
Abstract
The Budd-Chiari syndrome (BCS) is an uncommon and life-threatening disorder defined as the obstruction of hepatic venous outflow regardless of its causative mechanism or level of obstruction. The clinical presentation of BCS is highly variable and can range from asymptomatic cases to fulminant hepatic failure with encephalopathy. In the vast majority of cases, it is possible to identify an inherited or acquired prothrombotic risk factor as the underlying cause of thrombosis being chronic myeloproliferative diseases the most frequent etiological cause of BCS. In many cases, multiple factors are present. Anticoagulation is mandatory in patients with BCS. The need for an additional intervention, such as hepatic vein angioplasty, thrombolysis, transjugular intrahepatic portosystemic shunt, surgical shunts, or liver transplantation, depends on the severity of symptoms and response to treatment.
Similar content being viewed by others


Keywords
- Chronic myeloproliferative disease
- JAK2V617F mutation
- Prothrombotic disorder
- Anticoagulation
- Surgical shunt
- Transjugular intrahepatic portosystemic shunt
- Liver transplantation
Introduction
The Budd-Chiari syndrome (BCS) is an uncommon and life-threatening disorder defined as the obstruction of hepatic venous outflow regardless of its causative mechanism or level of obstruction. This obstruction can be traced from the small hepatic venules up to the entrance of the inferior vena cava (IVC) into the right atrium [1]. Hepatic outflow obstruction related to cardiac disease, pericardial disease, or sinusoidal obstruction syndrome is excluded from this definition [2].
The BCS is classified as primary or secondary depending on the cause of the obstruction: primary, when it originates from the vein, such as thrombosis or phlebitis; and secondary, when the cause of obstruction originates outside the vein (compression or invasion by tumors, abscess) [2, 3]. Obstruction leads to sinusoidal congestion, followed by ischemia and hepatocellular necrosis. This can result in centrilobular fibrosis, nodular regenerative hyperplasia, and finally cirrhosis [4].
In the vast majority of cases, it is possible to identify an inherited or acquired prothrombotic risk factor as the underlying cause of thrombosis. Furthermore, in many patients a combination of several risk factors is present.
The clinical presentation of BCS is highly variable and can range from asymptomatic cases to fulminant hepatic failure with encephalopathy [1, 5]. Typical symptoms are the triad of abdominal pain, hepatomegaly, and ascites. The diagnosis can only be established upon unequivocal radiological confirmation of hepatic venous outflow obstruction.
Anticoagulation is mandatory in all patients with BCS. The need for an additional intervention, such as hepatic vein (HV) angioplasty, thrombolysis, TIPS, surgical shunts, or liver transplantation depends on the severity of symptoms and response to treatment.
Epidemiology
There are limited data available regarding the prevalence and incidence of BCS. Interestingly, it varies according to geographic distribution with wealthy countries demonstrating much lower incidence rates [2, 3]. For example, studies from France and Spain have shown incidence rates of 1 in 2.5 million inhabitants per year [6], and 0.41 per million inhabitants per year [7], respectively. Conversely, BCS is the leading cause of hospitalization due to liver disease in third world countries [8, 9].
These geographic differences between developing and developed countries also apply to the type and level of obstruction to hepatic outflow. In Asia a pure IVC or combined IVC/HV block predominates, often related to membrane–web occlusion, that is thought to be the consequence of a previous thrombus [10, 11]; whereas in the West, pure HV block caused by HV thrombosis is most common [2, 3].
Differences also exist according to gender and age. While there is a slight male predominance in Asia, with a median age at diagnosis of 45 years, in Europe, BCS has a greater female prevalence [3] and a younger median age (35–38 years) at diagnosis [7]. This female predominance has decreased in recent BCS series [7, 12], attributed, at least in part, to the lower estrogen content of newer oral contraceptives.
Etiology
The BCS can be classified into primary or secondary disease. Given the different therapeutic and prognostic implications of secondary BCS, in the present review we will refer only to primary BCS.
In approximately 90% of BCS patients, at least one predisposing thrombophilic factor is identified. In 25–46% of cases, several factors coexist [7, 13–15]. Therefore, it is advisable to perform a complete etiological study even after a candidate thrombophilic factor has been identified [7]. Prothrombotic causes can be acquired or inherited (Table 13.1 ).
Acquired
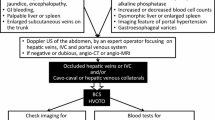
Chronic myeloproliferative diseases (MPD) are the most frequent etiological cause of BCS, found in up to 50% of cases [7, 14, 16]. The characteristic increase of blood cells in MPD is often, however, masked by the presence of portal hypertension with its consequent expansion of plasma volume [17] and hypersplenism [18]. Hence, all patients with BCS should be evaluated for MPD, even in the absence of typical clinical findings. The diagnosis of MPD has recently been facilitated by genetic testing for the V617F mutation of the JAK2 gene. This is recognized as a diagnostic criterion of MPD according to the most recent World Health Organization guidelines [19]. By including the analysis of this mutation in the etiological workup of patients with BCS, the probability of diagnosing underlying MPD increases by up to 20% [20]. However, even though over 90% of patients with polycythemia vera, 50–70% of patients with essential thrombocytosis and 40–50% of patients with primary myelofibrosis carry the V617F mutation of JAK2 gene [16, 18], the presence of this mutation does not define the phenotype of MPD. Hence, it is often necessary to perform additional hematological studies, including morphological evaluation of the bone marrow. Figure 13.1 details the proposed diagnostic algorithm for MPD in BCS.

Diagnostic algorithm of myeloproliferative disorders in Budd-Chiari syndrome
Pregnancy and the use of oral contraceptives are recognized prothrombotic risk factors in patients with BCS [21]. The prevalence of oral contraceptive use in BCS patients is estimated at up to 50–60%. Although common, both of these risk factors are weak and therefore almost always occur in conjunction with another predisposing thrombophilic condition [21, 22]. Apparently, pregnancy and contraceptive use are not sufficient to induce BCS in the absence of an underlying prothrombotic factor. Therefore, they behave as triggers or facilitators of thrombosis in these cases. Hence, both should be considered as risk factors for thrombotic diseases but not as primary prothrombotic disorders [23].
