
Abstract
MicroRNAs as critical regulators of gene expression important for functions including neuronal development, synapse formation, and synaptic plasticity have been linked with the regulation of neurobiological systems that underlie anxiety processing in the brain. In this chapter, we give an update on associative evidence linking regulation of microRNAs with anxiety- and trauma-related disorders. Moving beyond correlative research, functional studies have emerged recently that explore causal relationships between microRNA expression and anxiety-like behavior. It has been demonstrated that experimental up- or downregulation of the candidate microRNAs in important nodes of the anxiety neurocircuitry can indeed modulate anxiety-related behavior in animal models. Improved methodologies for assessing microRNA-mediated modulation have aided such functional studies, revealing a number of anxiety-regulating microRNAs including miR-15a, miR-17-92, miR-34, miR-101, miR-124, miR-135, and miR-155. Important functional target genes of these identified microRNAs are associated with specific neurotransmitter/neuromodulator signaling, neurotrophin (e.g., BDNF) expression and other aspects of synaptic plasticity, as well as with stress-regulatory/hypothalamic-pituitary-axis function. Furthermore, microRNAs have been revealed that are regulated in distinct brain regions following various anxiety-attenuating strategies. These include pharmacological treatments such as antidepressants and other drugs, as well as non-pharmacological interventions such as fear extinction/exposure therapy or positive stimuli such as exposure to environmental enrichment. These are first indications for a role for microRNAs in the mechanism of action of anxiolytic treatments. As research continues, there is much hope that a deeper understanding of the microRNA-mediated mechanisms underlying anxiety-related disorders could open up possibilities for future novel biomarker and treatment strategies.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Physiological anxiety is a beneficial emotion in response to real or potential threats, but patients with pathological anxiety display a broad range of exaggerated and/or enduring symptoms, such as increased arousal/vigilance and excessive fear- and anxiety-related responses (for detailed discussion into differences and characteristics between fear and anxiety, see Perusini and Fanselow 2015) including autonomic changes such as heart rate increases and sweating, often in the absence of any real threat or danger (Dell’osso et al. 2010). Many of these symptoms are shared by different anxiety disorders, which are classified in the tenth edition of the International Classification of Diseases (ICD-10) (WHO 1993; Craske et al. 2017) and the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) (American Psychiatric Association 2013). They include specific phobias, panic disorder (with or without agoraphobia), social phobia, and generalized anxiety. Anxiety- and trauma-related disorders, such as post-traumatic stress disorder (PTSD), are grouped together in the ICD-10 (WHO 1993; Craske et al. 2017). The current diagnostic systems are based on subjective measures of client self-report, clinical observation, and clinical judgment, while the recently proposed Research Domain Criteria (RDoC) system defines psychopathologies as phenomena of multilevel neurobiological characteristics underlying dimensions of behavior from normal to abnormal (Anderzhanova et al. 2017). The RDoC system holds promise for better characterization of complex psychiatric disorders that are commonly comorbid, as in the case of anxiety often comorbid with depression and alcohol/drug dependence, and aims to improve translatability of studies from animals to humans by supporting the endophenotype-based comparison on a neurobiological basis across behavioral dimension (Anderzhanova et al. 2017). Anxiety-related disorders are the most common mental disorders, affecting up to 14% of Europeans each year (Wittchen et al. 2011). In developed countries, the lifetime prevalence of these disorders is up to 28% (Kessler et al. 2012). Anxiety disorders impact the quality of life and place a high emotional and financial burden on families and government bodies (Olesen et al. 2012; Wittchen et al. 2011). Furthermore, the high comorbidity that exists between anxiety and other psychiatric disorders, such as depression and bipolar disorder, makes treating these disorders even more complex (Pavlova et al. 2017; McMillan et al. 2017).
Both psychotherapy and pharmacotherapy are currently used to treat anxiety-related disorders and have been shown to be more effective than placebo. However, a considerable percentage of patients only exhibit partial long-term benefits following therapy, failing to achieve complete remission (Bandelow et al. 2007, 2012; Farach et al. 2012). Pharmacological treatments include selective serotonin reuptake inhibitors (SSRIs) and other antidepressants (Baldwin et al. 2014). Benzodiazepines are not recommended as first-line treatments due to their potential side effects such as increased risk of drug dependence and should be used only transiently. Disadvantages of the current first-line drug treatments include a delayed period before the onset of therapeutic benefits, suicidal tendencies, and risk of sexual dysfunction, often leading to discontinuation of treatment (Maron and Nutt 2015; Farb and Ratner 2014; Ravindran and Stein 2010). Hence, we are in need of novel interventions targeting causal factors and resulting in long-lasting benefits with lower incidence of side effects. Despite this clear need for improved treatments, the development of novel drugs for anxiety-related disorders has been rather unsuccessful (Singewald et al. 2015; Griebel and Holmes 2013). One of the most important factors involved in this is the still limited knowledge that exists about underlying mechanisms and pathways mediating and modulating anxiety-related behavior, although impressive advances have been made.
2 Neurobiology and Molecular Basis of Anxiety
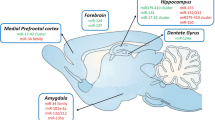
Key to understanding anxiety processing in the brain is the elucidation of the neuronal correlates that are involved. Regulation of fear and anxiety behavior depends on neuronal activity and communication between relevant brain areas, and there is evidence that cellular plasticity is vital for appropriate inter-regional communications (Hill and Martinowich 2016). It has been found that there is a high degree of overlap between humans and animals in the neural circuitry underlying anxiety-related behaviors (Calhoon and Tye 2015). The use of modern methodologies such as optogenetics, among others, in rodents has led to the improved and more detailed characterization of the intricate interplay between the different brain loci forming circuits to mediate and modulate anxiety-like behavior (Tovote et al. 2015; Orsini and Maren 2012; Dias et al. 2013; Calhoon and Tye 2015). At the center of this circuitry is the extended amygdala, important for the emotional interpretation of environmental information as it arises, determining whether downstream fear (see below) or reward pathways (e.g., via the nucleus accumbens, NA) will be activated. The extended amygdala includes different amygdala subregions, including the basolateral amygdala (BLA) and the central amygdala (CeA; composed of the centro-lateral (CeL) and centro-medial (CeM) amygdala), as well as the bed nucleus of the stria terminalis (BNST), which coordinates inputs from the amygdala and hippocampus. Together with other regions, such as the hypothalamus (HYP), they also modulate the neuroendocrine stress response. Other brain regions implicated in the modulation of anxiety-related behaviors include the medial prefrontal cortex (mPFC); the highly interconnected hippocampus, which encodes contextual aspects important for anxiety, and important modulatory monoaminergic cell body areas; the locus coeruleus (LC); the raphe nucleus (RN); and the ventral tegmental area (VTA) (Fig. 1, top panel) (for detailed review, see Tovote et al. 2015). The signal processing between these brain areas and neuronal populations is driven by a finely tuned interplay of different neurotransmitters and neuromodulators, including neurotransmitters such as gamma-aminobutyric acid (GABA); glutamate; monoamines such as serotonin, norepinephrine, and dopamine; and a number of neuropeptides, including neuropeptide Y (NPY), neuropeptide S (NPS), endocannabinoids, cholecystokinin (CCK), and calcitonin gene-related peptide (CGRP), which have been associated with anxiety regulation and considered as drug targets (Bowers et al. 2012; Wang and Pereira 2016; Millan 2003). Many anxiety disorders are considered stress-related, and thus regulation of the corticotropin-releasing factor (CRH) system and of the hypothalamic-pituitary-adrenal (HPA) axis has been found to be important players, particularly in stress-induced anxiety (Shin and Liberzon 2010; Bartlett et al. 2017). Aberrant anxiety reactions are thought to arise from altered processing and deficiencies in such circuitries, including imbalances in neurotransmitter regulation and downstream signaling pathways (Calhoon and Tye 2015; Millan 2003). Mechanisms that can orchestrate and restore this delicate balance are sought in the quest for more rational therapeutic targets in anxiety treatment. MicroRNAs are such candidates (see below), having been shown to interact with such regulatory neurotransmitters and signaling cascades (see below and Martinetz 2016, for review).

Brain areas investigated for a (functional) role of microRNAs in anxiety. (a) Schematic of proposed anxiety circuitry based on references (Calhoon and Tye 2015; Tovote et al. 2015). (b) Schematic representation summarizes the most recent studies assessing microRNAs in the brain for regional, functional, and therapeutic implications in anxiety-related behaviors. Studies that assessed the implications of candidate microRNA expression using functional approaches are indicated with an asterisk (∗), while red (knockdown), green (overexpression/enhancement), and blue (both) indicate the direction of regulation. Bed nucleus of the striatum terminalis (BNST), medial prefrontal cortex (mPFC), hippocampus (Hip), amygdala (Amy; amygdala subregions: basolateral amygdala (BLA) and central amygdala (CeA)), nucleus accumbens (NA), raphe nucleus (RN), hypothalamus (HYP), locus coeruleus (LC), periaqueductal gray (PAG), parabrachial nucleus (PB), ventral tegmental area (VTA), and lateral septum (LS)
The mechanisms leading to deficiencies in the networks processing anxiety are not understood in detail. Family and twin studies have indicated that anxiety disorders are multifactorial in cause, involving an interaction between genetic and environmental factors (Hettema et al. 2001, 2005; Klengel and Binder 2015; Sharma et al. 2016). Heritability in anxiety disorders is rather moderate but can amount to 30–48%, for example, in generalized anxiety/panic disorder (Hettema et al. 2001; Kendler et al. 1999; Shimada-Sugimoto et al. 2015). Evidence suggests that multiple genes with small effects are involved, with single gene variation insufficient to produce full anxiety pathology. Genome-wide association studies (GWAS) assessing the presence of single nucleotide polymorphisms (SNPs) have revealed a number of chromosomal risk loci, including genes associated with neurotransmitter and HPA axis regulation, such as serotonin receptor 1A (5HT-1A), catechol-O-methyltransferase (COMT), monoamine oxidase A (MAOA) genes, neuropeptide S receptor (NPSR), brain-derived neurotrophic factor (BDNF), and HPA axis genes corticotropin-releasing hormone receptor 1 (CRHR1) and FK506 binding protein 5 (FKBP5) (Smoller 2016; Bandelow et al. 2016; Domschke et al. 2012; Autry and Monteggia 2012).
