
Abstract
Peripheral nerve tumors include the benign neural sheath origin and other benign tumors that are not of neural sheath origin but involve the peripheral nerves. Malignant nerve tumors are divided into those of neural sheath origin and those of non-neural origin compressing or directly involving nerve by either direct extension or metastasis. Each of these categories of nerve tumors is discussed in this chapter.
Peripheral nerve tumors encompass neoplasms originating in peripheral nerves and those originating from tissues extrinsic to the peripheral nerves with consequent neural compression. Although much less common than nerve entrapment or nerve injuries, these peripheral nerve tumors are not rare, as several thousand cases are diagnosed annually [1–4]. Physicians undertaking the management of these lesions must have experience with the anatomy of the area involved by the tumor as well as a fundamental understanding of the internal structure of the involved nerve itself [5, 6]. Surgical resection is the treatment for most peripheral nerve tumors and can be quite complex with the risk of serious neurologic deficit. The pathologic diagnosis of these lesions can be difficult as well, and misinterpreted histology can result in incorrect treatment and serious disability. Physicians who are not comfortable in the diagnosis and treatment of these lesions should refer peripheral nerve tumors to a medical center with surgeons and pathologists with experience in such cases in order to maximize patient satisfaction and good outcomes.
Diagnosis
Peripheral nerve tumors have a broad and complex range of histopathology. The differential diagnosis of a mass or pain encompasses a wide range of etiologies, and peripheral nerve tumor is frequently overlooked. Commonly encountered neoplasms, such as schwannomas, neurofibromas, plexiform neurofibromas, and neurogenic sarcomas, have classic clinical presentations and characteristic gross and microscopic features [7]. The prototypically clinical presentation of a peripheral nerve tumor is a mass which can be mobilized perpendicular to the course of the nerve but not longitudinal to the nerve (Fig. 43.1a) [8]. Patients may also report induced paresthesia in the nerve distribution upon percussion of the nerve by the examiner. Once a peripheral nerve tumor is suspected, it is imperative to seek out evidence of other nerve tumors and von Recklinghausen’s disease (neurofibromatosis 1-NF1). Though MRI is not particularly helpful in managing patients with nerve injuries, MRI together with computed tomographic (CT) scanning is most useful in delineating the true extent of the tumor or identifying additional lesions diagnostic of NF1 (Fig. 43.1b). Angiography and myelography are occasionally required to fully evaluate complex lesions [9]. Any tumor located in close proximity to the spine should have the medial extent of the tumor carefully defined by such studies. Unfortunately, no imaging modality can conclusively differentiate schwannoma from neurofibroma or malignant versus benign peripheral nerve tumors with certainty. The diagnosis depends on histopathological evaluation of tumor tissue.

(a) Photo of a patient showing left posterior sciatic nerve mass (arrow) and (b) axial MRI scan showing a left sciatic nerve schwannoma
Once the diagnosis of a peripheral nerve tumor is made on clinical grounds, the clinician cannot determine at that point whether the diagnosis is a schwannoma, a neurofibroma, or a malignant nerve tumor. Depending on the physician’s experience with nerve tumors, it may be best to refer a patient to an appropriate medical center with experience in managing these complex patients where a definitive diagnosis can be made and, if necessary, definitive surgery can be performed. A partial or suboptimal resection may lead to clinical worsening.
Peripheral nerve tumors may be classified into benign and malignant tumors. The benign tumors may further be divided into those that are of neural sheath origin and those that involve the peripheral nerves but are not of neural sheath origin. Malignant tumors can be categorized as those of neural sheath origin, including neurogenic sarcomas, and malignancies of non-neural origin compressing or directly involving nerve by either direct extension or metastasis.
Benign Neural Sheath Tumors
Schwannomas
Schwannomas are the most common benign neural sheath tumor. The incidence of schwannomas is higher in females than males for reasons not fully understood [10]. The typical presentation of a schwannoma is a painless but palpable mass which may occur throughout the upper or lower extremities (Tables 43.1, 43.2, and 43.3). Percussing over the mass usually generates paresthesias in the distribution of the affected nerve (Tinel’s sign). In one series of 85 schwannomas, all patients but one had a Tinel’s sign [11]. Rarely, a schwannoma may present with a neurologic deficit or abnormal electrodiagnostic studies [12]. If the patient presents after a biopsy or an attempted resection of a schwannoma, symptoms then are more likely to be pain, paresthesias, neurologic deficit, or a combination of these symptoms. Occasionally, schwannomas can develop in patients with NF1 although neurofibromas are the most likely diagnosis in this population [13]. Schwannomatosis has recently been proposed as a separate syndrome in patients with multiple schwannomas but no stigmata of NF2 such as vestibular schwannomas[14]. Symptomatic tumors should be removed, provided that the patient is an acceptable surgical candidate.
Schwannomas most likely originate from Schwann cells, but the exact origin of these tumors has previously been disputed [15–17]. Histopathology reveals a tumor composed only of Schwann cells without fibroblasts or perineural cells. The tumor consists of two distinct cellular patterns. Antoni A tissue is characterized by hypercellular compact elongated cells which may demonstrate Verocay bodies made up of alternating parallel rows of tumor cells forming palisading region. Antoni B tissue is characterized by loosely arranged, hypocellular regions (Figs. 43.2 and 43.3). Lipid-laden cells may be found in either Antoni A or B tissue. The vasculature of schwannomas is often thickened, hyalinized, and may even be thrombosed. The compact Antoni type A tissue of a schwannoma must be differentiated from the less compact and more myxomatous matrix of a neurofibroma. Antoni type B tissue has a somewhat loose stroma and may be distinguished from a neurofibroma by mucopolysaccharide stain. Schwannoma matrix does not stain for mucopolysaccharide but neurofibroma matrix does. Schwannomas will demonstrate strong S 100 positivity and frequently also show positivity for podoplanin, calretinin, and SOX 10 [18–21].