Less commonly acquired diseases (e.g., antiphospholipid syndrome, paroxysmal nocturnal hemoglobinuria, Behçet disease, hypereosinophilic syndrome, granulomatous venulitis, etc.) have also been associated with BCS [2, 24].
Inherited
Other inherited prothrombotic disorders have been identified as candidate causes of BCS including factor V Leiden mutation, G20210A prothrombin gene mutation, and protein C, S, or antithrombin III deficiencies [13, 14]. The diagnosis of protein C, protein S, and antithrombin III deficiencies can be complicated by impaired synthesis due to hepatic dysfunction. In this situation, one diagnostic strategy is to compare the levels of the candidate deficient factor with the levels of other vitamin K-dependent factors. If these levels are globally reduced, it is reasonable to assume that the deficiency is secondary to hepatic dysfunction. Family history or genetic testing of relatives can be helpful to determine whether a specific deficit is primary or secondary.
The measurement of anticardiolipin antibodies and homocysteine levels are also affected by liver disease as they are commonly elevated in these patients, regardless of the etiology of hepatic dysfunction [25, 26]. Hyperhomocysteinemia is a relatively weak risk factor for thrombosis [18] and, in most cases, homocysteine levels are highly influenced by diet and by vitamin B12 or folic acid deficiencies [2].
Recently, genetic variations in the gene inhibitor of thrombin-activated fibrinolysis (TAFI) [27] and the plasminogen activator inhibitor 1 (PAI-1) [28] have been described as candidate etiological factors associated with both portal vein thrombosis (PVT) and BCS. Further studies are needed to assess the possible etiological role of these factors before their inclusion in routine diagnostic algorithms of BCS can be considered.
Clinical Manifestations
The clinical presentation of BCS is extremely heterogeneous and can range from the absence of symptoms to the development of fulminant hepatic failure with encephalopathy [1, 5]. The classical clinical triad of BCS consists of abdominal pain, hepatomegaly, and ascites. In a recent multicenter prospective European study of 163 patients with BCS, ascites was present at diagnosis in 83% of patients, hepatomegaly in 67%, and abdominal pain in 61% [7]. Esophageal varices were present at diagnostic endoscopy in 58% of these patients and gastrointestinal bleeding in 5% of cases [7].
The severity of presentation of BCS is influenced by (1) the number and type of affected veins; (2) the extension of thrombosis; and (3) the efficiency of compensatory mechanisms (hepatic collaterals and spontaneous intrahepatic porto-caval shunts) [3, 29, 30]. An asymptomatic presentation occurs in anywhere from 3 [7] to 15–20% of patients [3, 29] and is often associated with the presence of hepatic venous collaterals [29].
In other cases thrombosis is extensive and develops rapidly, leading to more severe symptoms and in rare cases to presentation with acute liver failure leading to renal impairment, coagulopathy, and death. Fever, jaundice, edema of lower limbs, hepatic encephalopathy, and hepatorenal syndrome, although infrequent, are other possible symptoms. When BCS is associated with thrombosis of the IVC collateral circulation, nephrotic syndrome may appear (proteinuria and edema in lower limbs).
In approximately 15% of cases, BCS and PVT occur simultaneously [7, 31]. In these patients there is a higher prevalence of multiple etiologies (58% of cases), the number increasing significantly with the extent of thrombosis [31]. Prognosis is worse than that for isolated PVT (5-year survival 59% vs. 85%, respectively). Likewise, therapeutic options and prognosis tend to be worse in BCS-PVT patients with additional splenic and/or superior mesenteric vein thrombosis compared to those without (5-year survival 48% vs. 76%, respectively) [31]. Therefore, patients with BCS and portal venous system thrombosis constitute an exceptional group with limited therapeutic options.
Another clinical finding in 60–80% of patients with BCS is the presence of benign hepatic regenerative nodules [4, 32]. The reported time for detection ranges between 1 and 14 years from diagnosis [33]. On computed tomography (CT) or magnetic resonance (MR) imaging, these nodules are characteristically small, in most cases under 4 cm in diameter, multiple (frequently more than 10 lesions), hypervascularized, and disseminated throughout the liver, with a periportal distribution [33]. Although there is no pathognomonic pattern, these nodules are frequently homogeneous; hyperechogenic on ultrasound, hyperattenuating on unenhanced CT [33]; markedly and homogeneously hyperattenuating on arterial phase and slightly hyperattenuating on portal-venous phase images [34]. On MR images benign nodules are characteristically bright on unenhanced T1-weighted and enhance strongly following endo-venous administration of gadolinium-based contrast agents [35]. On T2-weighted images they are predominantly isointense or hypointense relative to the normal liver [35]. Histologically, nodules are composed of moderately enlarged hepatocyte plates without nuclear atypia [33]. The presence of a central scar, found in less than 50% of patients, is a characteristic feature in nodules larger than 1 cm in diameter [36]. The pathogenesis of these nodules remains unclear, but they seem to be the result of focal defects of portal perfusion combined with hypervascularized areas of preserved venous outflow. A recent study conducted in BCS patients using serial multiphase contrast-enhanced multidetector CT has shown that benign hepatic nodules may increase in number and size over time [37].
Since the radiological characteristics of these benign hepatic nodules may be very similar to those found in hepatocellular carcinomas (HCC) nodules, imaging criteria for the diagnosis of HCC recommended in the AASLD-EASL guidelines are not applicable to patients with BCS [38, 39]. The distinction between benign nodules and HCC is especially relevant, because in a recent study by Valla et al., the cumulative incidence of HCC in BCS patients was 4% (after a median follow-up of 5 years) [40]. In this study, male gender, presence of factor V Leiden mutation, and IVC thrombosis were the most frequent factors associated with HCC [40]. Biopsy was suggested in patients with less than or equal to three nodules, nodules with a diameter more than or equal to 3 cm, heterogeneity or washout on the venous phase, changes in two consecutive imaging techniques, or increase in AFP levels [40]. Furthermore, an AFP above a cutoff value of 15 ng/mL had a positive predictive value of 100% and a negative predictive value of 90% for the diagnosis of HCC [40]. These findings need to be validated in larger cohort studies. It must also be kept in mind that liver biopsy in this population could be complicated by the risk of bleeding with ongoing anticoagulation therapy and with the risk of thrombotic events after discontinuation of anticoagulation.