The remaining variability associated with the development of anxiety-related disorders is accounted for predominantly by environmental factors (Hettema et al. 2005; Vieland et al. 1996; Norrholm and Ressler 2009; Schiele and Domschke 2017). Stress is particularly well studied in this regard (Dirven et al. 2017). For example, stress during pregnancy, neonatal stress, or traumatic experiences during childhood, such as parental loss or physical/emotional neglect, affect the function of various neurobiological mechanisms relevant for anxiety processing, including the HPA axis (Maccari et al. 2014), and are factors that have been linked with increased vulnerability to the development of anxiety disorders later in life (Stein et al. 2014; Drury et al. 2016; Fernandes and Osorio 2015; Klauke et al. 2010). Research suggests changes in gene expression of multiple known candidate genes such as genes involved in DNA transcription and translation play a role. The discovery in recent years of epigenetic mechanisms and their role in the control of gene expression made important contributions to our understanding of the complex development of anxiety-related disorders. It is thought that these mechanisms may explain how the combination of an accumulation of affected risk genes and environmental insults (G X E interaction) lead to the onset of anxiety-related disorders (Sharma et al. 2016) (Fig. 2).

Genetic/environmental/epigenetic influences in anxiety-related disorders. Schematic representation of gene × environment × epigenetics in the development of anxiety-related disorders. Genetic and environmental factors can lead to long-lasting changes in epigenetic mechanisms, resulting in changes in brain structure and also functional brain processing. These changes can subsequently lead to the development of anxiety-related disorders
3 Epigenetics in Anxiety: MicroRNAs
“Epigenetics” is the term given to mechanisms that together control gene expression in the absence of any alterations to the DNA base pair composition (Bjornsson et al. 2004). Epigenetic marks involved in the modulation of gene expression can be influenced by environmental factors, can be inherited to influence subsequent generations, but, importantly, can also be reversed (Jaenisch and Bird 2003), suggesting a dynamic modulation mediated by these marks. These mechanisms exert multiple levels of control over gene expression, ranging from DNA and histone modifications to posttranscriptional regulation via noncoding RNAs, including microRNAs (Dias et al. 2015; Cholewa-Waclaw et al. 2016), and they have emerged as critical regulators in nearly all neurobiological processes (Sevignani et al. 2006; Ong and Corces 2011). Environmental insults, such as stress, diet, or toxin exposure, can affect epigenetic mechanisms, which could explain their profound and long-lasting effects on multiple neurobiological systems and networks. Epigenetically mediated mechanisms could also shed light on individual variations in the development and treatment of some of the most common central nervous system (CNS) disorders (Yehuda and Bierer 2009; Landgrave-Gomez et al. 2015), including anxiety disorders (Nieto et al. 2016; O’Connor et al. 2016; Bartlett et al. 2017). Environmental influences associated with the development of anxiety disorders have been linked with distinct changes in epigenetic mechanisms, such as histone acetylation, DNA methylation, and noncoding RNA expression, which regulate the long-lasting expression of specific genes (Schiele and Domschke 2017). Genes targeted are, for example, associated with neurotransmission/neuromodulation, including GABAergic, glutamatergic, dopaminergic and serotonergic signaling, or genes associated with HPA axis functioning, many of them known to be aberrantly regulated in anxiety (Fass et al. 2014; Hunter 2012).
MicroRNAs are one of the best-studied posttranscriptional modifiers of gene expression, revealing their critical role in CNS development and homeostasis. As such, they have emerged as interesting candidates for studying molecular mechanisms underlying anxiety and anxiety disorders (Scott et al. 2015). Furthermore, the standardization of methods of examining microRNA-mediated effects (Baker 2010) and the recent increase in functional studies assessing causal roles of microRNAs have generated an opportunity to reassess where we stand with regard to microRNA-mediated mechanisms and anxiety. In the next sections, we briefly summarize microRNA function in the brain and then focus on an update on the current status of the role microRNAs are thought to play in the development, progression, and treatment of anxiety-related disorders.
3.1 MicroRNAs in the Central Nervous System
MicroRNAs are short (19–24-nucleotide) single-stranded ncRNAs that are genomically encoded and that mediate the posttranscriptional regulation of target genes via complementary binding to the messenger RNA seed sequence (Bartel 2009). There have been over 5,000 microRNA transcripts annotated in humans so far, together comprising 1–2% of the human genome (Londin et al. 2015). It is well documented that microRNAs can target hundreds of genes often within distinct functional networks via partial complementary binding and, furthermore, any given gene may have multiple target sites for different microRNAs (Lim et al. 2005; Wang et al. 2011). Originally thought of as “junk RNA”, such as leftover remnants of past viral infections, it has now been revealed that microRNAs may control the expression of up to 60% of the protein-coding genes in the human genome in a highly complex network-like fashion (Krol et al. 2010b; Bartel 2004). The first microRNA to be discovered was lin-4, which was found in Caenorhabditis elegans using a genetic screening tool for gene variations associated with deficits in the temporal control of postembryonic development (Lee et al. 1993). In 2001 the true potential of these small noncoding RNAs was revealed when their role as regulators of gene expression was demonstrated (Lau et al. 2001). Since this initial breakthrough discovery, the emergence of microRNAs and their downstream effects on target genes has revealed them as “fine tuners” of gene expression, and this has subsequently become one of the best-studied mechanisms of posttranscriptional regulation.
3.2 MicroRNA Induction, Biogenesis, Turnover, and Function
Induction, biogenesis, turnover, and general function of microRNAs have been the subject of specific reviews; for a detailed discussion on this topic, please see Ruegger and Grosshans (2012), Vidigal and Ventura (2015), Krol et al. (2010b), Bartel (2004, 2009), and Sim et al. (2014). Here, we will give a brief account of these processes (summarized in Fig. 3).

MicroRNA induction, biogenesis, turnover, and function. MicroRNA expression is induced in a number of ways including via neuronal activation whereby (1: orange) Ca2+ influx influences the expression of transcriptions factors such as MEF2 and CREB, which subsequently have been shown to mediate the expression of a wide range of microRNAs. Following transcriptional induction, RNA polymerase II transcribes microRNAs as precursor microRNAs (pri-miRNA) from specific microRNA genes or from within the introns of protein-coding genes. These pri-miRNAs are then processed by a member of the RNase III enzyme family, Drosha, and along with other cofactors, including DiGeorge syndrome critical region gene 8 protein (DGCR8), cleave the pri-miRNA into a 70–100-nucleotide-long pre-miRNA. (2: orange) microRNA turnover is in part controlled by the regulation of Drosha where decreased expression reduces the ability of a cell to transcribe new microRNAs. Exportin-5 then transports the pre-miRNA to the cell cytoplasm where Dicer processes it into a microRNA duplex. (3: orange) Ca2+ influx is also associated with increased Calpain expression, which releases Dicer from the postsynaptic densities (PSD). Along with Argonaute 2 (Ago2) and transactivation-responsive RNA-binding protein (TRBP), Dicer incorporates the microRNA “guide” strand, based on the thermodynamic asymmetry rule, into the microRNA-induced silencing complex (miRISC). The co-processed “passenger” strand is degraded at this point. The microRNA and miRISC complex then induce (1 and 2: yellow) mRNA degradation/destabilization and/or (3: yellow) translational repression via target gene binding through a “seed sequence” located between nucleotides 2 and 8 on the microRNA guide strand (enclosed panel). PABP, poly(A)-binding protein. CCR4, C-C motif chemokine receptor 4
In the nervous system, the induction of microRNA expression can occur via a number of different mechanisms of which the most studied is via neuronal activity (Sim et al. 2014). Generally speaking, Ca2+ influx following neuronal activation can lead to various downstream effects including the induced expression of transcription factors such as MEF2 and BDNF, which have subsequently been shown to mediate the expression of specific microRNAs such as miR-132 (Nudelman et al. 2010). Following the induction, precursor microRNA molecules (pri-miRNAs) are transcribed by RNA polymerase II, with approximately half of the microRNA genes being transcribed from their own nonprotein-coding transcripts and the other half from within the intergenic regions of protein-coding genes (Saini et al. 2007). Pri-miRNA transcripts are then subject to processing whereby they fold into hairpin structures that are subsequently bound by a member of the RNase III enzyme family, Drosha. Drosha itself is subject to modulation, which is another rate-limiting step on the biogenesis of mature microRNAs. Drosha, with cofactors including DiGeorge syndrome critical region gene 8 protein (DGCR8), cleaves the pri-miRNA into a 70–100-nucleotide-long pre-miRNA, which is subsequently exported to the cell cytoplasm by exportin-5. In the cytoplasm, another RNase III family enzyme, Dicer, removes the hairpin structure and processes the pre-miRNA into a microRNA duplex. In combination with the RNA-binding proteins, Argonaute 2 (Ago2) and the transactivation-responsive RNA-binding protein (TRBP), Dicer, incorporate the microRNA guide strand, based on the thermodynamic asymmetry rule, into the microRNA-induced silencing complex (miRISC), while the other “passenger” strand is degraded. The thermodynamic asymmetry rule implies that the microRNA strand with the least stable 5′ terminus will be chosen as the “guide” strand and incorporated into the miRISC complex, while the other “passenger” strand is degraded. Any given microRNA has a 5p and a 3p, which is dependent on what side of the hairpin each microRNA strand is situated. In principle, both the 5p and the 3p strand can act as the guide strand. Once incorporated into the miRISC complex, microRNAs then induce translational repression and/or mRNA degradation via binding with target genes through a “seed sequence” located between nucleotides 2 and 8 on the microRNA guide strand (Fig. 3, enclosed panel). A microRNA whose seed sequence has perfect complementarity with its target gene will have the greatest effect on the expression levels of this target.
While some microRNAs are expressed ubiquitously, such as let-7b, miR-17, and miR21, there are a number of microRNAs that are enriched in the brain, such as miR-128b (Lin et al. 2011), as well as microRNAs that are expressed in a temporal-, regional-, and cell-type-specific manner (Taguchi 2013), as shown for miR-7, miR-34a, and miR-132, among others, in the brain (Olsen et al. 2009). MicroRNA-mediated control over mechanisms such as gene expression regulation in the nervous system makes them essential mediators of important processes in the brain, including neuronal plasticity and learning and memory (Ruegger and Grosshans 2012). MicroRNA expression/turnover within neurons can be relatively fast. In the mouse retina, for example, light-sensitive microRNAs such as miR-204 and miR-211 and the microRNA-cluster miR-183/96/182 undergo rapid transcriptional repression, e.g., 30 min following adaption of the mouse to the dark chamber (Krol et al. 2010a). This microRNA regulation has subsequently been found to play a functional role, preventing retinal degeneration (Lumayag et al. 2013). A number of studies have further characterized microRNA turnover in the brain and have shown that microRNA expression is dynamic and under the control of a number of different mechanisms, including those involved in biogenesis, maturation, and degradation (Marzi et al. 2016) (Fig. 3). For example, one approach blocking the biogenesis of all new microRNAs, via ablation of Dicer, led to the discovery of varying half-lives of investigated microRNAs, ranging from 5 to 12 days (Gantier et al. 2011). Thus, microRNAs in general can be induced within minutes (e.g., by neuronal activity in neurons (Sim et al. 2014)) and can remain within cells in a stable condition for at least several days and maybe much longer (Gantier et al. 2011).