Schwannoma: low-power view (a) showing compact (Antoni A) and loose (Antoni B) type areas. (b) High-power view showing Antoni A (spindle-shaped cells in left upper corner) and Antoni B type (pale loose stroma and more stellate-shaped cells in right lower corner)

Verocay bodies: regimented/palisaded, spindle-shaped nuclei and intervening nuclear free zones
Schwannomas are well encapsulated and displace adjacent nerve fascicles laterally. Larger schwannomas tend to stretch and elongate the fascicles but do so at such a slow rate that neurological function is typically preserved [10]. Generally, one or two nerve fascicles enter the tumor proximally and emerge distally. Electrical stimulation across these fascicles does not produce a nerve action potential (NAP); thus, surgical resection is therefore possible without interrupting functional nerve fibers. Operative removal is usually indicated based upon expectant improvement of pain, improvement or stabilization of motor symptoms [9], or to prevent the tumor from enlarging to the point at which such symptoms would develop. MRI can provide information regarding the relationship of the tumor to the surrounding structures [22, 23] but has not been reliably able to distinguish between schwannomas and neurofibromas [11].
Operative Procedure
The patient is positioned so that the involved limb’s response to electrophysiologic stimulation can be observed (Fig. 43.4). The anesthesia providers should be instructed to avoid muscle relaxant medications. Surgical exposure includes nerve and related structures both proximal and distal to the lesion itself (Fig. 43.5). Exposure may be limited if resecting a schwannoma involving the pelvic plexus or the proximal spinal nerve level of the brachial plexus where exposure either proximal or distal to the lesion may be difficult. The schwannoma is usually found surrounded by nerve fascicles [16]. As a schwannoma increases in size, it may bulge out of the nerve in a concentric manner, leaving fascicles asymmetrically arrayed around the tumor itself. Schwannomas are encapsulated; thus, by dissecting longitudinally along the mass, the fascicles can then be gently dissected away from the tumor capsule (Fig. 43.5). Further dissection at the proximal and distal poles of the tumor usually reveals a relatively small nerve fascicle entering and leaving the tumor. When these fascicles are stimulated across the tumor, no NAP is conducted yielding a flat recording [11]. Stimulation of the proximal or, rarely, distal tumor fascicle alone may generate muscle contraction in the distribution of the nerve. This is the result of retrograde stimulation of healthy fascicles not of conduction across the fascicles entering the tumor. The entering and exiting nerve fascicles are divided which allows the en bloc removal of the tumor with its capsule intact as a single mass (Fig. 43.6). When unable to perform an en bloc resection, the capsule of the schwannoma can be opened longitudinally, and its relatively soft, usually homogenous, sometimes cystic contents can be evacuated. Following internal debulking of the tumor, the capsule can be gently dissected away from the fascicles. If this method is utilized, it should be done thoroughly to reduce the incidence of recurrence. Large schwannomas may extend beyond the immediate nerve tissue making resection more difficult and increasing the possibility of recurrence [24]. Following careful resection, the presenting symptoms of schwannomas usually resolve. Eighty-seven percent of patients had improved or stable motor function in one series, and 85 % of patients who presented with pain had either total or partial resolution of this symptom [11].

Positioning and incision for left sciatic nerve tumor resection. Note leg is positioned to allow observation of motor response upon stimulation

Surgical exposure of right sciatic schwannoma with both proximal and distal anatomy exposed

Surgical resection of sciatic nerve schwannoma. (a) Dissection of tumor with proximal and distal involved nerve, (b) dissection along longitudinal axis of nerve, (c) continuation of dissection, and (d) removal of tumor en bloc with capsule intact
Solitary Neurofibromas
Local or radicular pain is more likely to occur as a presenting symptom with solitary neurofibromas than with schwannomas [16]. In one large series, pain was associated with neurofibromas in 47 % of cases versus 31 % of schwannoma cases [11]. Solitary neurofibromas are less common than neurofibromas associated with NF1. As with schwannomas, these tumors can generally be displaced perpendicular to the course of the nerve of origin but not longitudinally. A neurologic deficit is slightly more likely to be present at the time of presentation than with a schwannoma but is greatly increased in neurofibroma patients who have had a prior biopsy or attempted but incomplete removal [25]. Solitary neurofibromas produce symptoms due to local axonal compression and are likely to be fusiform rather than plexiform in non-NF1 patients [10]. Tinel’s sign is present in most cases; one series of 197 neurofibromas reported a positive test in all patients [11].
For unclear reasons, solitary neurofibromas are found more frequently on the right side of the body than the left [10]. As with schwannomas, neurofibromas can occur throughout the upper or lower extremities (Tables 43.1, 43.2, and 43.3), but unlike schwannomas, they are more likely to arise from the motor portion of the nerve than the sensory portion [10]. The female predominance of neurofibromas is even greater than that of schwannomas by mechanisms that are not well understood [10].
Neurofibromas originate from cells believed to be more primitive than a Schwann cell, likely arising from the perineural fibroblast [26]. Compared to a schwannoma, the tumor’s histological background is less compact and more myxomatous, contains mast cells and lymphocytes, and stains more positively with a mucopolysaccharide stain, such as Alcian blue (Figs. 43.7 and 43.8). The myxo-collagenous background of a neurofibroma also usually stains intensely with a reticulum stain as opposed to a schwannomas where this is not seen. Fewer Schwann cells are present in neurofibromas than schwannomas and are mixed together with distorted myelinated and unmyelinated axons. The vasculature in neurofibromas is less likely to be thickened and hyalinized than in schwannomas. Neurofibromas can occur as a localized mass or can involve longer segments of nerves. No histologic difference exists between a solitary neurofibroma and one associated with NF1. Unlike schwannomas, neurofibromas involve the entire cross-section of the nerve, resulting in the absence of cleavage planes between normal nerve and tumor. This makes gross total resection of the tumor without removal of normal nerve impossible.