Diagnosis
In most cases, the diagnosis of BCS can be established with noninvasive radiological techniques (e.g., Doppler ultrasound, computerized tomography – CT, and magnetic resonance – MRI) (Chap. 9). Doppler ultrasound has a diagnostic sensitivity of more than 75% and should be the first line of investigation [1]. The absence of retrograde flow in the first phase of the triphasic HV waveform, failure to visualize HV, presence of intrahepatic or subcapsular collaterals, or obstruction of the intrahepatic IVC are the main sonographic findings in BCS (Fig. 13.2). As suggested by recent guidelines, MRI and CT evaluation can be considered for the purpose of diagnostic confirmation or if an experienced sonographer is not available [2]. In addition to obstruction of the hepatic venous outflow tract, both CT and MRI may demonstrate caudate lobe hypertrophy (75% of patients), rapid clearance of dye from the caudate lobe, and patchy hepatic enhancement due to uneven portal perfusion [2].

(a) Doppler ultrasound studies of a BCS patient showing the presence of multiple venous intrahepatic and subcapsular collaterals. Also note the absence of visualization of the hepatic veins. The association of both features is a distinctive characteristic of BCS, and it is present in 80% of cases. (b) Hepatic venography of another BCS patient demonstrating a fine spider-web network pattern without filling hepatic veins
Due to the increasing sophistication of these noninvasive radiological techniques, invasive diagnostic methods (e.g., venography of the IVC and HV) are nowadays limited to a small number of patients. As per recent guidelines, this gold standard technique is recommended if the diagnosis remains uncertain despite the above investigations or for the characterization of anatomy prior to treatment [2]. Venography classically demonstrates a spiderweb pattern formed by a rich collateral circulation between the tributaries of the HV [41] (see Fig. 13.2). Liver biopsy is infrequently required in the diagnostic algorithm for BCS. Its utility is hampered by sample variation and the risk of delaying therapeutic anticoagulation. Guidelines suggest that biopsy should be considered when imaging has failed to demonstrate obstruction of the large HVs and IVC but the diagnosis of BCS remains suspect [2].
Hemodynamic studies in patients with BCS demonstrate a normal cardiac index and normal mean systemic and cardiopulmonary pressures. Despite this, these patients have activation of the neurohumoral vasoactive systems, as evidenced by increased plasma renin activity, aldosterone and norepinephrine levels, and plasma volume expansion [17].
During pregnancy, in cases where a definitive diagnosis remain unclear after Doppler ultrasound, gadolinium MRI may be used, according to the guidelines of the European Society of Radiology, based on the probable safety of gadolinium during the pregnancy and importance of diagnosis for the mother’s health [22].
Treatment
The management of BCS rests on three pillars: (1) supportive care, (2) anticoagulation and control of the underlying thrombophilic disease, and (3) correction of hepatic venous outflow obstruction. Unfortunately, to date there are no prospective randomized controlled clinical trials comparing different treatment options in BCS. Therefore, recommendations are based on retrospective cohorts and prospective series of patients. Figure 13.3 summarizes the currently accepted therapeutic algorithm of BCS [2, 3].

Proposed therapeutic algorithm in Budd-Chiari syndrome
Supportive Care
Patients with BCS will often require therapy for ascites and varices. Since there are no specific guidelines for the management of portal hypertension in BCS patients, it is reasonable to follow the same treatment recommendations as for portal hypertension in cirrhosis.
Anticoagulation and Control of Underlying Thrombotic Disorders
All patients with BCS, even those without an underlying prothrombotic condition (<10%), and those who are asymptomatic, should receive anticoagulant therapy as soon as possible and for an indefinite period of time [42]. This recommendation aims to reduce the risk of clot extension and new thrombotic episodes. Although there are no prospective randomized controlled studies evaluating the efficacy of anticoagulants, indirect evidence suggests that this practice has significantly improved the prognosis of the disease [43, 44]. After initial management with low molecular weight heparin, treatment with coumarin derivatives (acenocoumarol or warfarin) is recommended [2]. Although there has been little research into the optimal level for chronic anticoagulation in patients with BCS, most studies aim for an international normalized ratio (INR) between 2 and 3, the conventional target for thrombosis in other vascular beds [2]. In addition, any supplemental therapies for coexisting MPD should be initiated and underlying prothrombotic risk factors such as oral contraceptives should be avoided.
Correction of Hepatic Venous Outflow Obstruction
Thrombolysis
The experience of thrombolysis in BCS is limited. Recombinant tissue plasminogen activator, streptokinase, or urokinase have been used. These agents can be instilled through a peripheral vein or locally after catheterization of the thrombosed vein. There are no studies comparing the efficacy of local vs. systemic infusion. A review by Sharma et al. indicates that the best results are achieved in patients with a recent and incomplete thrombosis who are treated with local and early infusion combined with another interventional procedure (e.g., angioplasty, stenting) to restore venous outflow [45]. Complications of thrombolysis can be fatal. Hence, this therapeutic option is contraindicated in patients with a potentially hemorrhagic condition, or patients who have had an invasive procedure, including paracentesis, in the previous 24 h. In summary, thrombolysis should only be attempted in select cases with acute or subacute BCS, and at experienced centers.