3.3 MicroRNAs as Modulators of Neuronal Plasticity and Learning and Memory
Adaptive responses to environmental stimuli are mediated via neuronal activity at a cellular level and involve a number of molecular changes altering synaptic dynamics, including local synaptic expression of plasticity-associated proteins. This expression is controlled by a number of synaptically located microRNAs, induced by neuronal activity, which ensures fine-tuned control over synaptic plasticity (Lugli et al. 2008; Hu et al. 2014; Schratt 2009; Gu et al. 2015) and learning and memory mechanisms (Wang et al. 2012). For example, miR-132 has emerged as an interesting candidate. High-frequency stimulation-induced long-term potentiation in the dentate gyrus reflecting enhanced synaptic plasticity is associated with increased expression levels of miR-132 (Wibrand et al. 2010). It was suggested that miR-132, depending on brain region and/or behavioral-specific signals, can act as a “proteome switch” and mediate different mechanisms of neuronal plasticity through its target genes, including the N-methyl-D-aspartate receptor subunit 2A (Bredy et al. 2011). The critical role played by microRNAs in these processes, which include learning and memory and related disorders, has been the topic of detailed reviews (please see Saab and Mansuy 2014; Hu and Li 2017; Aksoy-Aksel et al. 2014).
In relation to anxiety disorders, it should be mentioned that disturbances in learning and memory mechanisms are common in these disorders (Hemstedt et al. 2017), which are increasingly targeted by specific therapeutic interventions (e.g., Singewald et al. 2015). There is mounting evidence linking microRNAs to these processes (see below).
3.4 MicroRNAs in Anxiety and Anxiety-Related Disorders: Associative Studies
There is increasing evidence of microRNA dysregulation in disease. By far the best-studied field so far is cancer (for more detailed reviews, please see Kong et al. 2012; Nana-Sinkam and Croce 2013; Garzon et al. 2009). Concerning the brain, dysregulation of microRNAs has also been demonstrated in brain tumors, neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease, and, more recently, psychiatric disorders (Spadaro and Bredy 2012; Wang et al. 2012; You et al. 2016; Cao et al. 2016; O’Connor et al. 2016). In particular, the investigation of microRNAs in psychiatric disorders has gained momentum due to their potential application as molecular biomarkers and/or their use in microRNA-specific therapies for psychiatric illness (Kichukova et al. 2015; Scott et al. 2015).
Because of the inaccessibility of brain tissues in living patients, human studies investigating the role of microRNAs in psychiatric patients rely on postmortem brain tissues obtained from patients with higher mortality rates due to higher risk-taking and/or higher suicide rates accompanying disorders such as bipolar disorder, schizophrenia, and major depressive disorder (Rao et al. 2013). Little is known regarding brain microRNA changes in anxiety disorders on their own, but there are studies in comorbid patients suffering from anxiety disorders and depression showing altered microRNA regulation (see below).
The majority of human studies examining anxiety-related disorders are limited to the assessment of differential microRNA expression in the periphery – mostly in blood and its constituents – and the correlation of these findings with the presence of disease (Table 1). For example, in generalized anxiety disorder (GAD) patients, circulating miR-663 and miR-4505 levels correlated with anxiety symptoms (Chen et al. 2016), and miR-29c expression increased in response to social anxiety in a social stress task, corresponding both to the stress experience and to alterations in ventromedial prefrontal cortex functional connectivity (Vaisvaser et al. 2016). Other insights came from postmortem samples of patients who had suffered from depression comorbid with anxiety, which revealed a downregulation of miR-135a in the raphe nucleus (Issler et al. 2014). Introducing further complexity, single nucleotide polymorphisms (SNPs) in the precursors or mature microRNAs could interfere with balanced processing in anxiety (for review see Malan-Muller et al. 2013). For example, the search for SNPs associated with specific microRNA candidates revealed a number of microRNA variations, including miR-22, miR-138-2, miR-148a, miR-339, and miR-488, which are associated with anxiety-related disorders such as panic disorder (Muinos-Gimeno et al. 2011). Some microRNA candidates with SNPs, such as miR-22, were also identified in a different cohort of panic disorder patients (Kim et al. 2015). As another example, polymorphisms in the Dicer1 gene have been linked with PTSD (Wingo et al. 2015). These studies demonstrated aberrant microRNA expression and possibly regulation in anxiety and provided important indication of a role of microRNAs in anxiety-related disorders. For more detailed discussion of these findings, please see recent reviews (Snijders et al. 2018; Martinetz 2016; Scott et al. 2015; Issler and Chen 2015; Malan-Muller et al. 2013; O’Connor et al. 2012, 2016; Hommers et al. 2015). These correlative studies are very important but have the disadvantage that it is hard to conclude whether microRNA expression and its subsequent posttranscriptional regulation causes the anxiety disorder or is a consequence of it. This is where the use of relevant animal models of anxiety is invaluable. Indeed, there are preclinical studies emerging that attempt to provide causal evidence for a functional role of microRNAs in anxiety. In the next sections, we focus primarily on these studies.
3.5 MicroRNAs in Anxiety and Anxiety-Related Disorders: Studies Elucidating Functional/Causal Evidence
As mentioned above, microRNAs are abundant in the brain, where they appear to be differentially distributed, including the anxiety circuitry (Fig. 1), and even differently within neurons and distinct neuronal compartments. Functional implications of microRNAs in anxiety have been revealed using animal tests and models showing that down- or up-regulation of the candidate microRNAs in important nodes of the anxiety circuitry can indeed modulate anxiety-related behavior. The results of these studies are discussed below and are summarized in Table 2. In Fig. 1 (bottom panel), an overview is given of where in the brain such functional relationships have been investigated so far.
The first to explore the role of microRNAs in anxiety with a more functional approach were Haramati and colleagues, who locally depleted the microRNA-processing enzyme, Dicer, in the central amygdala of adult mice. As predicted, this depletion led to a drastically reduced ability for central amygdalar neurons to generate new mature microRNAs, which was found to be associated with increased anxiety-like behavior in the light/dark and open-field tests (Haramati et al. 2011). Since stress, as mentioned, is a major contributor to the development and triggering of anxiety-related behavior, stress-associated microRNA regulation has been particularly studied as a means of bridging stress with downstream pathology (Issler and Chen 2017). For example, in investigating mice following acute and chronic stress, miR-34c was revealed as a prominent stress-induced microRNA in the central amygdala. The authors suggest this microRNA plays a role in stress resilience and the development of anxiety. It was revealed that its upregulation reduced anxiety-like behavior via downregulating the expression of its target gene, corticotropin-releasing hormone receptor 1 (CRHR1), a key player in stress-induced anxiety. Furthermore, miR-34c expression was decreased in the central amygdala of Dicer-ablated neurons, which was associated with increased anxiety-like behavior (Haramati et al. 2011). Together these data highlight miR-34c expression in the central amygdala as a prominent microRNA involved in resilience to anxiety following stress. Attempting to further characterize the functional role of this microRNA, overexpression of miR-34c in the central amygdala produced an anxiolytic effect, demonstrating that manipulation of a single microRNA in one important node of the anxiety circuitry can drive a behavioral response (Haramati et al. 2011). Somewhat in contrast to these findings, Andolina and colleagues demonstrated that knockout (rather than overexpression) of miR-34 in the BLA is associated with resilience against stress-induced anxiety and with the facilitation of fear extinction (Andolina et al. 2016). Discrepancies in miR-34-mediated effects on anxiety behavior may be attributed to a number of reasons, including the fact that two different amygdalar subnuclei were investigated. Haramati et al. performed a lenti-virally mediated overexpression of miR-34c in the central amygdala, whereas Andolina et al. knocked out miR-34 in the BLA. This could suggest distinct regionally specific roles played by specific candidate microRNAs such as miR-34 in the central amygdala vs the BLA. Further underlining the importance of this microRNA, members of the miR-34 precursor family have emerged as critical regulators in a number of amygdalar-dependent learning paradigms, including fear learning. For example, it has been demonstrated that miR-34a expression is increased in the amygdala 30 min following auditory fear conditioning, while knockdown of this microRNA in the basolateral amygdala impairs fear learning (Dias et al. 2014). These data suggest that the miR-34 precursor family plays a critical role in amygdalar-dependent memory formation, with dissociated effects on fear and extinction learning, whereas knockdown of miR-34 in the basolateral amygdala can facilitate fear extinction and at the same time impairs the formation of fear memory.
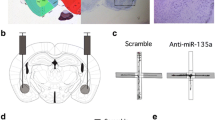
As discussed above, the RDoC initiative is fostering trans-diagnostic research investigating endophenotypes, to evaluate neurobiological mechanisms that may cut across traditional diagnoses. In line with the high comorbidity in anxiety disorders, involving multiple anxiety disorder categories and other comorbid conditions such as mood disorders, a number of studies assessing the functional role of microRNAs in animal models have investigated both anxiety- and depressive-like behaviors. Fonken and colleagues demonstrated that a genetic knockout of miR-155 in the hippocampus was associated with reduced anxiety- and depressive-like behavior (Fonken et al. 2016). Using a similar methodology, knockout of miR-17-92 in adult neural progenitor cells led to decreased neurogenesis in the dentate gyrus of the hippocampus and increased anxiety- and depressive-like behavior, while overexpression produced the opposite effects on neurogenesis and behavior (Jin et al. 2016). This is in line with reports linking enhanced trait anxiety with reduced hippocampal neurogenesis (Sah et al. 2012). The functional relationship between anxiety-related behavior and microRNA-mediated modulation has so far been demonstrated in three different brain areas in the anxiety circuitry: the amygdala, the hippocampus, and the raphe nucleus (Fig. 1, bottom panel). For example, overexpression of miR-135a in the raphe nucleus led to a reduction in anxiety- and depressive-like behavior elicited by social defeat, while miR-135a knockdown was associated with increased anxiety-like behavior (Issler et al. 2014). The authors found these effects to be mediated via interaction of miR-135a with the serotonergic system, targeting specifically the serotonin transporter (SERT) and serotonin receptor 1a (5HT-1a) genes in the dorsal raphe nucleus. As stated in the previous sections, microRNAs can target numerous genes and have varying effects in different cell types and tissues/brain areas due to distinct expression patterns and induction pathways. In agreement with this, a recent study demonstrated that knockdown of miR-135a in the basolateral amygdala also increased anxiety-like behavior, but in this brain area, it was found that miR-135a regulates anxiety via interaction with glutamatergic neurotransmission, targeting in particular complexin-2 (Mannironi et al. 2017).