Neurofibroma involving dorsal root ganglion: neoplasm composed of Schwann, perineurial-like and fibroblastic cells (neurons surrounded by satellite cells are residual from the infiltrated dorsal root ganglion)

Diffuse neurofibroma (a) low-power view and (b) high-power view. There is a disordered proliferation of relatively small spindle to stellate-shaped tumor cells, infiltrating adipose tissue and skeletal muscle
The optimal management for neurofibromas is guided by patient symptoms. Patients without symptoms with known neurofibromas can be managed expectantly with observation. Resection of neurofibromas is reserved for tumors causing pain, progressive neurologic deficits, or those in which the diagnosis is uncertain or malignancy is suspected. MRIs are useful to define the anatomic location and extent of the tumor [23]. In cases where the neurofibroma is on a proximal nerve, imaging studies of the spine should be performed to assess for intraspinal tumor extension [27].
Operative Procedure
The initial surgical approach is similar to that for a schwannoma. In most cases, the nerve fascicles are “basketted” around the main tumor mass and must be carefully dissected away. Neurofibromas usually have a capsule which is often more adherent to the central mass of the tumor than a schwannoma. The key steps for removal of a neurofibroma include dissecting out the fascicles at both ends of the tumor. If solitary, the fascicle entering and leaving the neurofibroma is larger than that seen in a comparable sized schwannoma. In other lesions, several fascicles approach and leave the tumor. It is more efficacious at times to section an entering or leaving fascicle or fascicles at one or the other pole and then gently elevate the mass away from the tumor and dissect beneath it to clear the mass away from the fascicles and the capsule left on the underside. NAP recording obtained across the tumor by stimulating the entering fascicle(s) is usually flat, indicating nonconducting fascicles that can be sectioned just as in a schwannoma [11]. NAP recording after the mass is removed can be used to confirm maintenance of most function just as in a schwannoma. Occasionally, the neurofibroma requires partial graft repair after resection, particularly when loss of function is a concern or when functional fascicles were sacrificed to gain complete tumor resection. Donner et al. reviewed their series of 123 neurofibromas occurring in 121 patients without NF1, noting that 90 % had stable or improved motor function and 88 % had partial or complete resolution of pain [11].
Neurofibromas Associated with Neurofibromatosis 1 (NF1)
Neurofibromatosis 1 (NF1), also known as von Recklinghausen disease, is a phakomatosis defined in part by multiple neurofibromas. NF1 is inherited on chromosome 17 in an autosomal dominant pattern with full penetrance but variable expression. The overall incidence of NF1 is approximately one per 300 individuals, with many afflicted patients showing mild symptoms with limited number of tumors [2]. NF1-associated neurofibromas are more evenly divided between males and females and are seen equally on both sides of the body [10]. Additionally, NF1-associated neurofibromas are more likely to present at an earlier stage than solitary neurofibromas [10]. The range of neurofibromas in NF1 includes tumors in small cutaneous nerves to larger lesions in larger proximal nerves [10]. Most cases of NF1-associated solitary neurofibroma can be excised without serious deficit, even when the tumor involves a major nerve. The technique is the same as that for a non-NF1 neurofibroma described above. In NF1 patients, there is a 15 % risk of malignant degeneration of the neurofibroma [28, 29]; thus, every attempt should be made to remove all tumor fragments intraoperatively.
Patients with NF1-associated neurofibromas do not have as high a rate of resolution of symptoms as patients with sporadic neurofibromas due to the more difficult dissection of neurofibromas in patients with NF1. A series of 48 patients with NF1 who had neurofibromas resected noted that 83 % had stable or improved motor function and 74 % had partial or complete resolution of pain symptoms [11]. However, 16 % of patients experienced new but usually mild pain syndromes. Since neurofibromas in NF1 patients are more likely to be plexiform, the incidence of total resection is 60 % as compared to 80 % for solitary neurofibromas [11].
Plexiform Neurofibromas
Plexiform neurofibromas are histologically identical to solitary neurofibromas (Fig. 43.9) but involve a length of nerve in an interweaving fashion [11]. Plexiform tumor masses lack a capsule and can be both intrafascicular and extrafascicular. These tumors present an operative challenge, but resection is possible, though complete resection is generally not feasible without incurring a neurological deficit. Indications for surgery include symptomatic tumors involving sensory nerves or branches. Even an attempt at a partial resection of a plexiform tumor may lead to a neurological deficit. Nerve section, both proximal and distal to such a lesion and repair of the gap, which is usually lengthy, does not usually restore function [30, 31]. Decompression and debulking of the tumor may provide a benefit in patients with severe pain as a dominant symptom or with large tumors. In selected cases, it may be possible to reduce pain by decompressing a nerve through a neurolysis. Tumors involving less important sensory nerves or branches, such as antebrachial cutaneous, superficial sensory radial, sural, or saphenous nerves, can be removed in total, along with the nerve of origin.

