
Abstract
Nowadays, a huge claim for personalized medicine is progressively growing, and, along this way, genetic studies represent one of the most representative steps.
Dilated cardiomyopathy (DCM) can be the consequence of clearly defined external etiologic factors, such as viral infections, toxins, drugs, metabolic disorders, etc., but at least 30–40% of cases (and maybe more) have a prevalent genetic origin, and in the remaining part, genetics may still play an important role.
With the expansion of clinical genetic testing, using high-quality next-generation sequencing (NGS) extended panels, these genetic causes of DCM have been increasingly identified. More than 50 genes, mapping to multiple biological pathways, are currently considered disease related, and causative variants can be identified in up to 35% of cases.
This growing amount of genetic informations, however, is still not followed by a parallel advance toward tailored clinical management. The reasons behind this gap are currently under investigation in the scientific community: the aim of this chapter is to provide a guide through the complexity of the genotype-phenotype interaction, analyzing (1) the most frequently encountered genes in DCM, (2) technical issues in NGS, (3) controversies beyond sequencing data interpretation, (4) the contribution of environmental modifiers, and (5) evidence-based genotype-phenotype correlations in DCM.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others
Keywords
- Precision medicine
- Genotype-environment interaction
- Environmental modifiers
- Next-generation sequencing
- Allelic heterogeneity
- Variant actionability
5.1 DCM-Associated Genes
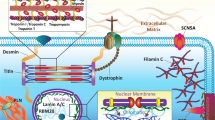
Nowadays, genetic laboratories from the USA and Europe offer different panels of genes related to DCM, ranging from 30 to more than 150 genes, with a great part of them only anecdotally associated with the disease or with a putative link on the basis of biological relationship with known genes. A detailed analysis of each different gene is far beyond the aim of this chapter, which will be focused in the complexity of the interpretation of “evidence-based” DCM genetic background. Here below is presented a brief list of the most investigated and evidence-based genes, grouped according to functional intracellular similarity. Cardiac sarcomeric and cytoskeletal genes (TTN overall) are the most frequently encountered. Other involved genes spread all over cardiomyocyte biological pathways and cell compartments, encoding components of desmosome, structural cytoskeleton, nuclear lamina, mitochondria, and ion flux-handling proteins [1] (Fig. 5.1).

Cardiomyocytes’ immunofluorescence, with schematic representation of gene clusters involved in DCM pathogenesis (courtesy of the authors)
We must premise that in these years times are rapidly changing, and this list may be no more representative of the entire genetic landscape of the disease in the next years.
5.1.1 Titin
(See Sect. 5.5.1)
5.1.2 Lamin A/C
(See Sect. 5.5.2)
5.1.3 Structural Cytoskeleton Z-Disk Genes
Cardiomyocyte’s structural integrity, sarcomeric orientation and contraction, and mechano-sensing transductions depend on cytoskeleton and Z-disk correct function. DES, DMD, FLNC, NEXN, NEBL, LDB3, and VCL encode for component of both sarcolemmal and sarcoplasmatic intermediate filaments, co-localizing to sarcolemmal membrane, sarcoplasmic membrane, and Z-disk structure. Notably, no or only a mild ATPase activity is known for these genes; thus all belongs to non-motor actin-binding protein group inside Z-disk structure. Mutations in these genes accounted for 5–10% of familial DCM, but this prevalence could increase after the inclusion of the recently discovered Filamin C (FLNC) gene.
Desmin ( DES ): Desmin is a cytoskeletal protein which forms muscle-specific intermediate filaments.
Mutations in the gene encoding Desmin cause a wide spectrum of phenotypes of different cardiomyopathies, skeletal myopathies, and mixed skeletal and cardiac myopathies. Desmin mutations account for 1–2% of all cases of DCM. Cardiac manifestations include restrictive cardiomyopathy (RCM), DCM, conduction system diseases, arrhythmias, and sudden death. Isolate cardiac phenotype is reported, or it can precede skeletal muscle involvement [2,3,4]. Truncating DES variants are associated with anticipated and more severe forms of DCM with diffuse LV fibrosis (unpublished data from Heart Muscle Disease Registry of Trieste, HMDR).
Dystrophin ( DMD ): The Dystrophin gene is located on the short arm of the X chromosome and consequently shows an x-linked pattern of inheritance. The dystrophin protein, in conjunction with the dystrophin glycoprotein complex, has an important role in force transmission, being integral to the mechanical link between the intracellular cytoskeleton and the extracellular matrix. Cardiac involvement is present in approximately 90% of the cases of Duchenne’s muscular dystrophy and 70% of Becker’s muscular dystrophy. Abnormal Q waves (“pseudonecrosis”) in lead I, aVL, and V6 or in lead II, III, and aVF have been described. Right bundle branch block, atrioventricular block, and supraventricular arrhythmias can be present. About 10% of female carriers of DMD mutations (Duchenne or Becker type) may develop a DCM in the absence of clinical involvement of skeletal muscle and, although in anecdotal forms, missense and truncating variants of DMD may present with isolated cardiac involvement in males, with DCM, and no signs of muscular dystrophy [5,6,7,8].
Filamin C (See Sect. 5.5.3)
Vinculin ( VCL ): This gene encodes a cytoskeletal protein (Vinculin) involved in cell-matrix and cell-cell adhesion. Specifically, Vinculin is involved in the linkage of integrin adhesion molecules to the actin cytoskeleton. Mutations in this gene, especially in cardiac-specific isoform metavinculin, are very rarely found (less than ten variants described so far) and have been mainly related to DCM but also to hypertrophic cardiomyopathy (HCM). Nowadays, only a limited number of cases sustain these associations, and segregation studies were no or only marginally in support of it. Moreover, some of the described families harbored a second mutation that explained the phenotype [9, 10].