Investigating microRNAs in trait anxiety in rats, Cohen and colleagues found that rats selectively bred for high novelty responding – which is associated with low trait anxiety – exhibit lower expression of miR-101a-3p in the amygdala compared with rats bred for low novelty responding that show high trait anxiety (Cohen et al. 2017). Supporting the relation of miR-101-3p regulation to anxiety-like behavior, overexpression of miR-101-3p in the basolateral amygdala was indeed associated with enhanced anxiety-like behavior. In this relation it should be mentioned that recent results suggest that the gut microbiome is necessary for appropriate regulation of microRNA expression in brain regions implicated in anxiety-like behaviors including the amygdala and prefrontal cortex, adding an additional level of complexity (Hoban et al. 2017). Also in hippocampal areas, functional microRNA regulation was investigated. Using a model of neonatal isolation-induced anxiety, Bahi revealed that expression of the brain-enriched miR-124a was increased in the dentate gyrus of neonatally isolated Wistar rats (Bahi 2016). Overexpression of miR-124a in the dentate gyrus further exacerbated the anxiety-like behavior, demonstrating that the intensity of the anxiety response correlates with the expression level of miR-124a in the dentate gyrus. Supporting these findings, the author more recently demonstrated that virally mediated reduction of miR-124a expression in the dentate gyrus is associated with decreased anxiety-like behavior, which inversely correlates with the expression of its target gene, brain-derived neurotrophic factor (BDNF), in this brain area (Bahi 2017). BDNF is an activity-dependent neurotrophic factor that has been extensively implicated in fear and anxiety regulation (Andero et al. 2014; Andero and Ressler 2012). The increase in functional evidence linking microRNA regulation with anxiety supports a crucial role played by these “fine tuners” of gene expression and highlights them as possible novel therapeutic targets for the treatment of anxiety disorders.
3.6 MicroRNAs in the Treatment of Anxiety-Related Disorders
MicroRNAs have been investigated as therapeutic targets for numerous disorders, including in particular the treatment of cancer, with the first cancer-targeted microRNA, MRX34, entering clinical trials in patients with advanced hepatocellular carcinoma (Ceman and Saugstad 2011; Kong et al. 2012; Ling et al. 2013). Miravirsen, which targets miR-122, was recently demonstrated in a phase II trial to have no long-term safety issues and sustained its virological response, which is associated with reduced occurrence of liver failure, in the treatment of chronic hepatitis C (van der Ree et al. 2014). More recently, microRNAs have also been studied as potential targets in psychiatric disorders (Tardito et al. 2013; O’Connor et al. 2012, 2016). One idea here is that microRNA-mediated regulation of relevant biological processes is linked with the therapeutic effect of anxiety disorder treatments. As studies functionally assessing microRNAs in the therapeutic action of drugs are scarce, here we have also included correlative studies that specifically focused on drug-induced changes in microRNA expression associated with a therapeutic decrease in anxiety-like behavior.
3.7 MicroRNA Regulation in Established and Experimental Pharmacological Interventions Used in Anxiety Treatment
A number of studies have assessed the possible contribution of specific microRNAs to the therapeutic action of currently used interventions for anxiety disorders (summarized Fig. 1 (bottom panel)). Indeed, medications used in anxiety and affective disorders have been shown to modulate microRNA levels in the brain, although microRNAs were not their original target. One of the first of such studies demonstrated that long-term administration of different mood stabilizers led to altered expression of microRNAs in the hippocampus of rats when compared with saline-treated controls (Zhou et al. 2009). Following chronic oral treatment with valproate (VPA) or lithium, a number of microRNAs, including miR-29a, Let-7b, Let-7c, miR-128a, miR-24a, miR-30c, miR-34a, and miR-221, exhibited decreased hippocampal expression, while miR-144 expression was increased. Interestingly, VPA has shown promise in the treatment of some forms of anxiety, such as social anxiety disorder (Kinrys et al. 2003), and has previously also been shown to be useful as an adjunct facilitating the effect of fear extinction, the central mechanism of exposure-based cognitive behavioral therapy (Kuriyama et al. 2011; Whittle et al. 2013). Further investigating functional downstream target genes of miR-34a in primary hippocampal cultures, the authors revealed that administration of a miR-34a inhibitor, as well as lithium or VPA, increased the expression of miR-34a’s target gene, metabotropic glutamate receptor 7 (GRM7). Furthermore, administration of an miR-34a precursor led to decreased expression levels of GRM7, suggesting a functional relationship between miR-34a and GRM7 expression levels and pointing toward one novel possibly common neurobiological effect of lithium and VPA with putative therapeutic significance.
Since this breakthrough study, the number of microRNAs linked with relevant downstream effects of therapeutic drugs commonly used in the treatment of anxiety and affective disorders has steadily increased. Antidepressants, such as selective serotonin reuptake inhibitors (SSRI) used as first-line treatments in different anxiety disorders (Baldwin et al. 2014), have been particularly investigated in this regard. Baudry and colleagues were the first to investigate SSRI effects following different modes of administration and revealed increased expression of miR-16 in the raphe nucleus and decreased expression in the locus coeruleus of mice (Baudry et al. 2010). Interestingly, the expression patterns of miR-16 in the raphe nucleus and locus coeruleus were inversely correlated with the expression levels of one of its target genes, the serotonin transporter (SERT). These data indicate that the therapeutic effect of fluoxetine could in part be mediated via differential miR-16 expression levels in monoaminergic neurons targeting SERT in the raphe nucleus and locus coeruleus (Baudry et al. 2010). Using a mouse model of PTSD, oral treatment with fluoxetine for 31 days reduced anxiety-related behavior in these mice, and this was associated with long-term effects on microRNA expression (Schmidt et al. 2013): the expression of miR-1971 was even after 74 days decreased in the prefrontal cortex in comparison with control groups, suggesting that the long-lasting beneficial effects on anxiety-related behaviors in fluoxetine-treated mice may be mediated in part by reduced miR-1971 expression. Taken together, these studies implicate the regulation of specific microRNA candidates (e.g., miR-16, miR-1971) in distinct brain regions in the therapeutic action of fluoxetine (see also Fig. 1).
Other SSRIs have also been investigated for a possible relation between their behavioral effects and microRNA regulation. It has been shown that maternal separation or chronic unpredictable stress-induced increase in depression-/anxiety-related behavior was associated with decreased levels of miR-326 expression in the nucleus accumbens and striatum (Zhang et al. 2015). Following chronic escitalopram treatment, the levels of miR-326 in both brain areas increased, thus returning to levels comparable with those of non-stressed or saline-treated rats. In investigating effects following tricyclic antidepressant treatment, it was shown that mice treated chronically with imipramine for 18–21 days display lower social avoidance, which was associated with increased expression of miR-135a in the raphe nucleus. As mentioned above, miR-135a targets the serotonergic system directly via binding to the serotonin transporter and 5HT-1a receptor gene (Issler et al. 2014). Demonstrating the functional involvement of miR-135a expression, its overexpression in serotonergic neurons was associated with decreased anxiety- and depression-like behavior following social defeat, while its knockdown led to increased anxiety- and depression-like behaviors (Issler et al. 2014).
Information concerning the effects of antidepressants on microRNA expression in the human brain is scarce. Interestingly, in patient’s peripheral blood, the baseline expression of miR-1202 was predictive of treatment success, as patients who responded to treatment exhibited lower blood expression of miR-1202, which subsequently increased following an 8-week treatment with citalopram (Lopez et al. 2014). This brain-enriched microRNA was found to target the GRM4 gene, which shows decreased expression following treatment with antidepressants (Cruceanu et al. 2016). This observation suggests that miR-1202 contributes to antidepressant action via modulation of glutamatergic neurotransmission. Using human postmortem tissues from patients, Lopez and colleagues were able to demonstrate that major depressive disorder is associated with decreased expression of miR-1202 in the prefrontal cortex and in blood (Lopez et al. 2014). To our knowledge, this is the only study that links the dysregulation of a specific microRNA in the brain of psychiatric patients with the expression in blood and that shows dynamic regulation with treatment, thus demonstrating its possible usefulness as a biomarker of treatment success. Importantly, this study demonstrates long-term changes in specific microRNA expression in the brain associated with an affective disorder. In a recent follow-up study, the authors further explored this relationship between peripheral miR-1202 expression levels and brain activity in patients, following an 8-week treatment with desvenlafaxine (Lopez et al. 2017). Using functional magnetic resonance imaging, the authors found that lower peripheral levels of miR-1202 in desvenlafaxine-treated subjects were associated with increased neural connectivity between the posterior cingulate and the parietal, occipital, and prefrontal cortices.
Therapeutic drugs targeting primarily other neurotransmitter and hormone systems besides the serotonergic system have also been examined for the involvement of microRNA candidates in their mode of action. In one study, treatment with the antidepressants agomelatin, which targets melatonin receptors, and tianeptine, which targets glutamatergic, adenosine, and opioid systems, normalized stress-induced changes in microRNA levels in the dentate gyrus, including the expression of miR-181b, miR-9, and miR-411, among others (Patrício et al. 2015). Treatment with the CRHR1 antagonist NBI-27914 was shown to decrease anxiety-like behavior of mice in the elevated plus maze and open-field test. It was found that miR-34b targets the CRHR1 gene and it was thus demonstrated that administration of an agomir for miR-34b into the paraventricular nucleus of the hypothalamus mimicked the behavioral effect of the CRHR1 antagonist and resulted in reduced anxiety-like behavior (Zhu et al. 2017). Also by targeting GABAergic neurotransmission, microRNA expression changes. Administration of the GABA agonist gaboxadol was associated with reduced anxiety-like behavior and increased miR-33 expression in the dorsal hippocampus (Jovasevic et al. 2015). The pharmacological enhancement of therapy-relevant cognitive mechanisms (see also next chapter) via “cognitive enhancers” including the partial NMDA agonist D-cycloserine (DCS) is a more recent avenue for the treatment of anxiety-related disorders (for recent review see Singewald et al. 2015). In exploring a possible involvement of microRNAs in this effect, the expression of 32 microRNAs in the left dorsal hippocampus was found to be regulated after treatment with DCS. This interesting first finding needs to be followed up in more detail also in relation to a possible facilitation of extinction learning (Malan-Muller et al. 2017).