Plexiform schwannoma, low-power view (a): multiple, well-circumscribed tumor nodules involving dermis. (b) High-power view showing well-circumscribed tumor nest, spindle cells arranged in a typical fascicles and palisades
Plexiform neurofibromas are very likely to be associated with NF1 but rarely may be also encountered as sporadic, solitary non-NF1 associated lesions. A sizable plexiform tumor can be accompanied by hundreds of smaller neurofibromas involving the nerve of origin of the larger lesion. These associated tumors involve the nerve proximal to and distal to the large lesion and, occasionally, other nerves in the same limb, a syndrome called regionalized NF1, or neurofibromatosis type 5 [32]. Only a palliative operative procedure can be performed in these situations. If the plexiform tumor is large and firm, removal can be considered to ensure that malignant transformation is not present, a process that occurs in 5 % of plexiform neurofibromas, usually marked by significant pain and rapid growth [33]. Secondary operations for neural sheath tumors for which repair are necessary require frozen section biopsy of nerves or elements of origin to ensure that residual tumor is not incorporated in the repair.
In one series, nine patients with plexiform neurofibromas were treated either by partial excision (n = 5), internal neurolysis with gross total excision (n = 1), internal neurolysis with subtotal excision (n = 1), or division of the nerve both proximal and distal to the tumor with nerve grafts (n = 2) [13]. Tumors recurred in four patients with the time to symptomatic recurrence being 4–19 months. All four of these patients had worse motor exams, and three of the four developed new pain syndromes. MRI is useful in monitoring the rate of growth of plexiform neuromas as rapid growth and development of pain may herald malignant degeneration [34].
Neurothekeoma
Neurothekeomas are benign nerve sheath tumors that generally occur in the first three decades of life [35]. They are soft mobile tumors often based in the dermis. The cell of origin for these tumors was believed to occur in either the Schwann cell line or the perineural cell line, and tumor cells are often arranged in fascicles bounded by myxoid stroma. Recent investigations have shown that these tumors are positive for vimentin, CD10, NKI/C3, and at times focal reactivity for smooth muscle actin, yet are negative for S 100 and GFAP [36]. It is postulated that neurothekeomas may actually be of fibroblastic origin [36]. Treatment is symptomatic, with most neurothekeomas removed suspected to be schwannomas peroperatively. A gross total resection can almost always be performed with only rare recurrences.
Other Benign Peripheral Nerve Tumors
Several types of tumors occur less frequently than lesions of neural sheath origin but can be responsible for pain and, in some instances, loss of nerve function (Table 43.4). Some of these are favorable lesions as they tend to cause symptoms from nerve compression rather than originating within neural tissue [10]. On occasions, these lesions may be adherent to the epineurium making resection difficult and recurrence rate high [37]. The following is a discussion of the more common lesions.
Desmoids and Myositis Ossificans
Desmoids are lesions believed to arise from muscle and can involve soft tissue and compress, incorporate, or adhere to major nerves. They originate from a mesenchymal origin and are benign in the sense that they do not metastasize to other parts of the body, but they are rarely cured due to their invasiveness of soft tissue. Recurrence after a presumed gross total resection is common [4]. Desmoids are most common on the abdominal wall, but they can be present in the neck, shoulder, or extremities as well. Operative treatment involves a relatively wide exposure of the lesion and early identification of any nerve or nerves involved. The desmoid usually needs to be sharply dissected away from the nerve and involved epineurium usually requires resection. Both the firmness of these lesions and their adherence to nerves and vessels make removal without deficit difficult unless great care is taken during the dissection. As a result, recurrence is common. Desmoids are associated with patients with familial adenomatous polyposis, a condition caused by a mutation of the APC gene, and studies are showing that certain APC mutations will generate a desmoid phenotype [38, 39].
Myositis ossificans is a poorly defined disorder that may be related to previous trauma or surgery. It usually produces a firm to hard mass of tissue with calcification and, like a desmoid, can envelope contiguous soft tissues, including bone. Symptoms develop due to neural compression, vascular compression, or both. Removal of these masses can be technically difficult. Complete resection is rarely possible and usually not indicated as neurolysis of the nerve often dramatically improves symptoms.
Myoblastomas and Lymphangiomas
These two pathologic entities have similar behavior and somewhat similar appearance during surgery. Myoblastomas consist of plump and somewhat angular cells in a compact arrangement with acidophilic granules, whereas lymphangiomas have cells of a lymphoid nature. These tumors tend to spread as a sheet of tumorous tissue and are less likely to form a globular mass than are a desmoid tumor or a hemangioma [4, 32]. Both tumors can also become adherent to or envelope nerves leading to neurological symptoms. Treatment involves a wide surgical exposure to determine normal anatomy distal and proximal to the lesion prior to skeletonizing the involved nerve away from the lesion.
Lipomas and Lipohamartomas
Lipomas are fatty lesions that usually occur solely in the subcutaneous region thus rarely involve the major nerves. Rarely, a large lipoma envelops or compresses nerve, or originates deep to the subcutaneous tissues in a limb and thus compresses or entraps nerve producing neurologic symptoms [4]. Common sites include the forearm or popliteal region, with resultant posterior interosseous or fibular (peroneal) nerve palsy, respectively. Larger lipomas in the supraclavicular fossa can involve the brachial plexus, while those in the buttock, leg, or arm can potentially compress the median, sciatic, or ulnar nerves.
Lipohamartomas are also composed of fatty tissue and are intrinsic to nerve and usually involve the median nerve at the wrist or palmar levels [32]. When median nerve lesions present with pain and paresthesias, treatment is sectioning of both the transverse carpal ligament in the palm, its extension covering the nerve and the lipomatous mass at the wrist level. When serious loss of median function occurs, more extensive surgery consisting of internal neurolysis with reduction of the bulk of the tumor from around individual fascicles can be performed. More focal lipohamartomas can be treated with resection and repair.
Hemangiomas, Hemangiopericytomas, and Vascular Malformations