Angioplasty/Stenting of Short-Length Hepatic Veins Stenosis
Partial or segmental stenosis is present in 60% of patients with IVC obstruction and 25–30% of those with HV obstruction [46]. Since angioplasty or stenting of this short-length stenosis could reestablish the physiological drainage of portal and sinusoidal blood through the HVs [47], their existence must be properly evaluated in all patients with BCS. Postangioplasty, restenosis may occur that necessitates subsequent angioplasties. Restenosis appears to occur more frequently when angioplasty is performed alone as in comparison to in combination with a stent [2]. As misplacement of a stent may compromise the subsequent performance of a portosystemic intrahepatic shunt, these procedures should only be attempted by highly experienced personnel.
Portosystemic Shunt
For many years surgical procedures, together with liver transplantation, have been the only therapeutic alternative for patients with BCS who have not responded to medical treatment or have not been candidates for angioplasty/stenting. The aim of these derivative techniques is to transform the portal system into the outflow tract [48]. The most frequent shunt performed is the mesocaval shunt with a calibrated 8–10 mm polytetrafluoroethylene stent or autologous jugular vein interposition. It is preferable to a porto-caval side-to-side shunt because it is easier to perform when hypertrophy of the caudate lobe is present [49]. Surgical shunts are ineffective if there is associated IVC thrombosis or severe compression of the IVC by an enlarged liver. In the setting of IVC compression, the presence of an infrahepatic caval pressure of greater than 20 mmHg or a gradient between it and the right atrium of 15 mmHg is predictive of inadequate shunt function unless the stenosis/compression of the IVC is simultaneously corrected with the placement of a stent [1, 50]. In the setting of IVC thrombosis or severe stenosis not amenable to standard shunts, some groups have performed a mesoatrial shunt. The utility of this remains limited; however, as it is quite challenging and is associated with considerable morbidity and occlusion risk [51].
Overall, surgical shunts are associated with significant mortality (close to 25% of patients) and have not demonstrated an independent survival advantage in large cohorts of patients [52, 53]. This is likely related to the high inherent mortality rate of the patient population with severe BCS who meet criteria for this treatment as well as to the high rate of dysfunction/thrombosis of the shunts (approximately 30% of cases) [51, 54, 55]. In support of this concept, it has been shown that patients who survive a surgical procedure and who maintain shunt patency have excellent long-term outcomes [55, 56]. Regardless, since the introduction of the transjugular intrahepatic portosystemic shunt (TIPS), surgical shunts are considered a second line derivative alternative.
Transjugular Intrahepatic Portosystemic Shunt (TIPS)
In experienced hands, TIPS is an extremely useful therapy for most BCS patients in whom medical treatment or recanalization have failed [57]. TIPS has a lower morbidity and mortality than surgery, and, contrary to surgical shunts, it is feasible in most patients with IVC obstruction and in those with severe IVC stenosis. A recent multicenter European study that included 124 BCS patients treated with TIPS showed excellent 1- and 5-year OLT-free survival (88 and 78%, respectively [57]). Furthermore, TIPS is even feasible in pregnant patients, because the radiation received by the fetus during TIPS placement is below the threshold to induce fetal deleterious effects [22].
Early stent thrombosis is not uncommon, even during the release of the prosthesis. Therefore, a sodium heparin infusion should be initiated immediately after the puncture of the portal vein [58]. To reduce the recurrence of postprocedure TIPS obstruction or dysfunction due to stent occlusion, PTFE covered stents are suggested. Recent data has shown a primary patency rate of 67% at 1 and 2 years in PTFE-covered stents compared to 19% at 1 year and 9% at 2 years in bare stents [57, 59].
In some patients, TIPS obstruction is not accompanied by the reappearance of symptoms of portal hypertension due to the formation of collaterals during the time that it was permeable [58, 59].
The use of TIPS instead of OLT in patients in whom angioplasty/stenting is not an option can avoid the risk of long-term immunosuppression and potentially increase the number of donor organs available to transplant centers [57].
Orthotopic liver transplantation (OLT) remains an alternative in those cases in which TIPS fails to improve the clinical evolution. Although it has been suggested that previous TIPS placement can jeopardize OLT [50, 60], recent studies have shown that TIPS did not worsen prognosis after OLT in BCS patients [57, 61]. TIPS placement does, however, require specialized training and it can be significantly more complex than in patients with cirrhosis alone. In more than 45% of cases, a transcaval approach (direct puncture from the intrahepatic inferior vena cava) may be required due to complete thrombosis of the HVs [57]. It is therefore highly recommended to refer such patients to centers with expertise in this technique [57].
Orthotropic Liver Transplantation (OLT)
The BCS represents 1% of all OLT cases in American and European databases [62] and should be restricted to those patients in whom TIPS has failed (see Fig. 13.3) (Chap. 17). OLT in BCS patients is a technical challenge for the surgical team because of the presence of retroperitoneal fibrosis related to HV thrombosis and also because the liver is often large, firm, and difficult to mobilize during hepatectomy. In addition, the classical “piggyback” technique for anastomosis becomes more challenging due to the increased size of the caudate lobe and occlusion of the HV ostia. Living donor liver transplantation (LDLT) is technically even more difficult and thus far, the only experience is in Asian centers [63, 64].
Disease recurrence after OLT is variable and ranges from 0 to 11% in different series [44, 65–69]. Other postoperative thrombotic complications are also frequent [66] but may be prevented by initiating heparin in the first 6 hours after surgery. Although there is insufficient evidence, indefinite anticoagulation is recommended. An exception would be those patients in whom OLT is performed for antithrombin III, protein C or S deficiencies, or a factor V mutation, as all of these disorders are corrected by OLT [70].
The natural history of MPD must also be considered in the posttransplant course. There is a risk of myelofibrosis or leukemic transformation that increases over time, estimated at between 10% and 25% at 15 and 25 years, respectively [71]. Although isolated cases of leukemic transformation after OLT in MPD patients have been reported [66, 68, 72], the rate of malignant transformation of MPD over a 10-year posttransplant period is comparable to that of nontransplant patients [3, 66]. Posttransplant recurrence of BCS in patients with paroxysmal nocturnal hemoglobinuria may occur and has been shown to affect up to 40% of patients [73]. The combination of two underlying thrombophilic disease further increases the risk of recurrence.