3.8 Non-pharmacological Interventions
Exposure-based cognitive behavioral therapy is a frequently used psychotherapeutic intervention for anxiety disorders. It involves the process of fear extinction, which results in the gradual reduction of fear- and anxiety-related responses to stimuli perceived as threatening to the patient (Abramowitz 2013; Milad and Quirk 2012; Craske and Stein 2016). Meta-analyses of cognitive behavioral therapy used in treating anxiety disorders show that it is efficacious for a number of anxiety-related disorders but has its limitations, including the problem that it produces insufficient long-lasting effects, and thus symptoms can return (Hofmann and Smits 2008; Tolin 2010; James et al. 2015; Gould et al. 2012). Importantly, anxiety patients are often associated with fear extinction deficits (Milad et al. 2009, 2013; Jovanovic et al. 2012; Michael et al. 2007; Duits et al. 2015; Wessa and Flor 2007). Thus, understanding the underlying mechanisms of deficient fear extinction (Graham and Milad 2011; Hill and Martinowich 2016; Baker et al. 2016; Maren and Holmes 2016) and how to overcome this deficiency may hold potential for the development of novel treatment strategies (for review see Holmes and Singewald 2013, Bukalo et al. 2014). Exploring the role of microRNAs in the rescue of deficient fear extinction, our group recently revealed an increased expression of miR-144-3p in the basolateral amygdala 2 h following fear extinction training. This increase was not seen in the very first phase of extinction training involving mainly fear retrieval, indicating that the effect was specific for extinction learning (Murphy et al. 2017). MiR-144-3p was found to target a number of genes implicated in the activation of plasticity-associated signaling cascades, including MAPK/ERK, PI3K/AKT, and NOTCH, which have been implicated in extinction mechanisms (Herry et al. 2006; Yang and Lu 2005; Dias et al. 2014). Confirming the functional significance of this, viral overexpression of miR-144-3p in the basolateral amygdala was able to rescue impaired fear extinction in extinction-impaired 129S1 mice and to prevent the return of fear in extinction-competent 129S6 mice (Murphy et al. 2017). Regulation of miR-144 in the context of anxiety has also been observed in humans. A recent study in healthy students found that whole blood miR-144 expression increased during the period preceding a stressful event (an effect involving, it is thought, anticipatory anxiety) before returning to baseline levels in the days following (Katsuura et al. 2012). In another study, miR-144-5p expression levels in the blood plasma of patients suffering from depression/anxiety were found to be lower in comparison with healthy controls (Wang et al. 2015). Compellingly, following 8 weeks of psychotherapy, miR-144-5p expression levels increased and were normalized to expression levels seen in healthy controls, suggesting that miR-144-5p expression levels could possibly be useful as a biomarker of treatment success.
While fear extinction is associated with the formation of a new fear-inhibitory memory, it is also important to note that during this process the original fear memory is brought into a labile state where it is subject to updating mechanisms (Flavell et al. 2013). It is during this period that the reconsolidation of the original fear memory can be disrupted or modulated, allowing for the transition to the predominant expression of a newly formed fear-inhibitory memory (Hemstedt et al. 2017). Exploring the role of microRNAs in this process, Lin and colleagues assessed the expression of microRNAs in the medial prefrontal cortex (Lin et al. 2011). Following fear extinction training, the expression of miR-128b was shown to be increased in the infralimbic subdivision of the medial prefrontal cortex (ILPFC). Interestingly, the authors found that miR-128b targets a number of plasticity-associated genes known to be important for fear memory retrieval (Lin et al. 2011). The fear-associated genes targeted by miR-128b include the regulator of calmodulin signaling (RCS), which has been demonstrated to inhibit calmodulin and to negatively regulate protein phosphatase calcineurin activity overall, leading to the disruption of fear memory consolidation/reconsolidation. Thus, increased miR-128b expression during fear extinction was suggested to facilitate the transition from the expression of fear to the expression of this newly formed fear-inhibitory memory. Indeed, overexpression of miR-128b in the ILPFC was able to enhance the effects of fear extinction, thus indicating that increased miR-128b expression could enhance exposure-based cognitive behavioral therapy via enhancing the expression of fear-inhibitory memories.
Beneficial environmental influences have been shown to contribute to successful outcomes of anxiety treatments (Kheirbek et al. 2012). In preclinical models, environmental enrichment, for example, has been used to model positive environmental stimulation, producing anxiolytic effects (Hendriksen et al. 2014; Huttenrauch et al. 2016). Interestingly, Ragu Varman and colleagues demonstrated increased expression of miR-183 and miR-124 in the amygdala following 30 days of environmental enrichment (Ragu Varman et al. 2013). Expression of miR-124 was also regulated in the dentate gyrus in response to environmental enrichment, and this was associated with increased neurogenesis (Brenes et al. 2016). Another non-pharmacological means that can improve anxiety symptomology is physical exercise. Subjecting male mice to voluntary wheel running reduced anxiety-like behavior and suppressed reinstatement of fear measured in their juvenile offspring, demonstrating transgenerational transmission of changes in anxiety-like behavior. The sperm of the mice following voluntary wheel running was found to have decreased expression of miR-19b and miR-455, while miR-133a was increased (Short et al. 2017). Furthermore, long-term exercise in mice, which was associated with increased resilience to stress, led to the decreased expression of miR-124 in the hippocampus and the increased expression of glucocorticoid receptors Nr3c1 and Nr3c1-1f (Pan-Vazquez et al. 2015). Interestingly, a recent genome-wide association study of positive emotion in humans identified a genetic variant proposed to mediate positive emotionality via the nucleus accumbens and miR-181 (Wingo et al. 2017). We have learned from these findings, taken together, that the beneficial effects of established as well as experimental therapeutic interventions for the treatment of anxiety-related disorders are associated with the regulation of microRNAs. As our understanding of microRNA-mediated therapeutic effects increases, novel therapeutic interventions targeting such mechanisms can be developed to improve treatments in anxiety-related disorders.
4 Conclusion and Further Perspectives
Epigenetic mechanisms, in particular microRNA-mediated modulation of gene expression and its dysregulation in disease states, have become a topic of sustained interest in the search for novel understanding and/or development of therapies for some of the most common diseases. In this chapter we have summarized the evidence linking microRNA regulation with anxiety-related behaviors and anxiety disorders. It has been found that specific microRNAs may play various roles in the development, progression, and treatment of anxiety. First studies have provided functional/causal evidence for such a role. Identified microRNAs include miR-15a, miR-17-92, the miR-34 family of microRNAs, miR-101, miR-124, miR-135, and miR-155, which influence anxiety behavior in a brain area-dependent manner. As an interesting example, miR-124 expression was shown to be upregulated in stress-induced anxiety, and its experimental targeted downregulation in the dentate gyrus led to a reduction in anxiety-like behavior, mediated in part via a miR-124a/BNDF interaction. Such functional knowledge aids the interpretation of associative studies and the identification of false positives. In this context, it was reported that running-induced anxiolysis is associated with hippocampal downregulation of miR-124.
In addition, we have summarized studies that examined how pharmacological and non-pharmacological interventions affect the expression of microRNAs. It was found that SSRIs commonly used to treat many anxiety disorders are associated with altered expression of a number of microRNAs in the brain, such as miR-16 and miR-1971. Non-pharmacological interventions, such as fear extinction, modeling exposure-based CBT, are associated with distinct changes in miR-144 and miR-128b in the BLA and infralimbic cortex of the mPFC, respectively. This new understanding has been aided by recent methodological advancements in assessing microRNAs functionally. In general, the field has moved beyond correlative studies whereby the presence of disease is associated with the altered expression of microRNA(s) and toward a more functional/causal understanding of microRNAs in distinct brain regions and of their roles in anxiety. Hence, the current data suggest that microRNAs may (in part) mediate therapeutic effects of established drugs and psychotherapeutic approaches used currently to treat these disorders.
This relation suggests that microRNAs could possibly be specifically targeted in the treatment of anxiety, although the therapeutic use of microRNAs for psychiatric diseases is in its very early stages. The initial problem encountered in such an approach – the fact that microRNAs have multiple targets – may in fact be an advantage, given that deregulation in anxiety disorders involves networks rather than single effector molecules. Thus, by targeting a single “key” microRNA, it is possible to modulate entire relevant gene networks. Target specificity may be determined by the observed highly distinct regional, cellular, and temporal expression of some of the candidate microRNAs in relevant nodes of the affected network. Along these lines, microRNA-based interventions targeting candidates such as miR-128b and miR-144 have the potential to augment the efficacy of exposure-based CBT through targeting plasticity-associated genes in extinction learning relevant brain areas such as the infralimbic cortex and basolateral amygdala, respectively. However, there is much work to be done before any microRNA-based therapy for anxiety can be used in a clinical setting. Answers to questions regarding oral bioavailability, the ability of microRNA-based therapeutics to cross the blood-brain barrier, and the presence of off-target effects currently elude us and are required to be solved before a push for microRNA-based interventions can be attempted. Therapeutic methodologies targeting microRNAs, such as virally mediated gene therapy or synthetic locked nucleic acid (LNA) agomirs/antagomirs, are associated with increased risks and thus require careful safety studies that consider off-target effects and other potential problems (Scott et al. 2015). In an effort to circumvent the limitations of virally mediated interventions, targeting mRNAs via locked nucleic acid probes, which are chemically modified to increase thermal stability and hybridization affinity, is a novel and possibly less invasive method of microRNA administration.
It is important to note that while to date numerous microRNAs have been implicated in anxiety, there is often little overlap within animal studies or between animal and human studies. This is in part due to the various experimental setups, the different behaviors assessed, and the different time points investigated. An important step forward would be to elucidate emerging microRNA candidates that play a role across various experimental setups and behaviors. Furthermore, it is unlikely that a single microRNA candidate alone will be critical in the development, progression, or treatment of anxiety; rather, a specific microRNA “signature” is more likely to be of importance in the various stages (Scott et al. 2015). In support of this, studies have revealed microRNA signatures for cancer, and more recently such signatures have emerged in preclinical models of stress-related disorders (Balakathiresan et al. 2014; Jiang et al. 2015).