Lim Domain Binding 3 ( LDB3 , or Cypher Zasp): LDB3 interact with alpha-actinin-2 and to protein kinase C, maintaining the structure of the Z-disk during muscle contraction and contributing to signal transduction cascades including cardiac hypertrophy and ventricular remodeling pathways. Mutations in this gene have been associated with left ventricular non-compaction (LVNC), DCM, HCM, skeletal myopathy, and peripheral neuropathy. The evidence on the pathogenicity of many of the first described variants is actually weak, as some of them have been found with similar frequency in patients and controls [11]. Those variants that are more likely pathogenic are mainly located in some of the zinc-binding LIM domains of the protein [12].
5.1.4 Desmosomal Genes
Desmosome is a symmetric myocyte structure in which each part resides in the cytoplasm of one of a pair of adjacent cells, anchoring intermediate filaments in the cytoskeleton to the cell surface. In combination with the adherents and gap junctions, it connects myocardial cells maintaining both the mechanical and electrical integrity of the heart. Several desmosome genes have been identified in patients with DCM, usually inherited with an autosomal dominant pattern. Interestingly, desmosome genes (Plakophilin-2 (PKP2), Desmoplakin (DSP), Desmocollin-2 (DSC2), Desmoglein-2 (DSG2), and Plakoglobin (JUP)) were initially described as causing arrhythmogenic right ventricular cardiomyopathy (ARVC), but in 2010, Elliott et al. demonstrated a prevalence of 5% of desmosomal protein coding genes mutations among 100 unrelated DCM patients [13]: in relation to this aspect, it is now useful to introduce the concept of “overlapping, gene-driven phenotype” between different forms of cardiomyopathies (which turns out to be a recurrent feature in many genotypes)—even if originally described as linked to a peculiar phenotype (in the case of JUP and DSP genes with Naxos and Carvajal diseases and with ARVC), a specific genotype can manifest itself in different ways according to others, also non-genetic, modifiers.
Furthermore, the genetic overlap between ARVC and DCM has also been shown in most of non-desmosomal ARVC-related genes (e.g., LMNA, TMEM43), increasing the possibility of a clinical overlap between different forms of cardiomyopathy.
It is worth mentioning the similarity between specific cardiac and cutaneous desmosomal protein isoforms: Desmoplakin, plakoglobin, and plakophilin-2 are, in fact, constitutively expressed in desmosomes of both cardiomyocytes and keratinocytes, and a radical mutation in one of these two proteins often may result in cardio-cutaneous syndromes. Cadherins, conversely (DSC and DSG), have different isoforms preferentially expressed in the heart (isoform 2) or in the cutis (isoforms 1 and 3) [14, 15].
Desmoplakin ( DSP ): DSP codes for the protein desmoplakin, an intracellular obligate component of desmosomes that anchors intermediate filaments, such as desmin and filamins, to the inner desmosomal plaques, while the N terminus of the protein (extracellular domains) interacts with plakophilin and plakoglobin. DSP-related DCM is associated with increased ventricular arrhythmic burden and left ventricular fibrosis, with or without right ventricular involvement (arrhythmogenic cardiomyopathy). In general, frameshift and nonsense mutations in DSP are considered as disease causing, even when they have not been previously described, while missense variants must be evaluated case by case. As previously mentioned, DSP mutations, if present in homozygosity and with autosomal recessive inheritance pattern, have also been associated with a series of diseases characterized by cardiac and cutaneous involvement, such as Carvajal syndrome (woolly hair, keratoderma, DCM), keratosis palmoplantaris striata II, woolly hair, and lethal acantholytic epidermolysis bullosa. To date, large observational studies investigating the prognosis and the clinical manifestation related to DSP-DCM in respect to other genotypes are still lacking, but preliminary data from single-family studies and from HMDR of Trieste seems to confirm the increased risk of malignant ventricular arrhythmias.
5.1.5 Sarcomeric (Motor) Genes
Mutations in genes encoding for proteins that form sarcomeric thick and thin filaments have been largely recognized as DCM causing. These proteins (Myosin-heavy chain alpha and beta (MYH6 and MYH7, respectively), myosin-binding protein C3 (MYBPC3), troponins (TNNT2, TNNI3, TNNC1), tropomyosin 1 (TPM1), cardiac actinin 1 (ACTN1), myopalladin (MYPN)) share catalytic activity and are involved in sarcomeric contraction (MYPN shares also structural properties with Z-disk genes); comprehensively, these genes are involved in about 10% of cases of genetic DCM. Also this group of genes is characterized by a large overlapping of phenotypes: this is due to increased allelic heterogeneity, where different mutations resulting in different phenotypes are scattered and intercalated through the entire nucleotide sequence of a given gene, and, more interestingly, a single variant may express itself in different phenotypes inside the same family [16, 17]. Here below a brief list of most frequently encountered sarcomeric genes in DCM genotyping:
Myosin-heavy chain alpha ( MYH7 ): MYH6 codes for the alpha subunit of cardiac myosin heavy chain. It is the predominant isoform of myosin heavy chain at the embryonic myocardium. The ATPase activity and the shortening velocity of this isoform are higher than those of the adult beta-myosin isoforms. After birth, MYH6 expression decreases and represents on average 7% of ventricular myosin in the adult heart. Despite its low expression, the presence of alpha-myosin is important for ventricular function, and its expression in adult atrial myocardium remains elevated, being the main isoform in this tissue (MYH6 variants are also strongly associated with atrial septal defects). The characterization of this gene in DCM is representative of the evolving knowledge in cardiac genetics: previous studies have highlighted the importance of MYH6 mutations in DCM patients, elucidating also a possible negative prognostic effect [18]. These MYH6 mutations were distributed in highly conserved residues and were predicted to negatively affect protein function, but, nevertheless, the progression of knowledge of genetic databases has cast some doubts about the real contribute of this gene in DCM, since there seems to be no significant mutation excess in DCM patients in respect to controls. Variant is this gene should be evaluated carefully case by case [11].