Five-year posttransplant survival in patients with BCS ranges from 45 to 95% [44, 50, 60–68, 74–76]. This wide range is likely due to the high heterogeneity in the type of BCS patients listed for OLT. A recent European study showed an actuarial overall survival of 76, 71, and 68% at 1, 5, and 10 years, respectively [65], similar to the survival rates reported using TIPS (88 and 78% at 1 and 5-year OLT-free survival, respectively) [57]. The overall mortality rate has been estimated at up to 25% [65, 66] with approximately half of the patients dying within the first 2 months after OLT as a result of surgical complications, infections, or rejection. In addition, there have been some cases of early mortality associated with early recurrence of BCS. These have been attributed to inadequate anticoagulation in the immediate posttransplant period [44, 67, 69]. Late mortality is typically associated with recurrence or progression of the underlying disease. In summary, it is clear that careful, multidisciplinary management of these patients, including appropriate treatment for hematological disease, is essential.
Budd-Chiari and Pregnancy
The majority of patients with BCS in Western countries are at child-bearing age, and many express their desire to become pregnant. For this reason it is important to understand the evolution and the possible consequences of pregnancy on BCS, and vice versa.
Management of Budd-Chiari Syndrome Established Prior to Pregnancy
Anticoagulation should be maintained during pregnancy. Oral anticoagulants are associated with a high risk of miscarriage (14.6–56%) and congenital malformations (30%) [22]. Therefore, although it is not necessary to change to heparin before conception, clinical practice guidelines recommend a pregnancy test as early as possible to confirm pregnancy, and then a switch to heparin [77]. Low molecular weight heparins (LMWH) such as enoxaparin, dalteparin, or tinzaparin are recommended (all three are approved by the FDA for use during pregnancy). During pregnancy, increased glomerular filtration, increased plasma distribution volume, and the presence of placental heparinase may necessitate increasing doses of heparin, and therefore periodic monitoring of anti-Xa activity should also be carried out [22].
Epidural anesthesia is contraindicated if the last dose of LMWH was administrated less than 12 h before. Prophylactic anticoagulation must be restarted within the first 24 h after delivery and, if there are no contraindications, therapeutic doses in the first 48 h.
Impact on the Course of Pregnancy
A recent multicenter retrospective study that evaluated the impact of BCS on pregnancy demonstrated an excellent maternal outcome provided that patients were compensated with well-controlled disease. Fetal outcome was, however, concerning with a 29% rate of pregnancy loss before week 20 of gestation, as compared to a rate of 16.2% in age-matched controls [78]. Pregnancies reaching week 20 of gestation were associated with an acceptable fetal prognosis even though 76% had preterm delivery [78].
Vaginal delivery is recommended in the setting of BCS as cesarean sections may be complicated by the presence of ascites, bleeding from pelvic collaterals, and a higher rate of postoperative thromboembolic disease [78]. All women with BCS who wish to become pregnant should therefore be properly informed of maternal and fetal risks and prognosis and should be considered high-risk pregnancies.
Prognosis
Due to recent therapeutic advances, the survival of BCS patients has improved to over 80% at 5 years [2]. Hepatocellular carcinoma, portal vein axis thrombosis, and aggravation or leukemic transformation of myeloproliferative disease are relevant factors that may overshadow outcome in this complex group of patients [2, 3].
There have been various attempts to determine parameters or combinations of parameters that may predict prognosis in BCS patients. The first BCS-specific prognostic index was described in 1999 by Zeitoun et al. and showed that age, serum creatinine, refractory ascites, and the Child-Pugh score were inversely related independent factors that allowed the differentiation of patients with good and poor prognoses (95% vs. 62% 5-year survival) [52]. A revised index by the same group incorporated a novel clinicopathological classification and identified age, the Child-Pugh score, ascites, serum creatinine, and the presence of features indicating acute injury superimposed on chronic lesions as independent prognostic indicators. This stratified patients into “low” and “high risk” had an excellent correlation with short- and long-term survival [79]. Subsequently, Murad et al. proposed the Rotterdam score, a model based on the presence of hepatic encephalopathy, ascites, prothrombin time and bilirubin, classifying patients into three groups with an estimated 5-year survival of 89, 74, and 42%, respectively [80]. It must be kept in mind that all of these prognostic indices were developed in the pre-TIPS era. They remain useful to identify patients with a poor prognosis on anticoagulation and supportive care who should be considered for TIPS. Indeed, with the introduction of TIPS into the current therapeutic algorithm, the prognosis of BCS patients has improved.
Some patients, however, are too sick for TIPS and require early OLT. To identify this group, a new prognostic index has been developed, called the BCS-TIPS prognostic index score [57]. A BCS-TIPS prognostic index score over 7 correlates with a sensitivity and specificity of 99% for death or OLT 1 year after TIPS [57]. Other prognostic indices such as the Child-Pugh score and model for end-stage liver disease (MELD) score have also been used, but exhibit a suboptimal prognostic accuracy [81]. Accordingly, a recent study published by Ratou et al. suggests that although all of these prognostic indices are valid for the assessment of transplant-free survival and invasive therapy-free survival, their predictive accuracy is suboptimal for use in individual patients in day-to-day clinical practice [82].
References
Janssen HL, Garcia-Pagan JC, Elias E, Mentha G, Hadengue A, Valla DC. Budd-Chiari syndrome: a review by an expert panel. J Hepatol. 2003;38:364–71.
DeLeve LD, Valla DC, Garcia-Tsao G. Vascular disorders of the liver. Hepatology. 2009;49:1729–64.
Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis. 2008;28:259–69.
Tanaka M, Wanless IR. Pathology of the liver in Budd-Chiari syndrome: portal vein thrombosis and the histogenesis of veno-centric cirrhosis, veno-portal cirrhosis, and large regenerative nodules. Hepatology. 1998;27:488–96.
Hernandez-Guerra M, Garcia-Pagan JC. Recommendations for the diagnosis and treatment of patients with Budd-Chiari syndrome. Gastroenterol Hepatol. 2004;27:473–9.