In summary, we have provided an update on the most recent research functionally linking microRNAs with anxiety-related behaviors. In particular, it is through these more functional studies that we will be able to move forward in search of novel therapeutic interventions for psychiatric disorders such as anxiety disorders. A more complete regional, temporal, and subcellular picture of the role of microRNAs in anxiety will have the potential to open up novel ways of using this knowledge in the development of microRNA-based therapeutics for the improved treatment of patients suffering from anxiety-related disorders (Aten et al. 2019; Hommers et al. 2018; Sun et al. 2019; Snijders et al. 2018).
References
Abramowitz JS (2013) The practice of exposure therapy: relevance of cognitive-behavioral theory and extinction theory. Behav Ther 44:548–558
Aksoy-Aksel A, Zampa F, Schratt G (2014) MicroRNAs and synaptic plasticity--a mutual relationship. Philos Trans R Soc Lond Ser B Biol Sci 369:20130515
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Publishing, Washington
Andero R, Ressler KJ (2012) Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav 11:503–512
Andero R, Dias BG, Ressler KJ (2014) A role for Tac2, NkB, and Nk3 receptor in normal and dysregulated fear memory consolidation. Neuron 83:444–454
Anderzhanova E, Kirmeier T, Wotjak CT (2017) Animal models in psychiatric research: the RDoC system as a new framework for endophenotype-oriented translational neuroscience. Neurobiol Stress 7:47–56
Andolina D, DI Segni M, Bisicchia E, D’alessandro F, Cestari V, Ventura A, Concepcion C, Puglisi-Allegra S, Ventura R (2016) Effects of lack of microRNA-34 on the neural circuitry underlying the stress response and anxiety. Neuropharmacology 107:305–316
Aten S, Page CE, Kalidindi A, Wheaton K, Niraula A, Godbout JP, Hoyt KR, Obrietan K (2019) miR-132/212 is induced by stress and its dysregulation triggers anxiety-related behavior. Neuropharmacology 144:256–270
Autry AE, Monteggia LM (2012) Brain-derived Neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64:238–258
Bahi A (2016) Sustained lentiviral-mediated overexpression of microRNA124a in the dentate gyrus exacerbates anxiety- and autism-like behaviors associated with neonatal isolation in rats. Behav Brain Res 311:298–308
Bahi A (2017) Hippocampal BDNF overexpression or microR124a silencing reduces anxiety- and autism-like behaviors in rats. Behav Brain Res 326:281–290
Baker M (2010) MicroRNA profiling: separating signal from noise. Nat Method 7:687–692
Baker KD, Bisby MA, Richardson R (2016) Impaired fear extinction in adolescent rodents: behavioural and neural analyses. Neurosci Biobehav Rev 70:59–73
Balakathiresan NS, Chandran R, Bhomia M, Jia M, Li H, Maheshwari RK (2014) Serum and amygdala microRNA signatures of posttraumatic stress: fear correlation and biomarker potential. J Psychiatr Res 57:65–73
Baldwin DS, Anderson IM, Nutt DJ, Allgulander C, Bandelow B, den Boer JA, Christmas DM, Davies S, Fineberg N, Lidbetter N, Malizia A, McCrone P, Nabarro D, O’neill C, Scott J, van der Wee N, Wittchen HU (2014) Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol 28:403–439
Bam M, Yang X, Zhou J, Ginsberg JP, Leyden Q, Nagarkatti PS, Nagarkatti M (2016a) Evidence for epigenetic regulation of pro-inflammatory cytokines, interleukin-12 and interferon gamma, in peripheral blood mononuclear cells from PTSD patients. J Neuroimmune Pharmacol 11:168–181
Bam M, Yang X, Zumbrun EE, Zhong Y, Zhou J, Ginsberg JP, Leyden Q, Zhang J, Nagarkatti PS, Nagarkatti M (2016b) Dysregulated immune system networks in war veterans with PTSD is an outcome of altered miRNA expression and DNA methylation. Sci Rep 6:31209
Bandelow B, Seidler-Brandler U, Becker A, Wedekind D, Ruther E (2007) Meta-analysis of randomized controlled comparisons of psychopharmacological and psychological treatments for anxiety disorders. World J Biol Psychiatry 8:175–187
Bandelow B, Sher L, Bunevicius R, Hollander E, Kasper S, Zohar J, Moller HJ (2012) Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract 16:77–84
Bandelow B, Baldwin D, Abelli M, Altamura C, Dell’osso B, Domschke K, Fineberg NA, Grunblatt E, Jarema M, Maron E, Nutt D, Pini S, Vaghi MM, Wichniak A, Zai G, Riederer P (2016) Biological markers for anxiety disorders, OCD and PTSD - a consensus statement. Part I: neuroimaging and genetics. World J Biol Psychiatry 17:321–365
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Bartlett AA, Singh R, Hunter RG (2017) Anxiety and epigenetics. Adv Exp Med Biol 978:145–166
Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O (2010) miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science 329:1537–1541
Bjornsson HT, Fallin MD, Feinberg AP (2004) An integrated epigenetic and genetic approach to common human disease. Trends Genet 20:350–358
Bowers ME, Choi DC, Ressler KJ (2012) Neuropeptide regulation of fear and anxiety: implications of cholecystokinin, endogenous opioids, and neuropeptide Y. Physiol Behav 107:699–710
Bredy TW, Lin Q, Wei W, Baker-Andresen D, Mattick JS (2011) MicroRNA regulation of neural plasticity and memory. Neurobiol Learn Mem 96:89–94
Brenes JC, Lackinger M, Hoglinger GU, Schratt G, Schwarting RK, Wohr M (2016) Differential effects of social and physical environmental enrichment on brain plasticity, cognition, and ultrasonic communication in rats. J Comp Neurol 524:1586–1607
Bukalo O, Pinard CR, Holmes A (2014) Mechanisms to medicines: elucidating neural and molecular substrates of fear extinction to identify novel treatments for anxiety disorders. Br J Pharmacol 171:4690–4718
Calhoon GG, Tye KM (2015) Resolving the neural circuits of anxiety. Nat Neurosci 18:1394–1404
Cao DD, Li L, Chan WY (2016) MicroRNAs: key regulators in the central nervous system and their implication in neurological diseases. Int J Mol Sci 17:E842
Ceman S, Saugstad J (2011) MicroRNAs: meta-controllers of gene expression in synaptic activity emerge as genetic and diagnostic markers of human disease. Pharmacol Ther 130:26–37
Chen S-D, Sun X-Y, Niu W, Kong L-M, He M-J, Fan H-M, Li W-S, Zhong A-F, Zhang L-Y, Lu J (2016) Correlation between the level of microRNA expression in peripheral blood mononuclear cells and symptomatology in patients with generalized anxiety disorder. Compr Psychiatry 69:216–224
Cholewa-Waclaw J, Bird A, von Schimmelmann M, Schaefer A, Yu H, Song H, Madabhushi R, Tsai LH (2016) The role of epigenetic mechanisms in the regulation of gene expression in the nervous system. J Neurosci 36:11427–11434
Cohen JL, Jackson NL, Ballestas ME, Webb WM, Lubin FD, Clinton SM (2017) Amygdalar expression of the microRNA miR-101a and its target Ezh2 contribute to rodent anxiety-like behaviour. Eur J Neurosci 46:2241
Craske MG, Stein MB (2016) Anxiety. Lancet 388:3048
Craske MG, Stein MB, Eley TC, Milad MR, Holmes A, Rapee RM, Wittchen HU (2017) Anxiety disorders. Nat Rev Dis Primers 3:17024
Cruceanu C, Lopez JP, Tsai WT, Turecki G (2016) Dysregulation of the glutamatergic receptors after antidepressant treatment in human neural progenitor cells. Mol Psychiatry 22:1228–1229
Dell’osso B, Buoli M, Baldwin DS, Altamura AC (2010) Serotonin norepinephrine reuptake inhibitors (SNRIs) in anxiety disorders: a comprehensive review of their clinical efficacy. Hum Psychopharmacol 25:17–29
Dias BG, Banerjee SB, Goodman JV, Ressler KJ (2013) Towards new approaches to disorders of fear and anxiety. Curr Opin Neurobiol 23:346–352
Dias BG, Goodman JV, Ahluwalia R, Easton AE, Andero R, Ressler KJ (2014) Amygdala-dependent fear memory consolidation via miR-34a and Notch signaling. Neuron 83:906–918
Dias BG, Maddox SA, Klengel T, Ressler KJ (2015) Epigenetic mechanisms underlying learning and the inheritance of learned behaviors. Trends Neurosci 38:96–107
Dirven BCJ, Homberg JR, Kozicz T, Henckens M (2017) Epigenetic programming of the neuroendocrine stress response by adult life stress. J Mol Endocrinol 59:R11–r31
Domschke K, Tidow N, Kuithan H, Schwarte K, Klauke B, Ambree O, Reif A, Schmidt H, Arolt V, Kersting A, Zwanzger P, Deckert J (2012) Monoamine oxidase A gene DNA hypomethylation – a risk factor for panic disorder? Int J Neuropsychopharmacol 15:1217–1228
Drury SS, Sanchez MM, Gonzalez A (2016) When mothering goes awry: challenges and opportunities for utilizing evidence across rodent, nonhuman primate and human studies to better define the biological consequences of negative early caregiving. Horm Behav 77:182–192
Duits P, Cath DC, Lissek S, Hox JJ, Hamm AO, Engelhard IM, van den Hout MA, Baas JM (2015) Updated meta-analysis of classical fear conditioning in the anxiety disorders. Depress Anxiety 32:239–253
Farach FJ, Pruitt LD, Jun JJ, Jerud AB, Zoellner LA, Roy-Byrne PP (2012) Pharmacological treatment of anxiety disorders: current treatments and future directions. J Anxiety Disord 26:833–843
Farb DH, Ratner MH (2014) Targeting the modulation of neural circuitry for the treatment of anxiety disorders. Pharmacol Rev 66:1002–1032
Fass DM, Schroeder FA, Perlis RH, Haggarty SJ (2014) Epigenetic mechanisms in mood disorders: targeting neuroplasticity. Neuroscience 264:112–130
Fernandes V, Osorio FL (2015) Are there associations between early emotional trauma and anxiety disorders? Evidence from a systematic literature review and meta-analysis. Eur Psychiatry 30:756–764