Vascular lesions including hemangioma, hemangiopericytomas, as well as aneurysms and arteriovenous fistulae, originate close to nerves and can compress or elevate them [40]. An operation for these lesions, especially if the nerve is involved, is never easy. Preoperative angiography is of some help in planning surgery. Intraoperatively, it is imperative to isolate vessels on the periphery of the lesion and ligate or clip them if they are not the major supply to the extremity. Nerves are carefully dissected away from the lesion while protecting them as much as possible. Occasionally, a hemangioma, hemangioblastoma, venous aneurysm, or fistula directly involves nerve or appears to originate in it. If the clinical situation warrants proceeding with surgical resection, a careful interfascicular dissection is necessary for removal. Each fascicle or group of fascicles needs to be stripped of the abnormal vascular tissue to assure adequate resection of the vascular lesion.
Hemangiopericytomas, which usually arise in the mediastinum and secondarily involves the brachial plexus, can behave in a malignant fashion and metastasize to other sites, including, rarely, the brain. It cannot usually be removed entirely, and the surgeon must be content with a subtotal procedure to decompress the brachial plexus.
Ganglions and Epidermoid Cysts
Most ganglions arise from joints and at sites that do not usually involve nerves, such as the dorsum or the side of the wrist [41]. Occasionally, this tumor arises from a region of the wrist joint that compresses the thenar sensory branch of the median nerve, the ulnar palmar branch, or the main trunk of the median or ulnar nerves. Other sites where these ganglions are clearly of joint origin include the forearm, where they arise from the radioulnar joints and compress the posterior interosseous nerve; the knee, involving the fibular (peroneal) nerve [42]; the ankle, involving the posterior tibial nerve; and the hip, involving the sciatic nerve. Ganglions at the elbow level can involve radial and, less frequently, median and ulnar nerves, whereas those in the shoulder can involve the brachial plexus, particularly the suprascapular branch in the region of the scapular notch. Ganglion cysts usually present as a tender mass resulting in pain or paresthesias in the distribution of the nerve involved [10]. Occasionally, they result in a motor and sensory deficit or both. Another type of ganglion does not appear to have a connection with a joint, such as those found in the deep fibular (peroneal) nerve over the head of the fibula [43, 44]. It is likely that these lesions originate from the superior tibiofibular joint after disruption of its capsule, allowing dissection of the synovial fluid along the articular branch of the fibular nerve reaching the epineurium of the nerve. The previous connection with the joint is often then obliterated. Intraneural ganglia may extend great distances along the course of the nerve, such as the fibular (peroneal) division of the sciatic nerve, resulting in motor or sensory deficit [4].
Most localized lesions are resectable without serious loss of function. For ganglions extrinsic to nerve but compressing it, the surgeon must dissect around such lesions after carefully identifying, dissecting away, and protecting the involved nerve. The origin of the cystic lesion should be ligated or secured in some other fashion close to the joint to reduce recurrence. For intraneural ganglions, interfascicular dissection seems to work best because fascicles are cleared of the cyst and then its capsule. For larger lesions, it may be necessary to evacuate the synovial-like contents of the cysts and dissect the capsule away from the decompressed and split fascicles. Occasionally, larger lesions require several operations before they are obliterated.
Epidermoid cysts usually do not arise within nerve itself but may compress an adjacent nerve. Common locations for the epidermoid cysts involve the sciatic nerve close to the sciatic notch or behind the knee, involving the posterior tibial nerve [4]. These cysts can be resected as a solitary mass after performing neurolysis on and gently retracting involved nerves. Occasionally, a large lesion at the hip or pelvic level requires initial evacuation and then dissection of the capsule away from adjacent tissues, including nerves.
Hypertrophic Neuropathy or Onion-Whorl Disease
Hypertrophic neuropathy is a rare disease of unknown etiology and pathogenesis. Fascicles and individual nerve fibers become encased by connective tissue in a circular fashion, as if the endoneurium has proliferated. This results in a hypertrophic nerve with an “onion-whorl” appearance to its nerve fibers. Histologically, there is proliferation of perineural cells, endoneurial fibrosis, fibrotic replacement of the perineurium, and significant reduction in myelin [10].
Hypertrophic neuropathy primarily affects children or young adults with a predilection for the fibular (peroneal) nerve in the leg or the median nerve in the arm [45, 46]. Less commonly, it can also be found in the brachial plexus as well as the ulnar, radial, and sciatic nerves [4, 45, 47]. The lesion usually involves a lengthy segment of nerve resulting in a progressive loss of function. It does not spread to other nerves in the body. Some of its histologic features suggest either a contusive stretch injury or a chronically compressive etiology, but a history of significant trauma is usually absent, and no obvious entrapment or irritative environment is found on surgical exploration.
Pain or progressive loss of function warrants exploration. Function is usually partially spared distal to the lesion, and NAPs across the lesion are often present. External or external and internal neurolysis is usually performed. Manipulation of the lesion, particularly by internal neurolysis, may produce additional and even complete loss of function, despite the fact that neurolysis is less invasive than resection. An alternative is to proceed with resection, despite the attendant loss, and to replace the lost segment by grafts. Since the disease usually involves a significant segment of nerve, the grafts are likely to be long.
Malignant Neural Sheath Tumors
Malignant neural sheath tumors (MNST) are a group of neoplasms consisting of malignant schwannomas and malignant neurofibromas, commonly grouped as neurogenic sarcoma and fibrosarcomas (Table 43.5) [27, 48]. The differentiation between malignant schwannomas and malignant neurofibromas can be difficult, a positive mucopolysaccharide stain favoring the latter. Histopathologic analysis reveals mitotic activity, cellular pleomorphism, necrosis, and hemorrhage in most cases (Figs. 43.10, 43.11, 43.12, and 43.13). Heterogeneous cell lines are present in 15 % of MNSTs and are generally found in patients with neurofibromatosis [48]. Approximately half of MNST arise in patients with NF1 [48] but can occur as solitary lesions in patients without NF1 [49]. Spontaneous malignant degeneration of solitary benign schwannomas is rare except in patients with NF1 [7]. Malignant degeneration of a plexiform neurofibroma in a patient with NF1 is not uncommon [18]. There is no gender preference in MNST and no specific laterality. Prior treatment with radiation therapy is a risk factor in the development of approximately 10 % of cases of MNST [48, 50]. MNST can be suspected if a mass increases rapidly in size and is associated with a progressive loss of function [51]. These lesions are often firmer than benign neural sheath tumors and are also relatively adherent to surrounding structures.

