. Am J Ophthalmol. 2000;129(2):205–14.
Spaide RF, Curcio CA. Drusen characterization with multimodal imaging. Retina. 2010;30(9):1441–54.
Rudolf M, et al. Sub-retinal drusenoid deposits in human retina: organization and composition. Exp Eye Res. 2008;87(5):402–8.
Holz FG, et al. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol. 2007;143(3):463–72.
Pichi F, et al. Fundus autofluorescence imaging in hereditary retinal diseases. Acta Ophthalmol. 2018;96(5):e549–61.
DiCarlo JE, Mahajan VB, Tsang SH. Gene therapy and genome surgery in the retina. J Clin Invest. 2018;128(6):2177–88.
Moore NA, et al. Gene therapy for inherited retinal and optic nerve degenerations. Expert Opin Biol Ther. 2018;18(1):37–49.
Takahashi VKL, et al. Gene therapy in inherited retinal degenerative diseases, a review. Ophthalmic Genet. 2018;39(5):560–8.
Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34(5):1659–76.
Greenstein VC, et al. Structural and functional changes associated with normal and abnormal fundus autofluorescence in patients with retinitis pigmentosa. Retina. 2012;32(2):349–57.
Lewis RA, et al. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64(2):422–34.
Tanna P, et al. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2017;101(1):25–30.
Zinkernagel MS, MacLaren RE. Recent advances and future prospects in choroideremia. Clin Ophthalmol. 2015;9:2195–200.
Charbel Issa P, et al. Macular telangiectasia type 2. Prog Retin Eye Res. 2013;34:49–77.
Finger RP, et al. Reading performance is reduced by parafoveal scotomas in patients with macular telangiectasia type 2. Invest Ophthalmol Vis Sci. 2009;50(3):1366–70.
Clemons TE, et al. Baseline characteristics of participants in the natural history study of macular telangiectasia (MacTel) MacTel Project Report No. 2. Ophthalmic Epidemiol. 2010;17(1):66–73.
Charbel Issa P, Holz FG, Scholl HP. Metamorphopsia in patients with macular telangiectasia type 2. Doc Ophthalmol. 2009;119(2):133–40.
Helb HM, et al. Abnormal macular pigment distribution in type 2 idiopathic macular telangiectasia. Retina. 2008;28(6):808–16.
Delori FC, et al. Macular pigment density measured by autofluorescence spectrometry: comparison with reflectometry and heterochromatic flicker photometry. J Opt Soc Am A Opt Image Sci Vis. 2001;18(6):1212–30.
Zeimer MB, et al. Idiopathic macular telangiectasia type 2: distribution of macular pigment and functional investigations. Retina. 2010;30(4):586–95.
Choi RY, et al. Macular pigment distribution responses to high-dose zeaxanthin supplementation in patients with macular telangiectasia type 2. Retina. 2017;37(12):2238–47.
Sallo FB, et al. The prevalence of type 2 idiopathic macular telangiectasia in two African populations. Ophthalmic Epidemiol. 2012;19(4):185–9.
Klein R, et al. The prevalence of macular telangiectasia type 2 in the Beaver Dam eye study. Am J Ophthalmol. 2010;150(1):55–62.e2.
Parmalee NL, et al. Identification of a potential susceptibility locus for macular telangiectasia type 2. PLoS One. 2012;7(8):e24268.
Parmalee NL, et al. Analysis of candidate genes for macular telangiectasia type 2. Mol Vis. 2010;16:2718–26.
Gillies MC, et al. Familial asymptomatic macular telangiectasia type 2. Ophthalmology. 2009;116(12):2422–9.
Delaere L, Spielberg L, Leys AM. Vertical transmission of macular telangiectasia type 2. Retin Cases Brief Rep. 2012;6(3):253–7.
Scerri TS, et al. Genome-wide analyses identify common variants associated with macular telangiectasia type 2. Nat Genet. 2017;49(4):559–67.
Wong WT, et al. Fundus autofluorescence in type 2 idiopathic macular telangiectasia: correlation with optical coherence tomography and microperimetry. Am J Ophthalmol. 2009;148(4):573–83.