Flavell CR, Lambert EA, Winters BD, Bredy TW (2013) Mechanisms governing the reactivation-dependent destabilization of memories and their role in extinction. Front Behav Neurosci 7:214
Fonken LK, Gaudet AD, Gaier KR, Nelson RJ, Popovich PG (2016) MicroRNA-155 deletion reduces anxiety- and depressive-like behaviors in mice. Psychoneuroendocrinology 63:362–369
Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D, Irving AT, Behlke MA, Hertzog PJ, Mackay F, Williams BR (2011) Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 39:5692–5703
Garzon R, Calin GA, Croce CM (2009) MicroRNAs in cancer. Annu Rev Med 60:167–179
Gould RL, Coulson MC, Howard RJ (2012) Efficacy of cognitive behavioral therapy for anxiety disorders in older people: a meta-analysis and meta-regression of randomized controlled trials. J Am Geriatr Soc 60:218–229
Graham BM, Milad MR (2011) The study of fear extinction: implications for anxiety disorders. Am J Psychiatry 168:1255–1265
Griebel G, Holmes A (2013) 50 years of hurdles and hope in anxiolytic drug discovery. Nat Rev Drug Discov 12:667–687
Gu QH, Yu D, Hu Z, Liu X, Yang Y, Luo Y, Zhu J, Li Z (2015) miR-26a and miR-384-5p are required for LTP maintenance and spine enlargement. Nat Commun 6:6789
Haramati S, Navon I, Issler O, Ezra-Nevo G, Gil S, Zwang R, Hornstein E, Chen A (2011) MicroRNA as repressors of stress-induced anxiety: the case of amygdalar miR-34. J Neurosci 31:14191–14203
Hemstedt TJ, Lattal KM, Wood MA (2017) Reconsolidation and extinction: using epigenetic signatures to challenge conventional wisdom. Neurobiol Learn Mem 142:55
Hendriksen H, Olivier B, Oosting RS (2014) From non-pharmacological treatments for post-traumatic stress disorder to novel therapeutic targets. Eur J Pharmacol 732:139–158
Herry C, Trifilieff P, Micheau J, Luthi A, Mons N (2006) Extinction of auditory fear conditioning requires MAPK/ERK activation in the basolateral amygdala. Eur J Neurosci 24:261–269
Hettema JM, Neale MC, Kendler KS (2001) A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry 158:1568–1578
Hettema JM, Prescott CA, Myers JM, Neale MC, Kendler KS (2005) The structure of genetic and environmental risk factors for anxiety disorders in men and women. Arch Gen Psychiatry 62:182–189
Hill JL, Martinowich K (2016) Activity-dependent signaling: influence on plasticity in circuits controlling fear-related behavior. Curr Opin Neurobiol 36:59–65
Hoban AE, Stilling RM, Moloney GM, Moloney RD, Shanahan F, Dinan TG, Cryan JF, Clarke G (2017) Microbial regulation of microRNA expression in the amygdala and prefrontal cortex. Microbiome 5:102
Hofmann SG, Smits JA (2008) Cognitive-behavioral therapy for adult anxiety disorders: a meta-analysis of randomized placebo-controlled trials. J Clin Psychiatry 69:621–632
Holmes A, Singewald N (2013) Individual differences in recovery from traumatic fear. Trends Neurosci 36:23–31
Hommers LG, Domschke K, Deckert J (2015) Heterogeneity and individuality: microRNAs in mental disorders. J Neural Transm 122:79–97
Hommers LG, Richter J, Yang Y, Raab A, Baumann C, Lang K, Schiele MA, Weber H, Wittmann A, Wolf C, Alpers GW, Arolt V, Domschke K, Fehm L, Fydrich T, Gerlach A, Gloster AT, Hamm AO, Helbig-Lang S, Kircher T, Lang T, Pané-Farré CA, Pauli P, Pfleiderer B, Reif A, Romanos M, Straube B, Ströhle A, Wittchen HU, Frantz S, Ertl G, Lohse MJ, Lueken U, Deckert J (2018) A functional genetic variation of SLC6A2 repressor hsa-miR-579-3p upregulates sympathetic noradrenergic processes of fear and anxiety. Transl Psychiatry 8(1):226
Hu Z, Li Z (2017) miRNAs in synapse development and synaptic plasticity. Curr Opin Neurobiol 45:24–31
Hu Z, Yu D, Gu QH, Yang Y, Tu K, Zhu J, Li Z (2014) miR-191 and miR-135 are required for long-lasting spine remodelling associated with synaptic long-term depression. Nat Commun 5:3263
Hunter RG (2012) Epigenetic effects of stress and corticosteroids in the brain. Front Cell Neurosci 6:18
Huttenrauch M, Salinas G, Wirths O (2016) Effects of long-term environmental enrichment on anxiety, memory, hippocampal plasticity and overall brain gene expression in C57BL6 mice. Front Mol Neurosci 9:62
Issler O, Chen A (2015) Determining the role of microRNAs in psychiatric disorders. Nat Rev Neurosci 16:201–212
Issler O, Chen A (2017) The role of microRNAs in stress-induced psychopathologies A2. In: Fink G (ed) Stress: neuroendocrinology and neurobiology. Academic Press, San Diego
Issler O, Haramati S, Paul ED, Maeno H, Navon I, Zwang R, Gil S, Mayberg HS, Dunlop BW, Menke A, Awatramani R, Binder EB, Deneris ES, Lowry CA, Chen A (2014) MicroRNA 135 is essential for chronic stress resiliency, antidepressant efficacy, and intact serotonergic activity. Neuron 83:344–360
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–254
James AC, James G, Cowdrey FA, Soler A, Choke A (2015) Cognitive behavioural therapy for anxiety disorders in children and adolescents. Cochrane Database Syst Rev 6:Cd004690
Jiang X, Du L, Wang L, Li J, Liu Y, Zheng G, Qu A, Zhang X, Pan H, Yang Y, Wang C (2015) Serum microRNA expression signatures identified from genome-wide microRNA profiling serve as novel noninvasive biomarkers for diagnosis and recurrence of bladder cancer. Int J Cancer 136:854–862
Jin J, Kim SN, Liu X, Zhang H, Zhang C, Seo JS, Kim Y, Sun T (2016) miR-17-92 cluster regulates adult hippocampal neurogenesis, anxiety, and depression. Cell Rep 16:1653–1663
Jovanovic T, Kazama A, Bachevalier J, Davis M (2012) Impaired safety signal learning may be a biomarker of PTSD. Neuropharmacology 62:695–704
Jovasevic V, Corcoran KA, Leaderbrand K, Yamawaki N, Guedea AL, Chen HJ, Shepherd GM, Radulovic J (2015) GABAergic mechanisms regulated by miR-33 encode state-dependent fear. Nat Neurosci 18:1265–1271
Katsuura S, Kuwano Y, Yamagishi N, Kurokawa K, Kajita K, Akaike Y, Nishida K, Masuda K, Tanahashi T, Rokutan K (2012) MicroRNAs miR-144/144∗ and miR-16 in peripheral blood are potential biomarkers for naturalistic stress in healthy Japanese medical students. Neurosci Lett 516:79–84
Kendler KS, Karkowski LM, Prescott CA (1999) Fears and phobias: reliability and heritability. Psychol Med 29:539–553
Kessler RC, Petukhova M, Sampson NA, Zaslavsky AM, Wittchen HU (2012) Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res 21:169–184
Kheirbek MA, Klemenhagen KC, Sahay A, Hen R (2012) Neurogenesis and generalization: a new approach to stratify and treat anxiety disorders. Nat Neurosci 15:1613–1620
Kichukova TM, Popov NT, Ivanov HY, Vachev TI (2015) Circulating microRNAs as a novel class of potential diagnostic biomarkers in neuropsychiatric disorders. Folia Med 57:159–172
Kim B, Kim MK, Kim SW, Kim KM, Kim HS, An HJ, Kim JO, Choi TK, Kim NK, Lee SH (2015) Association of human microRNAs miR-22 and miR-491 polymorphisms with panic disorder with or without agoraphobia in a Korean population. J Affect Disord 188:118–126
Kinrys G, Pollack MH, Simon NM, Worthington JJ, Nardi AE, Versiani M (2003) Valproic acid for the treatment of social anxiety disorder. Int Clin Psychopharmacol 18:169–172
Klauke B, Deckert J, Reif A, Pauli P, Domschke K (2010) Life events in panic disorder-an update on “candidate stressors”. Depress Anxiety 27:716–730
Klengel T, Binder EB (2015) Epigenetics of stress-related psychiatric disorders and gene × environment interactions. Neuron 86:1343–1357
Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M (2012) microRNAs in cancer management. Lancet Oncol 13:e249–e258
Krol J, Busskamp V, Markiewicz I, Stadler MB, Ribi S, Richter J, Duebel J, Bicker S, Fehling HJ, Schubeler D, Oertner TG, Schratt G, Bibel M, Roska B, Filipowicz W (2010a) Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 141:618–631
Krol J, Loedige I, Filipowicz W (2010b) The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11:597–610
Kuriyama K, Honma M, Soshi T, Fujii T, Kim Y (2011) Effect of d-cycloserine and valproic acid on the extinction of reinstated fear-conditioned responses and habituation of fear conditioning in healthy humans: a randomized controlled trial. Psychopharmacology 218:589–597
Landgrave-Gomez J, Mercado-Gomez O, Guevara-Guzman R (2015) Epigenetic mechanisms in neurological and neurodegenerative diseases. Front Cell Neurosci 9:58
Lau NC, Lim LP, Weinstein EG, Bartel DP (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294:858–862
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433:769–773
Lin Q, Wei W, Coelho CM, Li X, Baker-Andresen D, Dudley K, Ratnu VS, Boskovic Z, Kobor MS, Sun YE, Bredy TW (2011) The brain-specific microRNA miR-128b regulates the formation of fear-extinction memory. Nat Neurosci 14:1115–1117
Ling H, Fabbri M, Calin GA (2013) MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov 12:847–865
Londin E, Loher P, Telonis AG, Quann K, Clark P, Jing Y, Hatzimichael E, Kirino Y, Honda S, Lally M, Ramratnam B, Comstock CES, Knudsen KE, Gomella L, Spaeth GL, Hark L, Katz LJ, Witkiewicz A, Rostami A, Jimenez SA, Hollingsworth MA, Yeh JJ, Shaw CA, McKenzie SE, Bray P, Nelson PT, Zupo S, van Roosbroeck K, Keating MJ, Calin GA, Yeo C, Jimbo M, Cozzitorto J, Brody JR, Delgrosso K, Mattick JS, Fortina P, Rigoutsos I (2015) Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci U S A 112:E1106–E1115