Myosin-heavy chain beta ( MYH7 ): β myosin heavy chain was the first sarcomeric protein to be linked with cardiomyopathy, and mutations in MYH7 are now common causes of HCM and are also associated with DCM, LVNC, and RCM. In respect to DCM, they are responsible for about 4–6% of cases of familial DCM. Truncating variants should generally be considered pathogenic. The converter region of the protein (amino acid: 700–790) represents a mutation hotspot which have been shown to correlate with possible overlapping phenotypes and severe prognosis [16, 17].
Troponin T type 2 ( TNNT2 ):The protein troponin T type 2 is the tropomyosin-binding subunit of the troponin complex, which is located on the thin filament of striated muscles and regulates muscle contraction in response to alterations in intracellular calcium ion concentration. Mutations in TNNT2 have also been associated with HCM, DCM, RCM, and LVNC. Patients with TNNT2 mutations generally exhibit a high frequency of premature sudden cardiac death. It accounts for 2–3% of DCM familial forms. Variant Arg173Trp has been clearly associated almost exclusively with dilated phenotype [19].
Myosin-binding protein C3 ( MYBPC3 ): This gene 3 encodes for a member of myosin-associated proteins, which localized in the cross-bridge-bearing zone (C region) of A bands in cardiac muscle. It is the most common mutated gene in HCM, and, as others sarcomeric genes, it has been associated also with dilated or non-compaction phenotype. The more recent evidences raise questions about its contribution to DCM phenotype, given the relatively similar prevalence of MYBPC3 rare variants in healthy and affected individuals of explored populations [11]. However, it must be underlined that some HCM that develop “burnout” physiology may turn in dilated phenotype: particular attention should be paid to this aspect when facing a DCM patient with a rare variant in MYBPC3.
5.1.6 Ion Channel-Related Genes
Genes encoding for ion-channel proteins are strongly associated with channelopaties, but, in the last years, a growing amount of studies extended the phenotypical spectrum of clinical entities related to a defect in one of these genes to also to structural (dilated or non-compaction) phenotypes. The mechanistic links behind these associations is still poorly understood, but it is potentially related to altered membrane stability (i.e., syntrophin-mediated interaction between SCN5A and DMD) or altered calcium handling leading to sarcomeric inefficiency (phospholamban (PLN) and RYR2 variants). HCN4 (hyperpolarization-activated cyclic nucleotide-gated potassium channel 4) mutations have also been recently shown to be associated with LVNC, with or without DCM overlap (NB: the association between HCN4 and DCM needs still to be demonstrated) [20,21,22,23].
SCN5A: This gene encodes the voltage-gated sodium channel known as tetrodotoxin-resistant Nav1.5 dependent. The protein expression is predominant at heart. It is responsible for the fast sodium current that causes phase 0 of the action potential. Mutations in this gene, with marked allelic heterogeneity, have been strongly associated with Brugada syndrome in case of loss of function effect and long QT type 3 in case of gain of function effect, both diseases with autosomal dominant transmission. The association with DCM has been, in proportion, very rarely reported; it is generally accepted that these mutations are located in two specific regions of the channel: in the voltage-sensitive domain (VSD) and intracellular loops. One of the best characterized mutations is Arg222Gln [20], which affects the VSD. This mutation is also associated with frequent ventricular arrhythmias, cardiac conduction disease, and, in some cases, atrial fibrillation. None of the carriers presented a prolonged QTc. Recently, especially for truncating variants, the association with DCM has been further confirmed [11,21,22,].
Ryanodine Receptor 2 ( RYR2 ): This gene encodes a ryanodine receptor found in cardiac muscle sarcoplasmic reticulum. The encoded protein is one of the components of calcium channel, mediating the release of Ca2+ from the sarcoplasmic reticulum into the cytoplasm and thereby playing a key role in triggering cardiac muscle contraction. Mutations (>95% missense) in this gene are known to result in catecholaminergic polymorphic ventricular tachycardia (CPVT), typically in the absence of structural heart disease. Some missense mutations have also been originally associated with the development of ARVC; however, it is now accepted that these carriers had not fulfilled current diagnostic criteria for the disease. Among missense variants, only one has been clearly associated with the development of structural (hypertrophic) heart disease in patients diagnosed with CPVT. A different variant (exon 3 deletion) has been demonstrated, in two families, to segregate with CPVT and progressive left ventricular dysfunction and/or cavity enlargement in some members [20]. Thus, the presence of DCM without CPVT phenotype related to RYR2 (radical) mutations is yet to be demonstrated.
5.1.7 Other Genes
BCL2-Associated Athanogene 3, BAG3: Members of the BAG family, including BAG3, are cytoprotective proteins that bind to and regulate Hsp70 family molecular chaperones. Heterozygous mutations in BAG3 have been associated with DCM. Mechanism of disease may, at least in part, depends on a decreased capability to compensate external stressors. The severity of DCM, in fact, has been shown to vary considerably between carriers. By the age of 70, the disease penetrance is apparently 100%. Both non-truncating and truncating BAG3 mutations are reported, with variable penetrance. A specific variant (Pro209Leu), typically a spontaneous de novo variant, is linked to pediatric myofibrillar myopathy [24, 25].
RNA-Binding Motif Protein 20, RBM20: This gene encodes a RNA-binding protein that acts as a regulator of mRNA splicing of a subset of genes involved in cardiac development, mainly sarcomeric genes (TTN, but also MYH7, TNNT2, and others). The association of this gene with DCM was firstly established in 2009 by genome-wide linkage analysis and progressively confirmed by subsequent studies. Remarkably, these mutations were located in exon 9, which appears to be a mutational hotspot. Nowadays, also mutations out of exon 9 are reported to be DCM causative, with similar penetrance and clinical manifestations. In respect to prevalence in DCM families, RBM 20 represents a rare genotype, accounting for 2–3% of cases. For this reason, so far, we should underline that evidence-based genotype-phenotype correlations are still lacking: only a small number of studies, in fact, with small numbers of index-patients or families, and short follow-up, reported a phenotype characterized by “severe heart failure, arrhythmia, and the need for cardiac transplantation” [26, 27], which still need to be confirmed in further studies.