Malignant neural sheath tumor, conventional: cellular (fibrosarcoma-like) spindle cell neoplasm with frequent mitoses and perivascular condensation of tumor cells

Malignant neural sheath tumor, epithelioid: discohesive round to polygonal cells with well-defined cytoplasmic borders (epithelioid), embedded in a loose/myxoid stroma. Mitotic activity is high

Malignant neural sheath tumor, with divergent (epithelial) differentiation: spindle cell neoplasm with focal gland formation

Malignant neural sheath tumor, with divergent (rhabdomyosarcomatous) differentiation (high-power view of H and E stained smear): atypical elongated cell with eosinophilic cytoplasm myofibrillar bands
MNST usually manifests as pain with or without progressive loss of neurological function. Other common findings of MNST include a mass with irregular borders, a rapid increase in size, and a large size at presentation [10]. On rare occasion, patients with MNST present with metastasis to lung, bone, liver, or other organs [10]. MR imaging (Fig. 43.14a, b) is useful in defining the anatomy of the tumor as well as to screen for metastasis [22]. The 5-year survival for patients with MNST can approach 50 % but is much lower in patients with NF1 [52].

MR imaging of malignant peripheral nerve sheath tumor of left sciatic nerve (a) coronal T1 pre-contrast image (b) axial T1 post-contrast image
Despite preoperative symptoms and findings suggesting malignancy, the clinician cannot be certain of the diagnosis without a biopsy, which usually requires an operation. If prior biopsy was performed with a diagnosis of MNST, then a metastatic evaluation needs to be performed including a chest x-ray, CT or MR scan, abdominal CT, and bone scan. Following this and based on the extent of metastatic disease, options include the following:
-
1.
Perform wide local resection, including removal of adjacent as well as adherent soft tissues (Fig. 43.15a, b). Resection should include a several-centimeter margin of entering and exiting nerve shown to be free of malignant changes on frozen and subsequent permanent sections. At that time, some surgical oncologists favor placement of tissue rods for irradiation locally, whereas others favor external radiation. Chemotherapy may also be added.
Fig. 43.15 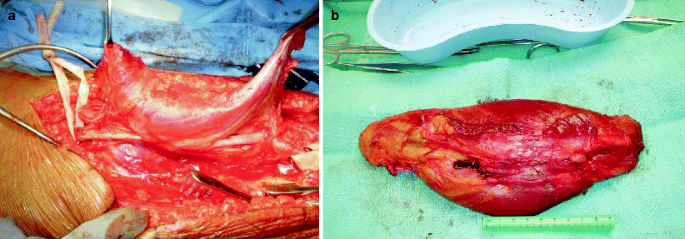
Surgical resection of malignant peripheral nerve sheath tumor of sciatic nerve. (a) Note adherent biceps femoris to tumor and wide local resection to remove adjacent and adherent tissue. (b) Resected tumor with wide margin or tissue and visible proximal and distal stump of sciatic nerve
-
2.
Amputate the limb well above the level of the lesion. Proximal upper limb lesions can also be managed by forequarter amputation of shoulder and arm above the level of the plexus or spinal nerves. Proximal lower limb lesions may require hip disarticulation with section of the proximal sciatic and femoral nerves.
-
3.
Irradiate the limb with or without adjuvant chemotherapy in patients who refuse surgery beyond an attempt to remove the lesion or in patients with widely metastatic disease.