Sallo FB, et al. Multimodal imaging in type 2 idiopathic macular telangiectasia. Retina. 2015;35(4):742–9.
Toto L, et al. Multimodal imaging of macular telangiectasia type 2: focus on vascular changes using optical coherence tomography angiography. Invest Ophthalmol Vis Sci. 2016;57(9):OCT268–76.
Surguch V, Gamulescu MA, Gabel VP. Optical coherence tomography findings in idiopathic juxtafoveal retinal telangiectasis. Graefes Arch Clin Exp Ophthalmol. 2007;245(6):783–8.
Sallo FB, et al. “En face” OCT imaging of the IS/OS junction line in type 2 idiopathic macular telangiectasia. Invest Ophthalmol Vis Sci. 2012;53(10):6145–52.
Chin EK, et al. Staging of macular telangiectasia: power-Doppler optical coherence tomography and macular pigment optical density. Invest Ophthalmol Vis Sci. 2013;54(7):4459–70.
Cogan DG, Kuwabara T. Capillary shunts in the pathogenesis of diabetic retinopathy. Diabetes. 1963;12(4):293–300.
Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52(2):1156–63.
Zhang X, et al. Diabetic macular edema: new concepts in patho-physiology and treatment. Cell Biosci. 2014;4(1):27.
Yu DY, et al. Pathogenesis and intervention strategies in diabetic retinopathy. Clin Exp Ophthalmol. 2001;29(3):164–6.
Tarr JM, et al. Pathophysiology of diabetic retinopathy. ISRN Ophthalmol. 2013;2013:343560.
Vlassara H, Palace M. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251(2):87–101.
Singh R, et al. Advanced glycation end-products: a review. Diabetologia. 2001;44(2):129–46.
Behl T, Kaur I, Kotwani A. Implication of oxidative stress in progression of diabetic retinopathy. Surv Ophthalmol. 2016;61(2):187–96.
Hammes H-P, et al. Differential accumulation of advanced glycation end products in the course of diabetic retinopathy. Diabetologia. 1999;42(6):728–36.
Glenn JV, Stitt AW. The role of advanced glycation end products in retinal ageing and disease. Biochim Biophys Acta. 2009;1790(10):1109–16.
Stitt AW. Advanced glycation: an important pathological event in diabetic and age related ocular disease. Br J Ophthalmol. 2001;85(6):746–53.
Stitt AW. AGEs and diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010;51(10):4867–74.
Kandarakis SA, et al. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Prog Retin Eye Res. 2014;42:85–102.
Calvo P, et al. Diabetic macular edema: options for adjunct therapy. Drugs. 2015;75(13):1461–9.
Eisma JH, Dulle JE, Fort PE. Current knowledge on diabetic retinopathy from human donor tissues. World J Diabetes. 2015;6(2):312.
Hernández C, et al. Neuroprotection as a therapeutic target for diabetic retinopathy. J Diabet Res. 2016;2016:9508541.
Yamagishi S-I, et al. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim Biophys Acta. 2012;1820(5):663–71.
Klemm M, et al. Effects of short term changes in the blood glucose level on the autofluorescence lifetime of the human retina in healthy volunteers. In: SPIE BiOS. SPIE; 2016.
de la Maza MP, et al. Fluorescent advanced glycation end-products (ages) detected by spectro-photofluorimetry, as a screening tool to detect diabetic microvascular complications. J Diabet Mellitus. 2012;2(02):221.
Vujosevic S, et al. Diabetic macular edema: fundus autofluorescence and functional correlations. Invest Ophthalmol Vis Sci. 2011;52(1):442–8.
Araki N, et al. Immunochemical evidence for the presence of advanced glycation end products in human lens proteins and its positive correlation with aging. J Biol Chem. 1992;267(15):10211–4.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this chapter

Cite this chapter
Bernstein, P. et al. (2019). Fluorescence Lifetime Imaging Ophthalmoscopy (FLIO). In: Bille, J. (eds) High Resolution Imaging in Microscopy and Ophthalmology. Springer, Cham. https://doi.org/10.1007/978-3-030-16638-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-16638-0_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16637-3
Online ISBN: 978-3-030-16638-0
eBook Packages: MedicineMedicine (R0)