5.2 Technical Issues in Genetic Sequencing
Over the last three decades, different approaches and technologies have been used to obtain genetic information in families or sporadic patients with hereditary diseases. Linkage analysis was the first method used to identify new disease genes, but this technique requires very large families or a large number of sporadic cases. The advent of “old” sequencing technology (Sanger method) has made genetic analysis much more effective, but with timing analysis and high costs, especially for pathologies with high genetic heterogeneity such as cardiomyopathies.
More recently we are witnessing a revolution in medical genetics and scientific research applied both to the identification of new disease genes and to the massive parallel study of a large number of genes. This is due to the discovery of high-efficiency instruments (NGS) that allowed the entry into what is called the era of the precision medicine; speed, reliability, and limited costs are the advantages peculiar of these techniques that allow the parallel analysis of a large number of genes.
NGS technologies can be applied in various formats, with the aim of sequencing the entire genome (including non-coding parts), or the exome, which includes only the coding regions of the genome, or a group (panel) of selected genes. Currently (but technologies are continuously improving), the latter application seems to offer the best compromise between costs, execution speed, and accuracy for certified diagnostic purposes, as it usually guarantees greater coverage of the analyzed genes [28, 29].
Different next-generation platforms have been proposed, differing from each other mainly in their methods of clonal amplification of short DNA fragments (50–400 bases) as a genomic library template and how these fragment libraries are subsequently sequenced through repetitive cycles to provide a nucleotide readout (see Table 5.1) [30].
However, the discovery of new single nucleotide variants (SNVs) using NGS still requires validation with Sanger sequencing methods because of the possible loss of precision in obtaining a really high number of short DNA fragments using the polymerase chain reaction (PCR) during library building. NGS platforms have in fact error rates of approximately ten times higher (1 in 1000 bases with 20× coverage) than Sanger sequencing (1 in 10,000 bases). Although the reading depth cutoff for NGS platforms is conventionally set at 20×, many studies indicate that average reading depths greater than 100× are required for the use of these platforms as independent tool for newly discovered variants, even under optimal conditions [31].
5.3 The Complexity in Variant Classification Process
Traditionally, a mutation is defined as a permanent change in the nucleotide sequence, whereas a polymorphism is defined as a variant with a frequency above 1%. These terms, however, which have been used widely, actually seem no longer suitable to describe the complexity of interindividual genetic variability. The Human Genome Project, culminating in 2001 with the determination of the complete sequence of human DNA [32], provided a first quantitative assessment of the interindividual genetic variability and the possible impact that this variability has on human health. Subsequent multiple international projects (like ESP and 1000 genomes, recently merged with other projects in the most comprehensive exome and genome database: gnomAD; http://gnomad.broadinstitute.org) led to the conclusion that about 1 in 1000 nucleotides in the human genome (three million in total) differs between people, and this variation is largely responsible for the physical, behavioral, and medical unique characteristics of each individual. In this line, the term “mutation” is no more strictly associated with the concept of pathogenicity, as the term polymorphism with the concept of benignity.
Taking into account the higher complexity of genetic information, the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines defined a new standard [33]; both terms, mutation and polymorphism, should now be replaced by the term “variant,” followed by one of these modifiers: (I) pathogenic, (II) likely pathogenic, (III) uncertain significance, (IV) likely benign, or (V) benign. Several stringent criteria are required to reach one of these different modifiers, which are defined by crosschecking the evidence that derives from different categories of evaluation: (a) population and disease-specific genetic databases, (b) in silico predictive algorithms, (c) biochemical characteristics, (d) literature evidences. A free access website, http://wintervar.wglab.org/results.php, released from ACMG, allows a guideline-based, point-by-point analysis of each—missense—variant of interest.
This classification approach is more stringent than the previous ones and may result in a larger proportion of variants being categorized as uncertain significance. It is hoped that this approach will reduce the substantial number of variants being reported as “causative” of disease without having sufficient supporting evidence for that classification. It is important to keep in mind that when a variant is classified as pathogenic, healthcare providers are highly likely to take that as “actionable,” i.e., to alter the treatment or surveillance of a patient or remove such management in a genotype-negative family member, based on that determination [11].
In recent years, in fact, genetic laboratories often showed a lack of uniformity in the definition of variants, especially for variants originally described in the past literature, which are still reported as pathogenic in older databases but were subsequently found to be too common in general population, so unlikely to be disease causing. This dis-homogeneity potentially led to different clinical management of similar variants.
A similar argument is related to new candidate genes: these genes are included in offered extended panel tests on the basis of a putative biological relationship with known disease-causing genes, but—still—in the absence of solid population or scientific supporting data. The actual net effect of extended gene panels is an increase in the amount of variants of unknown significance and a relative decrease in actionable variants.
It is important now to provide a brief mention to the mostly used of these “clinically oriented variant classification” databases: ClinVar and HGMD [34, 35]. The ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) is a public database that better represents the “historical” process that characterizes the classification of each variant: quoting, “ClinVar is a freely accessible, public archive of reports ‘coming from research and diagnostic laboratories’ of the relationships among human variations and phenotypes, with supporting evidence. ClinVar thus facilitates access to (…) the history of that interpretation.” The Human Gene Mutation Database (HGMD®, https://portal.biobase-international.com/hgmd/pro/start.php), available under subscription in the most updated version (last 3 years), is the other most reliable source of information about “known (published) gene lesions responsible for human inherited disease.” Since nowadays not all laboratories are active submitters to ClinVar or HGMD®, clinicians should still be careful in referring to them as a gold standard for variant classification: when a potentially disease related rare variant is found in a patient, these databases should be intended as a valuable source of informations to crosscheck with, but representing only a part of the multi-parametric approach that finally lead to definite variant classification.