A critical step in managing these lesions is to obtain as thorough a neuropathologic examination of as much tissue as possible. As a result, further treatment depends heavily on the nature of the malignancy as determined by permanent sections. The recurrence rate is high, even after wide local excision and irradiation, but some patients survive beyond 5 years with proper management [53]. Although patients have died despite forequarter amputation or hip disarticulation when results for metastasis were negative, the largest percentage of survivors beyond 5 years is in this category [27]. In recent years, however, limb-sparing procedures, including wide local resection, placement of x-ray rods, and sometimes external x-ray treatment, in conjunction with chemotherapy in carefully selected patients resulted in good to excellent survival rates [54–56]. Therefore, limb-sparing surgery is an accepted form of treatment in many patients with MNST.
Metastatic Carcinoma
This diverse group of tumors usually involves nerves by direct extension from the primary site. Occasionally, a malignancy may metastasize to nerve or tissues adjacent to it. Treatment plans must be individualized given the wide scope of variability in presentation and extent of involvement with each particular cancer. Breast carcinoma is the most common metastatic lesion involving nerve [4, 27]. Metastases to nerves have also been described with lung, melanoma, thymoma, pancreatic, and prostate cancer [8, 27, 57–59]. Most of these metastases occur in the brachial plexus. Metastasis located at an infraclavicular axillary level appears to present by direct extension, while metastasis located at the supraclavicular plexus is more likely with breast cancer disseminated through the lymph nodes. Most of these patients had prior mastectomy followed by irradiation, making differentiation between neoplastic plexopathy with metastatic breast cancer and radiation plexopathy more challenging (see Chap. 46) [60, 61].
If a mass is palpable or seen on scan in a patient with a history of breast cancer, then decompression of involved neural elements is reasonable. Unlike malignant neural sheath tumors, the nerve involved by metastasis can be treated with external neurolysis and careful removal of tumor from involved nerve. As much of the adjacent mass as possible is also removed. Metastases do not typically invade nerve beyond the epineural level, but exceptions exist. In cases of invasion of the neural elements by metastatic lesions, resection of the involved neural elements may be considered for pain control as a palliative procedure.
Metastatic lung cancer also commonly involves nerve and particularly the brachial plexus. Lung cancer may involve the brachial plexus by direct extension and most often produces a Pancoast syndrome. If pain is a severe problem, a posterior subscapular resection of the first rib and subtotal resection of the apical tumor to decompress the lower elements of the plexus may be performed as a palliative procedure. This procedure combined with cervical laminectomy for associated epidural metastatic disease is also palliative. Occasionally, a high contralateral open cervical cordotomy also helps control the pain associated with a Pancoast syndrome. The main focus for this palliative operation is adequate decompression of the compressed or entrapped nerve [62].
True metastatic disease involving nerve is seen with lymphoma, bladder cancer, and melanoma, although it is less common than breast or pulmonary cancer. With melanomas involving the plexus, removing the tumor from any epineural attachment has sufficed. The surgical procedure is then followed by local irradiation. A similar approach is also sufficient, at least for palliative purposes, for lymphoma.
References
Seddon HJ. Surgical disorders of the peripheral nerves. Baltimore: Williams and Wilkins; 1972.
Hajdu SI. Peripheral nerve sheath tumors. Histogenesis, classification, and prognosis. Cancer. 1993;72(12):3549–52.
Brooks D. Clinical presentation and treatment of peripheral nerve tumor. In: R.D Bunge P, Lambert E, Thomas P, editors. Peripheral neuropathy. Philadelphia: W.B. Saunders; 1984. p. 2236–51.
Kline DG, Hudson AR. Operative results of major nerve injuries, entrapments, and tumors. Philadelphia: W.B. Saunders; 1994.
Hudson AR. Peripheral nerve surgery. In: R.B Bunge P, Lambert E, Thomas P, editors. Peripheral neuropathy. Philadelphia: W.B. Saunders; 1984. p. 420–38.
Rizzoli HV, Horwitz NH. Peripheral nerve tumors. In: Horwitz NH, Rizzoli HV, editors. Postoperative complications of extracranial neurological surgery. Baltimore: Williams and Wilkins; 1987. p. 283–98.
Harkin JC, Reed RJ. Tumors of the peripheral nervous system. In: Atlas of tumor pathology, fasicle, vol. 3. Washington, D.C.: Armed Forces Institute of Pathology; 1969.
Campbell R. Tumors of the peripheral nerve. In: Youmans J, editor. Neurological surgery. Philadelphia: W.B. Saunders; 1990. p. 3667–75.
Craviato H. Neoplasms of peripheral nerve. In: Wilkins R, Rengachary S, editors. Neurosurgery. Baltimore: Williams and Wilkins; 1988. p. 1894–9.
Kline DG, Hudson A. Nerve injuries. Philadelphia: W.B. Saunders; 1995.
Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg. 1994;81(3):362–73.
Thomas JE et al. Neurogenic tumors of the sciatic nerve. A clinicopathologic study of 35 cases. Mayo Clin Proc. 1983;58(10):640–7.
Das Gupta TK et al. Benign solitary schwannomas (neurilemomas). Cancer. 1969;24(2):355–66.
MacCollin M et al. Schwannomatosis: a clinical and pathologic study. Neurology. 1996;46(4):1072–9.
Ehrlich HE, Martin H. Schwannomas (neurilemomas) in the head and neck. Surg Gynecol Obstet. 1943;76:577–83.
Woodhall B. Peripheral nerve tumors. The surgical clinics of North America. 1954. p. 1167–72.
Fisher ER, Vuzevski VD. Cytogenesis of schwannoma (neurilemoma), neurofibroma, dermatofibroma, and dermatofibrosarcoma as revealed by electron microscopy. Am J Clin Pathol. 1968;49(2):141–54.
Rodriguez FJ et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123(3):295–319.
Fine SW, McClain SA, Li M. Immunohistochemical staining for calretinin is useful for differentiating schwannomas from neurofibromas. Am J Clin Pathol. 2004;122(4):552–9.