Valla DC. Hepatic venous outflow tract obstruction etiopathogenesis: Asia versus the West. J Gastroenterol Hepatol. 2004;19:S204–11.
Darwish MS, Plessier A, Hernandez-Guerra M, Fabris F, Eapen CE, Bahr MJ, et al. Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med. 2009;151:167–75.
Shrestha SM, Okuda K, Uchida T, Maharjan KG, Shrestha S, Joshi BL, et al. Endemicity and clinical picture of liver disease due to obstruction of the hepatic portion of the inferior vena cava in Nepal. J Gastroenterol Hepatol. 1996;11:170–9.
Amarapurkar DN, Punamiya SJ, Patel ND. Changing spectrum of Budd-Chiari syndrome in India with special reference to non-surgical treatment. World J Gastroenterol. 2008;14:278–85.
Horton JD, San Miguel FL, Membreno F, Wright F, Paima J, Foster P, et al. Budd-Chiari syndrome: illustrated review of current management. Liver Int. 2008;28:455–66.
Okuda K. Inferior vena cava thrombosis at its hepatic portion (obliterative hepatocavopathy). Semin Liver Dis. 2002;22:15–26.
Valla DC. Primary Budd-Chiari syndrome. J Hepatol. 2009;50:195–203.
Denninger MH, Chait Y, Casadevall N, Hillaire S, Guillin MC, Bezeaud A, et al. Cause of portal or hepatic venous thrombosis in adults: the role of multiple concurrent factors. Hepatology. 2000;31:587–91.
Janssen HL, Meinardi JR, Vleggaar FP, van Uum SH, Haagsma EB, Der Meer FJ, et al. Factor V Leiden mutation, prothrombin gene mutation, and deficiencies in coagulation inhibitors associated with Budd-Chiari syndrome and portal vein thrombosis: results of a case-control study. Blood. 2000;96:2364–8.
Primignani M, Barosi G, Bergamaschi G, Gianelli U, Fabris F, Reati R, et al. Role of the JAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology. 2006;44:1528–34.
Bittencourt PL, Couto CA, Ribeiro DD. Portal vein thrombosis and Budd-Chiari syndrome. Clin Liver Dis. 2009;13:127–44.
Hernandez-Guerra M, Lopez E, Bellot P, Piera C, Turnes J, Abraldes JG, et al. Systemic hemodynamics, vasoactive systems, and plasma volume in patients with severe Budd-Chiari syndrome. Hepatology. 2006;43:27–33.
Primignani M, Mannucci PM. The role of thrombophilia in splanchnic vein thrombosis. Semin Liver Dis. 2008;28:293–301.
Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, Hanson CA, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110:1092–7.
Kiladjian JJ, Cervantes F, Leebeek FW, Marzac C, Cassinat B, Chevret S, et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood. 2008;111:4922–9.
Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology. 1986;90:807–11.
Perarnau JM, Bacq Y. Hepatic vascular involvement related to pregnancy, oral contraceptives, and estrogen replacement therapy. Semin Liver Dis. 2008;28:315–27.
Briere JB. Budd-Chiari syndrome and portal vein thrombosis associated with myeloproliferative disorders: diagnosis and management. Semin Thromb Hemost. 2006;32:208–18.
Hoekstra J, Leebeek FW, Plessier A, Raffa S, Murad SD, Heller J, et al. Paroxysmal nocturnal hemoglobinuria in Budd-Chiari Syndrome: Findings from a cohort study. J Hepatol. 2009;51:696–706.
Mangia A, Margaglione M, Cascavilla I, Gentile R, Cappucci G, Facciorusso D, et al. Anticardiolipin antibodies in patients with liver disease. Am J Gastroenterol. 1999;94:2983–7.
Bosy-Westphal A, Ruschmeyer M, Czech N, Oehler G, Hinrichsen H, Plauth M, et al. Determinants of hyperhomocysteinemia in patients with chronic liver disease and after orthotopic liver transplantation. Am J Clin Nutr. 2003;77:1269–77.
de Bruijne EL, Darwish MS, de Maat MP, Tanck MW, Haagsma EB, van HB, et al. Genetic variation in thrombin-activatable fibrinolysis inhibitor (TAFI) is associated with the risk of splanchnic vein thrombosis. Thromb Haemost. 2007;97:181–5.
Hoekstra J, Leebeek FW, Guimaraes A, Murad S, Malfliet J, Plessier A, et al. Impaired fibrinolysis as risk factor for Budd-Chiari syndrome. Blood. 2010;115:388–95.
Hadengue A, Poliquin M, Vilgrain V, Belghiti J, Degott C, Erlinger S, et al. The changing scene of hepatic vein thrombosis: recognition of asymptomatic cases. Gastroenterology. 1994;106:1042–7.
Valla D. Hepatic vein thrombosis (Budd-Chiari syndrome). Semin Liver Dis. 2002;22:5–14.
Darwish MS, Valla DC, de Groen PC, Zeitoun G, Haagsma EB, Kuipers EJ, et al. Pathogenesis and treatment of Budd-Chiari syndrome combined with portal vein thrombosis. Am J Gastroenterol. 2006;101:83–90.
Wanless IR. Benign liver tumors. Clin Liver Dis. 2002;6:513–26, ix.
Vilgrain V, Lewin M, Vons C, Denys A, Valla D, Flejou JF, et al. Hepatic nodules in Budd-Chiari syndrome: imaging features. Radiology. 1999;210:443–50.
Brancatelli G, Vilgrain V, Federle MP, Hakime A, Lagalla R, Iannaccone R, et al. Budd-Chiari syndrome: spectrum of imaging findings. AJR Am J Roentgenol. 2007;188:W168–76.
Erden A, Erden I, Karayalcin S, Yurdaydin C. Budd-Chiari syndrome: evaluation with multiphase contrast-enhanced three-dimensional MR angiography. AJR Am J Roentgenol. 2002;179:1287–92.