In respect to variants in DCM-related genes, a recent report [11] shed some light in this topic, helping the clinicians to reassess the classification of variants and genes offered by clinical laboratories according to the new guideline standards, in order to elucidate the common characteristics of true actionable variants. The authors found that in some genes, previously strongly associated with a given cardiomyopathy, a rare variant was not clinically informative because there is an unacceptably high likelihood of false positive interpretation, while, by contrast, in other genes, diagnostic laboratories may have been overly conservative when assessing variant pathogenicity. Interestingly, some genes proposed on the basis of several (but dated) studies as among the most common causes of DCM (e.g., MYBPC3, MYH6, and missense variants in SCN5A) showed no excess variation among affected cases, raising an important question about their contribution to DCM phenotype development. Identifying the frequency of the most common HCM pathogenic variant in the available population databases (c.1504C>T in MYBPC3: 2.5 × 10−5) as the conservative upper bound, this study clearly elucidated what is the major allele frequency (MAF) threshold for a rare variant to be considered pathogenic: 0.0001 in ExAC (ExAC is the first release version of gnomAD, composed by exome data).
The emerging concept is the odds ratio (OR) of a given variant, to be disease causing (e.g., LMNA-truncating variants (tv) reached an OR of ∼99 to develop DCM, TTN-truncating variant an OR ∼20 to ∼50, FLNC not tested): the higher OR corresponds to higher actionability.
To summarize, clinicians should be aware that the “pathogenicity” of a variant is a fluid and evolving definition that should be periodically re-evaluated with the evidence coming from database and scientific progress, in order to be continuously customized to the patient.
5.4 The External Modulation of Genotype: Environmental Triggers
In DCM, both in sporadic and in familial cases, the pathogenicity of a gene variant is modulated by interfering, non-genetic environmental factors: this interaction could be largely responsible for variability in disease phenotype and prognosis. It is important to keep in mind how the actual knowledge in this field (contribution of interfering factors) may still be invalidated by a different accuracy in underlying genetic characterization, with the oldest reports being published before the release of 2015 ACMG standard. Below is reported a brief summary of known interfering environmental factors: inflammation, toxic exposure, hormones, and metabolic profile. Notably, in this field the research is currently very active, and all the following statements are susceptible to possible modifications in the next future (Table 5.2).
To conclude, we may say that the phenotypically normal heart with a pathogenic variant (definition that should be constantly re-evaluated) represents a model of failing but compensated heart, which is no or less able to sustain a second, environmental, failing hit [48]: all these potential “second hits” must be taken into account in DCM treatment and prognosis stratification.
5.5 Evidence-Based Genotype-Phenotype Correlations
As previously mentioned, the key factor for a correct genotype-phenotype analysis is the accuracy of the underlying variant classification: reliable genotype-dependent phenotypic informations are in fact achievable only if driven by a solid pathogenicity assessment.
Then, as patient’s phenotype represents the final results of a long-lasting process of interactions between genetic background and environment, clinicians are aware that discovering the net effect of the pathogenic variant requires a careful “pruning” of “confounding” factors. Furthermore, some correlations could also be outlined “a posteriori”, i.e., by the type of response to the medical therapy.
Finally, in assessing this correlation, it is important to focus on what is the best starting point: specific mutation versus specific gene versus specific clusters of genes with similar function inside the cardiomyocyte.
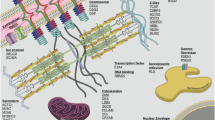
In this line, in respect to truly personalized medicine, the most correct approach should be the correlation between a specific pathogenic variant in a gene and its “private” phenotype, but, in order to achieve a more clinically meaningful classification, gene clustering attempts have been made and were shown to allow a rough, but functional, orientation, especially in therapeutic management [49]. At the current state of knowledge, a good compromise could be represented by the correlation between a specific gene and its phenotype, just preceded by a brief general distinction on the two main categories of variant (in respect to structural protein effect): missense and truncating (or radical). Generally speaking, the former is expected to affect protein morphology and/or function by changing a single amino acid in the protein sequence, while the latter is expected to cause a premature truncation of the amino acid sequence, leading to a decrease of total protein amount or effectiveness at the cellular level, mainly through nonsense-mediated decay (NMD). Consequently, truncating variants are generally considered less tolerated and linked to haploinsufficiency. Among all the human genes, the ones that are most conserved, expressed in early development, and highly tissue specific usually do not tolerate to be expressed in a single copy and are called haploinsufficient genes [50]. All cardiomyopathy-causing genes are included in this category, but they are not mutated with similar proportions of truncating and missense variants: for example, truncating variants on TTN have been discovered as the most frequent mutations in all DCM, whereas, in other DCM disease-causing genes, missense variants are the most frequently encountered (with, interestingly, similar actionability). With these principles in mind, among the several papers published on this topic, only few of them demonstrate evidence-based genotype-phenotype correlations that are helpful in the clinical management of patients with genetic DCM. To date, the best characterized correlations regard LMNA and TTN genes. Filamin C and other genes, in the next future, may reach a similar level of evidence (Fig. 5.2).

Schematic representation of genotype-phenotype correlations (adapted from [51])
5.5.1 Lamin A/C
LMNA represent the more investigated gene in DCM, and the natural history of LMNA-DCM has been outlined in several papers [52,53,54]. Comprehensively, with a confirmed mortality rate around 12% at 4 years (up to 30% at 12 years of follow-up), it could be considered the more aggressive genotype in DCM. Its phenotypic expression is characterized by a relatively high incidence of sudden cardiac death or major ventricular arrhythmias, even before the development of systolic left ventricular dysfunction. The median age at disease onset is between 30 and 40 years, and penetrance is almost complete at the age of 70 [52].
It is associated also to a primary disease of the conduction system, with supraventricular arrhythmias and atrioventricular block, by some authors called LMNA “atriopathy.” To date, LMNA pathogenic variants represent the only genetic background in DCM that is included in current guidelines, as it may change clinical choices such as the implantable cardioverter-defibrillator (ICD) therapy in primary prevention regardless of left ventricular ejection fraction values (Class IIa, level of evidence B, for ICD implantation in the presence of risk factors [55]: NSVT during ambulatory electrocardiogram monitoring, LVEF < 45% at first evaluation, male sex, and non-missense mutations).