Naber U et al. Podoplanin and CD34 in peripheral nerve sheath tumours: focus on neurofibromatosis 1-associated atypical neurofibroma. J Neurooncol. 2011;103(2):239–45.
Nonaka D, Chiriboga L, Rubin BP. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol. 2008;32(9):1291–8.
Suh JS et al. Peripheral (extracranial) nerve tumors: correlation of MR imaging and histologic findings. Radiology. 1992;183(2):341–6.
Cerofolini E et al. MR of benign peripheral nerve sheath tumors. J Comput Assist Tomogr. 1991;15(4):593–7.
Gyhra A et al. Schwannoma of the brachial plexus with intrathoracic extension. Thorax. 1980;35(9):703–4.
de Souza FM, Smith PE, Molony TJ. Management of brachial plexus tumors. J Otolaryngol. 1979;8(6):537–40.
Haraida S. Comparison of various basement membrane components in benign and malignant peripheral nerve tumours. Virchows Arch A Pathol Anat Histopathol. 1992;421(4):331–8.
Lusk MD, Kline DG, Garcia CA. Tumors of the brachial plexus. Neurosurgery. 1987;21(4):439–53.
Byrne JJ, Cahill JM. Tumors of major peripheral nerves. Am J Surg. 1961;102:724–7.
Hosoi K. Multiple neurofibromatosis (von Recklinghausen’s disease) with special reference to malignant transformation. Arch Surg. 1931;22:258–81.
Das Gupta TK. Tumors of the peripheral nerves. Clin Neurosurg. 1978;25:574–90.
Richardson RR et al. Neurogenic tumors of the brachial plexus: report of two cases. Neurosurgery. 1979;4(1):66–70.
Byrne JJ. Nerve tumors. In: Gelberman R, editor. Operative nerve repair and reconstruction. Philadelphia: J.B. Lippincott; 1991.
Belzerg AJ, Campbell JN. Neoplasms of peripheral nerves. In: Wilkins RH, editor. Neurosurgery. New York: McGraw-Hill; 1996. p. 3217–24.
Tucker T et al. Longitudinal study of neurofibromatosis 1 associated plexiform neurofibromas. J Med Genet. 2009;46(2):81–5.
Gallager RL, Helwig EB. Neurothekeoma–a benign cutaneous tumor of neural origin. Am J Clin Pathol. 1980;74(6):759–64.
Fetsch JF et al. Neurothekeoma: an analysis of 178 tumors with detailed immunohistochemical data and long-term patient follow-up information. Am J Surg Pathol. 2007;31(7):1103–14.
Woodruff JM. The pathology and treatment of peripheral nerve tumors and tumor-like conditions. CA Cancer J Clin. 1993;43(5):290–308.
Bertario L et al. Genotype and phenotype factors as determinants of desmoid tumors in patients with familial adenomatous polyposis. Int J Cancer. 2001;95(2):102–7.
Couture J et al. A germline mutation at the extreme 3′ end of the APC gene results in a severe desmoid phenotype and is associated with overexpression of beta-catenin in the desmoid tumor. Clin Genet. 2000;57(3):205–12.
Curtis RM, Clark GL. Tumors of the blood and lymphatic vessels. In: Gelberman R, editor. Operative nerve repair and reconstruction. Philadelphia: J.B. Lippincott; 1991.
Tindall SC. Ganglion cysts of peripheral nerves. In: Wilkins RH, Rengachary SS, editors. Neurosurgery. New York: McGraw-Hill; 1996. p. 3225.
Kim DH, Kline DG. Management and results of peroneal nerve lesions. Neurosurgery. 1996;39(2):312–9; discussion 319–20.
Nathan P, Young NP, Sorenson EJ, Spinner RJ, Daube JR. Clinical and electrodiagnostic correlates of peroneal intraneural ganglia. Neurology. 2009;72:447–52.
Hebert-Blouin MN, Amrani KK, Wang H. Tibialis anterior branch involvement in fibular intraneural ganglia. Muscle Nerve. 2010;41:524–32.
Bilbao JM et al. Perineurioma (localized hypertrophic neuropathy). Arch Pathol Lab Med. 1984;108(7):557–60.
Mitsumoto H, Wilbourn AJ, Goren H. Perineurioma as the cause of localized hypertrophic neuropathy. Muscle Nerve. 1980;3(5):403–12.
Hudson AR, Trammer B. Brachial plexus injuries. In: Rengachary S, Wilkins R, editors. Neurosurgery. New York: McGraw-Hill; 1985. p. 1817–32.
Ducatman BS et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006–21.
D’Agostino AN, Soule EH, Miller RH. Primary malignant neoplasms of nerves (malignant neurilemomas) in patients without manifestations of multiple neurofibromatosis (Von Recklinghausen’s disease). Cancer. 1963;16:1003–14.
Foley KM et al. Radiation-induced malignant and atypical peripheral nerve sheath tumors. Ann Neurol. 1980;7(4):311–8.
Vieta JO, Pack GT. Malignant neurilemmomas of peripheral nerves. Am J Surg. 1951;82(4):416–31.
Ghosh BC et al. Malignant schwannoma. A clinicopathologic study. Cancer. 1973;31(1):184–90.
Gentili FR, B. Malignant peripheral nerve tumors. Abstract at meeting. In: Eighth international congress of neurological surgery. Toronto, Canada. 1985;7–13.
Williard WC et al. Comparison of amputation with limb-sparing operations for adult soft tissue sarcoma of the extremity. Ann Surg. 1992;215(3):269–75.
Springfield DS. Introduction to limb-salvage surgery for sarcomas. Orthop Clin North Am. 1991;22(1):1–5.
Rooser B, Gustafson P, Rydholm A. Is there no influence of local control on the rate of metastases in high-grade soft tissue sarcoma? Cancer. 1990;65(8):1727–9.
Nagai H, Kuroda A, Morioka Y. Lymphatic and local spread of T1 and T2 pancreatic cancer. A study of autopsy material. Ann Surg. 1986;204(1):65–71.
Artico M et al. Late intraneural metastasis of the brachial plexus from mammary carcinoma. Report of a case. J Neurosurg Sci. 1991;35(1):51–3.
Ampil FL. Brachial plexus neuropathy from prostatic carcinoma metastasis. J La State Med Soc. 1986;138(9):38–40.
Kori SH, Foley KM, Posner JB. Brachial plexus lesions in patients with cancer: 100 cases. Neurology. 1981;31(1):45–50.
Lederman RJ, Wilbourn AJ. Brachial plexopathy: recurrent cancer or radiation? Neurology. 1984;34(10):1331–5.
Hudson AR, Berry H, Mayfield F. Chronic injuries of peripheral nerves by entrapment. In: Youmans J, editor. Neurological surgery. Philadelphia: W.B. Saunders; 1982. p. 2430–74.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Gressot, L.V., Islup, K., Kline, D.G., Kim, D.H. (2014). Peripheral Nerve Tumors. In: Katirji, B., Kaminski, H., Ruff, R. (eds) Neuromuscular Disorders in Clinical Practice. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6567-6_43
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6567-6_43
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6566-9
Online ISBN: 978-1-4614-6567-6
eBook Packages: MedicineMedicine (R0)