Maetani Y, Itoh K, Egawa H, Haga H, Sakurai T, Nishida N, et al. Benign hepatic nodules in Budd-Chiari syndrome: radiologic-pathologic correlation with emphasis on the central scar. AJR Am J Roentgenol. 2002;178:869–75.
Flor N, Zuin M, Brovelli F, Maggioni M, Tentori A, Sardanelli F, et al. Regenerative nodules in patients with chronic Budd-Chiari syndrome: A longitudinal study using multiphase contrast-enhanced multidetector CT. Eur J Radiol. 2010;73:588–93.
Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–36.
Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencioni R, Burroughs AK, et al. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol. 2001;35:421–30.
Moucari R, Rautou PE, Cazals-Hatem D, Geara A, Bureau C, Consigny Y, et al. Hepatocellular carcinoma in Budd-Chiari syndrome: characteristics and risk factors. Gut. 2008;57:828–35.
Kamath PS. Budd-Chiari syndrome: radiologic findings. Liver Transpl. 2006;12:S21–2.
Valla DC. The diagnosis and management of the Budd-Chiari syndrome: consensus and controversies. Hepatology. 2003;38:793–803.
Campbell Jr DA, Rolles K, Jamieson N, O’Grady J, Wight D, Williams R, et al. Hepatic transplantation with perioperative and long term anticoagulation as treatment for Budd-Chiari syndrome. Surg Gynecol Obstet. 1988;166:511–8.
Halff G, Todo S, Tzakis AG, Gordon RD, Starzl TE. Liver transplantation for the Budd-Chiari syndrome. Ann Surg. 1990;211:43–9.
Sharma S, Texeira A, Texeira P, Elias E, Wilde J, Olliff SP. Pharmacological thrombolysis in Budd Chiari syndrome: a single centre experience and review of the literature. J Hepatol. 2004;40:172–80.
Valla D, Hadengue A, El Younsi M, Azar N, Zeitoun G, Boudet MJ, et al. Hepatic venous outflow block caused by short-length hepatic vein stenoses. Hepatology. 1997;25:814–9.
Fisher NC, McCafferty I, Dolapci M, Wali M, Buckels JAC, Olliff SP, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting (see comments). Gut. 1999;44:568–74.
Tilanus HW, Metselaar HJ, Lameris JS. Budd-Chiari syndrome (see comments). Ned Tijdschr Geneeskd. 1995;139:161–5.
Mitchell MC, Boitnott JK, Kaufman S, Cameron JL, Maddrey WC. Budd-Chiari syndrome: etiology, diagnosis and management. Medicine (Baltimore). 1982;61:199–218.
Shaked A, Goldstein RM, Klintmalm GB, Drazan K, Husberg B, Busuttil RW. Portosystemic shunt versus orthotopic liver transplantation for the Budd-Chiari syndrome. Surg Gynecol Obstet. 1992;174:453–9.
Hemming AW, Langer B, Greig P, Taylor BR, Adams R, Heathcote EJ. Treatment of Budd-Chiari syndrome with portosystemic shunt or liver transplantation. Am J Surg. 1996;171:176–80.
Zeitoun G, Escolano S, Hadengue A, Azar N, El Younsi M, Mallet A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology. 1999;30:84–9.
Langlet P, Valla D. Is surgical portosystemic shunt the treatment of choice in Budd-Chiari syndrome? Acta Gastroenterol Belg. 2002;65:155–60.
Panis Y, Belghiti J, Valla D, Benhamou JP, Fekete F. Portosystemic shunt in Budd-Chiari syndrome: long-term survival and factors affecting shunt patency in 25 patients in Western countries. Surgery. 1994;115:276–81.
Bachet JB, Condat B, Hagege H, Plessier A, Consigny Y, Belghiti J, et al. Long-term portosystemic shunt patency as a determinant of outcome in Budd-Chiari syndrome. J Hepatol. 2007;46:60–8.
Madsen MS, Petersen TH, Sommer H. Segmental portal hypertension. Ann Surg. 1986;204(1):72–7.
Garcia-Pagan JC, Heydtmann M, Raffa S, Plessier A, Murad S, Fabris F, et al. TIPS for Budd-Chiari syndrome: long-term results and prognostics factors in 124 patients. Gastroenterology. 2008;135:808–15.
Perello A, Garcia-Pagan JC, Gilabert R, Suarez Y, Moitinho E, Cervantes F, et al. TIPS is a useful long-term derivative therapy for patients with Budd-Chiari syndrome uncontrolled by medical therapy. Hepatology. 2002;35:132–9.
Hernandez-Guerra M, Turnes J, Rubinstein P, Olliff S, Elias E, Bosch J, et al. PTFE-covered stents improve TIPS patency in Budd-Chiari syndrome. Hepatology. 2004;40:1197–202.
Turnes J, Garcia-Pagan JC, Gonzalez-Abraldes J, Real M, Moitinho E, Gilabert R, et al. Stenosis of the suprahepatic inferior vena cava as a complication of transjugular intrahepatic portosystemic shunt in Budd-Chiari patients. Liver Transpl. 2001;7:649–51.
Segev DL, Nguyen GC, Locke JE, Simpkins CE, Montgomery RA, Maley WR, et al. Twenty years of liver transplantation for Budd-Chiari syndrome: a national registry analysis. Liver Transpl. 2007;13:1285–94.
Adam R, McMaster P, O’Grady JG, Castaing D, Klempnauer JL, Jamieson N, et al. Evolution of liver transplantation in Europe: report of the European Liver Transplant Registry. Liver Transp. 2003;9:1231–43.
Yamada T, Tanaka K, Ogura Y, Ko S, Nakajima Y, Takada Y, et al. Surgical techniques and long-term outcomes of living donor liver transplantation for Budd-Chiari syndrome. Am J Transplant. 2006;6:2463–9.
Yan L, Li B, Zeng Y, Wen T, Zhao J, Wang W, et al. Living donor liver transplantation for Budd-Chiari syndrome using cryopreserved vena cava graft in retrohepatic vena cava reconstruction. Liver Transpl. 2006;12:1017–9.