The type of variant (missense versus truncating) and its site (before or after the nuclear lamina interacting domain) have also been addressed in respect to prognosis: actual evidence shows that mortality rates are similar, but truncating variants are related to anticipated penetrance of the disease. No clear effect is still demonstrated in respect to the site of variants [56].
5.5.2 Titin
Titin (TTN) is known as the largest sarcomeric protein that resides within the heart muscle. Due to alternative splicing of TTN, the heart expresses two major isoforms (N2B and N2BA) that incorporate four distinct regions termed the Z-line, I-band, A-band, and M-line. The amino terminus of Titin is embedded in the sarcomere Z-disk and participates in myofibril assembly, stabilization, and maintenance. The elastic I-band behaves as a bidirectional spring, restoring sarcomeres to their resting length after systole and limiting their stretch in early diastole. The inextensible A-band binds myosin and myosin-binding protein and is thought to be critical for biomechanical sensing and signaling. The M-band contains a kinase that may participate in strain-sensitive signaling and affect gene expression and cardiac remodeling in DCM.
Due to its higher prevalence in DCM population in respect to Lamin A/c (TTN 12–18% of whole DCM population, versus LMNA 4–6%), Titin is becoming the more broadly assessed genotype, despite its relatively recent discovery as a DCM-related gene [57]. To date, the evidence of pathogenicity is related almost exclusively to truncating variants. Since Titin-truncating variants (TTNtv) were reported also in 2–3% of general population without overt cardiomyopathy, many efforts have been made, firstly, to outline the characteristics that distinguish the disease-related truncating variants from the benign ones.
An important study by Roberts et al. elucidated the importance of the specific site of truncating variants: of the 364 exons of the entire gene, only a part of them is translated in cardiac isoforms N2B and N2BA [58]: Proportion (or percentage) of exons spliced in (PSI) is the concept that allows to correlate the exon site of the truncating variant with the molecular—and clinical—consequences of this truncation, with a PSI > 15% set as a lowest threshold to be penetrant and PSI > 90% describing exons sites with higher cardiac expression and higher association with fully penetrant DCM phenotype. The entire A-band and the proximal or terminal part of I-band contain exons with PSI proximal to 100%. Tv in M-band exons and Z-band exons should be evaluated case by case. This is the reason why the OR of a TTNtv varies between 20 and 50 according to the site involved by the mutation.
A second paper by the same group further demonstrates this concept, showing that also in general population without overt cardiomyopathy, the presence of TTNtv in sites with PSI > 15% mildly, but significantly, affects cardiac dimensions and function when assessed with 3D cardiac magnetic resonance [48].
Lower ventricular mass values, with lower ventricular wall thickness, have been recently outlined as a peculiar phenotypic manifestation of TTNtv [49, 59].
In respect to other clinical manifestations of TTN-related DCM, evidences are in favor of a relatively mild and treatable form of the disease in respect to LMNA-related one, with lower mortality rates, in line with the general DCM population. This could be true, especially in relatives that are diagnosed in a preclinical state [49, 59].
Clinicians must be aware that TTNtv, even if in small proportion of cases, could be linked to malignant ventricular arrhythmias especially in the presence of external modifiers: comprehensively, the sum of the actual evidences recommends a complete and continuous clinical follow-up of patients with TTNtv-related DCM and their relatives, even in the absence of overt cardiomyopathy [60].
Titin missense variants, on the contrary, nowadays are considered mostly as benign. This assumption has been tested in a recent multicenter study that sequenced TTN gene in a cohort of 147 DCM patients in which the outcome was not affected by the presence of Titin missense variants, confirming that most of these variants could be in fact benign (despite a highly conservative and accurate selection of variants: lowest population frequency, familial segregation, software predictions of pathogenicity) [61]. Recently, however, this “simple” classification has been questioned: a report in fact elucidated the pathogenicity of a specific TTN missense variant in DCM phenotype with non-compaction aspects, raising the threshold of complexity in TTN variant evaluation [62].
5.5.3 Filamin C
FLNC encodes filamin C, an intermediate filament that cross-links polymerized actin, contributing in anchoring cellular membrane proteins to cytoskeleton and in maintaining sarcomeric and Z-disk stability. It directly interacts with two protein complexes that link the subsarcolemmal actin cytoskeleton to the extracellular matrix: (1) the dystrophin-associated glycoprotein and (2) the integrin complexes, while, at intercalated disks, filamin C is located in the fascia adherens [63].
The association with DCM was initially reported by two separate studies [63, 64]. Ortiz et al. evaluated with NGS panels a cohort of 2877 patients referred for various cardiac diseases (including channelopathies and HCM, the latter representing almost one half of the cases) and identified 28 unrelated probands with FLNC-truncating variants, previously diagnosed mainly with DCM or, in minor part, with arrhythmogenic or RCM. Truncating variants in FLNC came out to cause an overlapping phenotype of dilated and left dominant arrhythmogenic cardiomyopathy complicated by frequent premature sudden death, with the phenotypic hallmark represented by subepicardial-transmural fibrosis in inferolateral LV wall. Interestingly, a small portion of probands (<5%) had prominent right ventricular involvement or restrictive phenotype.
The cumulative incidence of MVA or SD was found to be between 15 and 20% in a median follow-up of 5 years, and the mortality rate was about 6% for the same follow-up. We should underline that these data refer to a limited cohort of probands referred for genetic testing due to aggressive familial disease, representing a potential selection bias. Data on large cohorts of FLNCtv-related DCM patients are still lacking to confirm or modulate this aggressive phenotype.
Furthermore, it is worth mentioning that FLNC missense variants have been identified in a previous study also in families with HCM, although with a mild degree of LV hypertrophy. As for other cytoskeletal or sarcomeric genotypes with allelic heterogeneity, this fact suggests that filaminopathies can generate a spectrum of different cardiac disorders that at least in part may be related to the type of variant [65].
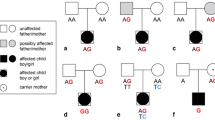
FLNC has only recently been included in the genetic screening of patients with inherited cardiomyopathies and sudden death, and its real prevalence in DCM has still to be elucidated. Figure 5.3 shows familial pedigrees of three families carrying FLNCtv.

Familial pedigrees of three families carrying FLNCtv
5.5.4 Insights from Clinical Presentation and Left Ventricular Reverse Remodeling (LVRR)
In clinical practice, especially in newly diagnosed DCM patients without familial history of cardiac disease, cardiologists may find useful to know peculiar findings that are representative of a specific genotype and, hopefully, able to guide disease treatment and prognostic assessment, at least in the short time.
A recent report from HMDR of Trieste tried to shed some light in this sense, differentiating genotypes on the basis of response to therapy: a different response, in fact, can be interpreted as the indirect evidence of different, mutation-driven, underlying pathogenic processes [49]. These mutation-dependent processes may not, or only marginally, be detectable otherwise.
Despite several limitations (possible selection bias in single referral center, limited number of patients partially grouped in gene clusters, thus introducing a possible heterogenic genetic background), this study allowed some interesting observations both in clinical presentation and LVRR rate in different genetic-based DCM, especially in relatively less investigated genotypes.
In respect to clinical presentation, most of the clinical and instrumental characteristics did not differ between the different genotypes. Except for a lower rate of left bundle-brunch block in both TTN and structural cytoskeleton Z-disk group and a trend toward a mild degree of LV dilation and dysfunction in LMNA mutation carriers (part of these findings have been subsequently confirmed in other studies) [59, 60], symptoms, electrocardiographic, and echocardiographic findings were grossly similar across different genotypes, being consistent with the hypothesis that DCM represents the final common phenotype of multiple genetic-based cardiac diseases and their relationship with environmental modifiers.
The most interesting finding was related to LVRR: a significant association was in fact demonstrated between lack of LVRR and specific genotypes (FLNC, DES, DMD, and other cytoskeletal Z-disk genes overall, followed by LMNAc). Conversely, TTN genotypes were most frequently associated with positive LVRR on optimal medical therapy (Fig. 5.4).

Association between LVRR and absence of LVRR according to different genotypes (data from HMDR) [49]
This kind of approach showed how phenotype correlations can be inferred also in this way, as an “ongoing” process, once more related to the interactions with external modifiers, in these cases represented by medications.
To conclude, the emerging concept elucidated in this chapter is that disease manifestation and prognosis are the results of the interaction between genotype and environment: the contribution of each factor to the patient’s clinical status is modulated by (1) genetic variant’s actionability and (2) type and severity of environmental factor(s). Summarizing, high actionable genotypes (with higher OR, as LMNAtv, or double pathogenic variants) may be per se the major determinants of disease manifestation/prognosis, while strong interfering environmental factors (e.g., chemotherapy) play a major role especially in cases with less actionable genotype.
Future perspectives in genetics will further investigate these aspects.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- DCM:
-
Dilated cardiomyopathy
- HCM:
-
Hypertrophic cardiomyopathy
- HF:
-
Heart failure
- HMDR:
-
Heart Muscle Disease Registry of Trieste
- LV:
-
Left ventricular
- LVEF:
-
Left ventricular ejection fraction
- LVNC:
-
LV non-compaction
- LVRR:
-
Left ventricular reverse remodeling
- RV:
-
Right ventricular
- MAF:
-
Major allele frequency
- NGS:
-
Next-generation sequencing
- PPCM:
-
Peripartum cardiomyopathy
- PSI:
-
Proportion or percentage (of exons) spliced in
- RCM:
-
Restrictive cardiomyopathy
References
McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121(7):731–48.
Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342(11):770–80.
Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999;100(5):461–4.
Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007;115(10):1244–51.
Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112(18):2799–804.
Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94(8):1023–31.
Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, et al. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation. 1996;94(12):3168–75.
Politano L, Nigro V, Nigro G, Petretta VR, Passamano L, Papparella S, et al. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA. 1996;275(17):1335–8.
Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation. 2002;105(4):431–7.
Wells QS, Ausborn NL, Funke BH, Pfotenhauer JP, Fredi JL, Baxter S, et al. Familial dilated cardiomyopathy associated with congenital defects in the setting of a novel VCL mutation (Lys815Arg) in conjunction with a known MYPBC3 variant. Cardiogenetics. 2011;1(1). pii: e10.
Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19(2):192–203.
Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42(11):2014–27.
Elliott P, O’Mahony C, Syrris P, Evans A, Rivera Sorensen C, Sheppard MN, et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3(4):314–22.
Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107(6):700–14.
Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart. 2011;97(7):530–9.
Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47.
Garcia-Giustiniani D, Arad M, Ortiz-Genga M, Barriales-Villa R, Fernandez X, Rodriguez-Garcia I, et al. Phenotype and prognostic correlations of the converter region mutations affecting the beta myosin heavy chain. Heart. 2015;101(13):1047–53.
Merlo M, Sinagra G, Carniel E, Slavov D, Zhu X, Barbati G, et al. Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin Transl Sci. 2013;6(6):424–8.
Campbell N, Sinagra G, Jones KL, Slavov D, Gowan K, Merlo M, et al. Whole exome sequencing identifies a troponin T mutation hot spot in familial dilated cardiomyopathy. PLoS One. 2013;8(10):e78104.
Marjamaa A, Laitinen-Forsblom P, Lahtinen AM, Viitasalo M, Toivonen L, Kontula K, et al. Search for cardiac calcium cycling gene mutations in familial ventricular arrhythmias resembling catecholaminergic polymorphic ventricular tachycardia. BMC Med Genet. 2009;10:12.
McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011;57(21):2160–8.
Schweizer PA, Koenen M, Katus HA, Thomas D. A distinct cardiomyopathy: HCN4 syndrome comprising myocardial noncompaction, bradycardia, mitral valve defects, and aortic dilation. J Am Coll Cardiol. 2017;69(9):1209–10.
van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14(11):1199–207.
Knezevic T, Myers VD, Gordon J, Tilley DG, Sharp TE III, Wang J, et al. BAG3: a new player in the heart failure paradigm. Heart Fail Rev. 2015;20(4):423–34.
Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88(3):273–82.
Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54(10):930–41.
Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest. 2014;124(8):3419–30.
Djemie T, Weckhuysen S, von Spiczak S, Carvill GL, Jaehn J, Anttonen AK, et al. Pitfalls in genetic testing: the story of missed SCN1A mutations. Mol Genet Genomic Med. 2016;4(4):457–64.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 2015;17(4):253–61.
Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–52.
Gullapalli RR, Desai KV, Santana-Santos L, Kant JA, Becich MJ. Next generation sequencing in clinical medicine: challenges and lessons for pathology and biomedical informatics. J Pathol Inform. 2012;3:40.
McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, et al. A physical map of the human genome. Nature. 2001;409(6822):934–41.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8.
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–81.
Merlo M, Anzini M, Bussani R, Artico J, Barbati G, Stolfo D, et al. Characterization and long-term prognosis of postmyocarditic dilated cardiomyopathy compared with idiopathic dilated cardiomyopathy. Am J Cardiol. 2016;118(6):895–900.
Belkaya S, Kontorovich AR, Byun M, Mulero-Navarro S, Bajolle F, Cobat A, et al. Autosomal recessive cardiomyopathy presenting as acute myocarditis. J Am Coll Cardiol. 2017;69(13):1653–65.
Frantz S, Falcao-Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M, et al. The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail. 2018;20(3):445–59.
Guzzo-Merello G, Cobo-Marcos M, Gallego-Delgado M, Garcia-Pavia P. Alcoholic cardiomyopathy. World J Cardiol. 2014;6(8):771–81.
Marichalar-Mendia X, Rodriguez-Tojo MJ, Acha-Sagredo A, Rey-Barja N, Aguirre-Urizar JM. Oral cancer and polymorphism of ethanol metabolising genes. Oral Oncol. 2010;46(1):9–13.
Renu K, V GA PBT, Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy - An update. Eur J Pharmacol. 2018;818:241–53.
Ware JS, Amor-Salamanca A, Tayal U, Govind R, Serrano I, Salazar-Mendiguchia J, et al. Genetic etiology for alcohol-induced cardiac toxicity. J Am Coll Cardiol. 2018;71(20):2293–302.
European Society of Gynecology, Association for European Paediatric Cardiology, German Society for Gender Medicine, Regitz-Zagrosek V, Blomstrom Lundqvist C, Borghi C, et al. ESC Guidelines on the management of cardiovascular diseases during pregnancy: the Task Force on the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). Eur Heart J. 2011;32(24):3147–97.
Hilfiker-Kleiner D, Haghikia A, Berliner D, Vogel-Claussen J, Schwab J, Franke A, et al. Bromocriptine for the treatment of peripartum cardiomyopathy: a multicentre randomized study. Eur Heart J. 2017;38(35):2671–9.
Merlo M, Pyxaras SA, Pinamonti B, Barbati G, Di Lenarda A, Sinagra G. Prevalence and prognostic significance of left ventricular reverse remodeling in dilated cardiomyopathy receiving tailored medical treatment. J Am Coll Cardiol. 2011;57(13):1468–76.
Ernande L, Derumeaux G. Diabetic cardiomyopathy: myth or reality? Arch Cardiovasc Dis. 2012;105(4):218–25.
Florwick A, Dharmaraj T, Jurgens J, Valle D, Wilson KL. LMNA sequences of 60,706 unrelated individuals reveal 132 novel missense variants in A-type lamins and suggest a link between variant p.G602S and type 2 diabetes. Front Genet. 2017;8:79.
Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49(1):46–53.
Dal Ferro M, Stolfo D, Altinier A, Gigli M, Perrieri M, Ramani F, et al. Association between mutation status and left ventricular reverse remodelling in dilated cardiomyopathy. Heart. 2017;103(21):1704–10.
Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348(6235):666–9.
Merlo M, Cannata A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228–39.
van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15(4):376–84.
Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal JM, Androulakis AF, et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol. 2016;68(21):2299–307.
Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41(5):771–80.
Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793–867.
Nishiuchi S, Makiyama T, Aiba T, Nakajima K, Hirose S, Kohjitani H, et al. Gene-based risk stratification for cardiac disorders in LMNA mutation carriers. Circ Cardiovasc Genet. 2017;10(6). pii: e001603.
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–28.
Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7(270):270ra6.
Jansweijer JA, Nieuwhof K, Russo F, Hoorntje ET, Jongbloed JD, Lekanne Deprez RH, et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail. 2017;19(4):512–21.
Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiaran Aizpurua A, Merken JJ, Wang P, et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J. 2018;39(10):864–73.
Begay RL, Graw S, Sinagra G, Merlo M, Slavov D, Gowan K, et al. Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc. 2015;4(11). pii: e002645.
Hastings R, de Villiers CP, Hooper C, Ormondroyd L, Pagnamenta A, Lise S, et al. Combination of whole genome sequencing, linkage, and functional studies implicates a missense mutation in titin as a cause of autosomal dominant cardiomyopathy with features of left ventricular noncompaction. Circ Cardiovasc Genet. 2016;9(5):426–35.
Begay RL, Tharp CA, Martin A, Graw SL, Sinagra G, Miani D, et al. FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Transl Sci. 2016;1(5):344–59.
Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68(22):2440–51.
Valdes-Mas R, Gutierrez-Fernandez A, Gomez J, Coto E, Astudillo A, Puente DA, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:5326.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this chapter

Cite this chapter
Dal Ferro, M., Severini, G.M., Gigli, M., Mestroni, L., Sinagra, G. (2019). Genetics of Dilated Cardiomyopathy: Current Knowledge and Future Perspectives. In: Sinagra, G., Merlo, M., Pinamonti, B. (eds) Dilated Cardiomyopathy . Springer, Cham. https://doi.org/10.1007/978-3-030-13864-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-13864-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-13863-9
Online ISBN: 978-3-030-13864-6
eBook Packages: MedicineMedicine (R0)