Mentha G, Giostra E, Majno PE, Bechstein WO, Neuhaus P, O’Grady J, et al. Liver transplantation for Budd-Chiari syndrome: a European study on 248 patients from 51 centres. J Hepatol. 2006;44:520–8.
Cruz E, Ascher NL, Roberts JP, Bass NM, Yao FY. High incidence of recurrence and hematologic events following liver transplantation for Budd-Chiari syndrome. Clin Transplant. 2005;19:501–6.
Ringe B, Lang H, Oldhafer KJ, Gebel M, Flemming P, Georgii A, et al. Which is the best surgery for Budd-Chiari syndrome: venous decompression or liver transplantation? A single-center experience with 50 patients. Hepatology. 1995;21:1337–44.
Srinivasan P, Rela M, Prachalias A, Muiesan P, Portmann B, Mufti GJ, et al. Liver transplantation for Budd-Chiari syndrome. Transplantation. 2002;73:973–7.
Melear JM, Goldstein RM, Levy MF, Molmenti EP, Cooper B, Netto GJ, et al. Hematologic aspects of liver transplantation for Budd-Chiari syndrome with special reference to myeloproliferative disorders. Transplantation. 2002;74:1090–5.
Karasu Z, Nart D, Lebe E, Demirbas T, Memis A, Kilic M, et al. Liver transplantation in a patient with Budd-Chiari Syndrome secondary to factor V Leiden mutation. Transplant Proc. 2003;35(8):3008–10.
Ganguli SC, Ramzan NN, McKusick MA, Andrews JC, Phyliky RL, Kamath PS. Budd-Chiari syndrome in patients with hematological disease: a therapeutic challenge. Hepatology. 1998;27:1157–61.
Au WY, Fung A, Liu CL, Fan ST, Ma SK, Liang R, et al. Serial analysis of JAK2 mutation in a patient who developed essential thrombocythemia after orthotopic liver transplantation. Am J Hematol. 2006;81:880–2.
Bahr MJ, Schubert J, Bleck JS, Tietge UJ, Boozari B, Schmidt RE, et al. Recurrence of Budd-Chiari syndrome after liver transplantation in paroxysmal nocturnal hemoglobinuria. Transpl Int. 2003;16:890–4.
Jamieson NV, Williams R, Calne RY. Liver transplantation for Budd-Chiari syndrome, 1976-1990. Ann Chir. 1991;45:362–5.
Rao AR, Chui AK, Gurkhan A, Shi LW, Al-Harbi I, Waugh R, et al. Orthotopic liver transplantation for treatment of patients with Budd-Chiari syndrome: a Singe-center experience. Transplant Proc. 2000;32:2206–7.
Ulrich F, Steinmuller T, Lang M, Settmacher U, Muller AR, Jonas S, et al. Liver transplantation in patients with advanced Budd-Chiari syndrome. Transplant Proc. 2002;34:2278.
Bates SM, Greer IA, Pabinger I, Sofaer S, Hirsh J. Venous thromboembolism, thrombophilia, antithrombotic therapy, and pregnancy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:844S–86.
Rautou PE, Angermayr B, Garcia-Pagan JC, Moucari R, Peck-Radosavljevic M, Raffa S, et al. Pregnancy in women with known and treated Budd-Chiari syndrome: maternal and fetal outcomes. J Hepaol. 2009;51:47–54.
Langlet P, Escolano S, Valla D, Coste-Zeitoun D, Denie C, Mallet A, et al. Clinicopathological forms and prognostic index in Budd-Chiari syndrome. J Hepatol. 2003;39:496–501.
Murad SD, Valla DC, de Groen PC, Zeitoun G, Hopmans JA, Haagsma EB, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology. 2004;39:500–8.
Darwish MS, Kim WR, de Groen PC, Kamath PS, Malinchoc M, Valla DC, et al. Can the model for end-stage liver disease be used to predict the prognosis in patients with Budd-Chiari syndrome? Liver Transpl. 2007;13:867–74.
Rautou PE, Moucari R, Escolano S, Cazals-Hatem D, Denie C, Chagneau-Derrode C, et al. Prognostic indices for Budd-Chiari syndrome: valid for clinical studies but insufficient for individual management. Am J Gastroenterol. 2009;104:1140–6.
Mohanty D, Shetty S, Ghosh K, Pawar A, Abraham P. Hereditary thrombophilia as a cause of Budd-Chiari syndrome: a study from Western India. Hepatology. 2001;34:666–70.
Mahmoud AE, Elias E, Beauchamp N, Wilde JT. Prevalence of the factor V Leiden mutation in hepatic and portal vein thrombosis [see comments]. Gut. 1997;40:798–800.
Bhattacharyya M, Makharia G, Kannan M, Ahmed RP, Gupta PK, Saxena R. Inherited prothrombotic defects in Budd-Chiari syndrome and portal vein thrombosis: a study from North India. Am J Clin Pathol. 2004;121:844–7.
Bayraktar Y, Balkanci F, Bayraktar M, Calguneri M. Budd-Chiari syndrome: a common complication of Behcet’s disease. Am J Gastroenterol. 1997;92:858–62.
Acknowledgments
Thanks to Dr. Cervantes, Dr. Reverter, Dra. Gilabert, and Dra. García-Criado for their precious and helpful collaboration. SSR is founded by “Río Hortega” Instituto de Salud Carlos III (CM08/00161). This work is supported in part by grants from Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación (FIS 06/0623, FIS 09/01261 and SAF 07/61298). CIBERehd is funded by the Instituto de Salud Carlos III.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer New York
About this chapter
Cite this chapter
Seijo-Ríos, S., Tandon, P., Bosch, J., García-Pagán, J.C. (2011). Budd-Chiari Syndrome. In: DeLeve, L., Garcia-Tsao, G. (eds) Vascular Liver Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-8327-5_13
Download citation
DOI: https://doi.org/10.1007/978-1-4419-8327-5_13
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-8326-8
Online ISBN: 978-1-4419-8327-5
eBook Packages: MedicineMedicine (R0)