Lopez JP, Lim R, Cruceanu C, Crapper L, Fasano C, Labonte B, Maussion G, Yang JP, Yerko V, Vigneault E, El Mestikawy S, Mechawar N, Pavlidis P, Turecki G (2014) miR-1202 is a primate-specific and brain-enriched microRNA involved in major depression and antidepressant treatment. Nat Med 20:764–768
Lopez JP, Pereira F, Richard-Devantoy S, Berlim M, Chachamovich E, Fiori LM, Niola P, Turecki G, Jollant F (2017) Co-variation of peripheral levels of miR-1202 and brain activity and connectivity during antidepressant treatment. Neuropsychopharmacology 42:2043
Lugli G, Torvik VI, Larson J, Smalheiser NR (2008) Expression of microRNAs and their precursors in synaptic fractions of adult mouse forebrain. J Neurochem 106:650–661
Lumayag S, Haldin CE, Corbett NJ, Wahlin KJ, Cowan C, Turturro S, Larsen PE, Kovacs B, Witmer PD, Valle D, Zack DJ, Nicholson DA, Xu S (2013) Inactivation of the microRNA-183/96/182 cluster results in syndromic retinal degeneration. Proc Natl Acad Sci U S A 110:E507–E516
Maccari S, Krugers HJ, Morley-Fletcher S, Szyf M, Brunton PJ (2014) The consequences of early-life adversity: neurobiological, behavioural and epigenetic adaptations. J Neuroendocrinol 26:707–723
Malan-Muller S, Hemmings SM, Seedat S (2013) Big effects of small RNAs: a review of microRNAs in anxiety. Mol Neurobiol 47:726–739
Malan-Muller S, Fairbairn L, Hart S, Daniels WMU, Jalali Sefid Dashti M, Kidd M, Seedat S, Gamieldien J, Hemmings SMJ (2017) The role of microRNAs in the therapeutic action of D-cycloserine in a post-traumatic stress disorder animal model: an exploratory study. Psychiatr Genet 27:139
Mannironi C, Biundo A, Rajendran S, de Vito F, Saba L, Caioli S, Zona C, Ciotti T, Caristi S, Perlas E, del Vecchio G, Bozzoni I, Rinaldi A, Mele A, Presutti C (2017) miR-135a regulates synaptic transmission and anxiety-like behavior in amygdala. Mol Neurobiol 55:3301–3315
Maren S, Holmes A (2016) Stress and fear extinction. Neuropsychopharmacology 41:58–79
Maron E, Nutt D (2015) Biological predictors of pharmacological therapy in anxiety disorders. Dialogues Clin Neurosci 17:305–317
Martin CG, Kim H, Yun S, Livingston W, Fetta J, Mysliwiec V, Baxter T, Gill JM (2017) Circulating miRNA associated with posttraumatic stress disorder in a cohort of military combat veterans. Psychiatry Res 251:261–265
Martinetz S (2016) MicroRNA’s impact on neurotransmitter and neuropeptide systems: small but mighty mediators of anxiety. Pflugers Arch 468:1061–1069
Marzi MJ, Ghini F, Cerruti B, de Pretis S, Bonetti P, Giacomelli C, Gorski MM, Kress T, Pelizzola M, Muller H, Amati B, Nicassio F (2016) Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res 26:554–565
McMillan KA, Asmundson GJG, Sareen J (2017) Comorbid PTSD and social anxiety disorder: associations with quality of life and suicide attempts. J Nerv Ment Dis 205:732–737
Michael T, Blechert J, Vriends N, Margraf J, Wilhelm FH (2007) Fear conditioning in panic disorder: enhanced resistance to extinction. J Abnorm Psychol 116:612–617
Milad MR, Quirk GJ (2012) Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol 63:129–151
Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL (2009) Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry 66:1075–1082
Milad MR, Furtak SC, Greenberg JL, Keshaviah A, Im JJ, Falkenstein MJ, Jenike M, Rauch SL, Wilhelm S (2013) Deficits in conditioned fear extinction in obsessive-compulsive disorder and neurobiological changes in the fear circuit. JAMA Psychiat 70:608–618; quiz 554
Millan MJ (2003) The neurobiology and control of anxious states. Prog Neurobiol 70:83–244
Muinos-Gimeno M, Espinosa-Parrilla Y, Guidi M, Kagerbauer B, Sipila T, Maron E, Pettai K, Kananen L, Navines R, Martin-Santos R, Gratacos M, Metspalu A, Hovatta I, Estivill X (2011) Human microRNAs miR-22, miR-138-2, miR-148a, and miR-488 are associated with panic disorder and regulate several anxiety candidate genes and related pathways. Biol Psychiatry 69:526–533
Murphy CP, Li X, Maurer V, Oberhauser M, Gstir R, Wearick-Silva LE, Viola TW, Schafferer S, Grassi-Oliveira R, Whittle N, Huttenhofer A, Bredy TW, Singewald N (2017) MicroRNA-mediated rescue of fear extinction memory by miR-144-3p in extinction-impaired mice. Biol Psychiatry 81:979–989
Nana-Sinkam SP, Croce CM (2013) Clinical applications for microRNAs in cancer. Clin Pharmacol Ther 93:98–104
Nieto SJ, Patriquin MA, Nielsen DA, Kosten TA (2016) Don’t worry; be informed about the epigenetics of anxiety. Pharmacol Biochem Behav 146:60–72
Norrholm SD, Ressler KJ (2009) Genetics of anxiety and trauma-related disorders. Neuroscience 164:272–287
Nudelman AS, Dirocco DP, Lambert TJ, Garelick MG, Le J, Nathanson NM, Storm DR (2010) Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus 20:492–498
O’Connor RM, Dinan TG, Cryan JF (2012) Little things on which happiness depends: microRNAs as novel therapeutic targets for the treatment of anxiety and depression. Mol Psychiatry 17:359–376
O’Connor RM, Gururajan A, Dinan TG, Kenny PJ, Cryan JF (2016) All roads Lead to the miRNome: miRNAs have a central role in the molecular pathophysiology of psychiatric disorders. Trends Pharmacol Sci 37:1029–1044
Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jonsson B, CDBE2010 Study Group; European Brain Council (2012) The economic cost of brain disorders in Europe. Eur J Neurol 19:155–162
Olsen L, Klausen M, Helboe L, Nielsen FC, Werge T (2009) MicroRNAs show mutually exclusive expression patterns in the brain of adult male rats. PLoS One 4:e7225
Ong CT, Corces VG (2011) Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 12:283–293
Orsini CA, Maren S (2012) Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev 36:1773–1802
Pan-Vazquez A, Rye N, Ameri M, McSparron B, Smallwood G, Bickerdyke J, Rathbone A, Dajas-Bailador F, Toledo-Rodriguez M (2015) Impact of voluntary exercise and housing conditions on hippocampal glucocorticoid receptor, miR-124 and anxiety. Mol Brain 8:40
Patrício P, Mateus-Pinheiro A, Irmler M, Alves ND, Machado-Santos AR, Morais M, Correia JS, Korostynski M, Piechota M, Stoffel R, Beckers J, Bessa JM, Almeida OFX, Sousa N, Pinto L (2015) Differential and converging molecular mechanisms of Antidepressants’ action in the hippocampal dentate Gyrus. Neuropsychopharmacology 40:338–349
Pavlova B, Perlis RH, Mantere O, Sellgren CM, Isometsa E, Mitchell PB, Alda M, Uher R (2017) Prevalence of current anxiety disorders in people with bipolar disorder during euthymia: a meta-analysis. Psychol Med 47:1107–1115
Perusini JN, Fanselow MS (2015) Neurobehavioral perspectives on the distinction between fear and anxiety. Learn Mem 22:417–425
Ragu Varman D, Marimuthu G, Rajan KE (2013) Environmental enrichment upregulates micro-RNA-183 and alters acetylcholinesterase splice variants to reduce anxiety-like behavior in the little Indian field mouse (Mus booduga). J Neurosci Res 91:426–435
Rao P, Benito E, Fischer A (2013) MicroRNAs as biomarkers for CNS disease. Front Mol Neurosci 6:39
Ravindran LN, Stein MB (2010) The pharmacologic treatment of anxiety disorders: a review of progress. J Clin Psychiatry 71:839–854
Ruegger S, Grosshans H (2012) MicroRNA turnover: when, how, and why. Trends Biochem Sci 37:436–446
Saab BJ, Mansuy IM (2014) Neuroepigenetics of memory formation and impairment: the role of microRNAs. Neuropharmacology 80:61–69
Sah A, Schmuckermair C, Sartori SB, Gaburro S, Kandasamy M, Irschick R, Klimaschewski L, Landgraf R, Aigner L, Singewald N (2012) Anxiety- rather than depression-like behavior is associated with adult neurogenesis in a female mouse model of higher trait anxiety- and comorbid depression-like behavior. Transl Psychiatry 2:e171
Saini HK, Griffiths-Jones S, Enright AJ (2007) Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci U S A 104:17719–17724
Schiele MA, Domschke K (2017) Epigenetics at the crossroads between genes, environment and resilience in anxiety disorders. Genes Brain Behav 17:e12423
Schmidt U, Herrmann L, Hagl K, Novak B, Huber C, Holsboer F, Wotjak CT, Buell DR (2013) Therapeutic action of fluoxetine is associated with a reduction in prefrontal cortical miR-1971 expression levels in a mouse model of posttraumatic stress disorder. Front Psych 4:66
Schratt G (2009) microRNAs at the synapse. Nat Rev Neurosci 10:842–849
Scott KA, Hoban AE, Clarke G, Moloney GM, Dinan TG, Cryan JF (2015) Thinking small: towards microRNA-based therapeutics for anxiety disorders. Expert Opin Investig Drugs 24:529–542
Sevignani C, Calin GA, Siracusa LD, Croce CM (2006) Mammalian microRNAs: a small world for fine-tuning gene expression. Mamm Genome 17:189–202
Sharma S, Powers A, Bradley B, Ressler KJ (2016) Gene x environment determinants of stress- and anxiety-related disorders. Annu Rev Psychol 67:239–261
Shimada-Sugimoto M, Otowa T, Hettema JM (2015) Genetics of anxiety disorders: genetic epidemiological and molecular studies in humans. Psychiatry Clin Neurosci 69:388–401
Shin LM, Liberzon I (2010) The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology 35:169–191
Short AK, Yeshurun S, Powell R, Perreau VM, Fox A, Kim JH, Pang TY, Hannan AJ (2017) Exercise alters mouse sperm small noncoding RNAs and induces a transgenerational modification of male offspring conditioned fear and anxiety. Transl Psychiatry 7:e1114
Sim SE, Bakes J, Kaang BK (2014) Neuronal activity-dependent regulation of MicroRNAs. Mol Cells 37:511–517
Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ (2015) Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther 149:150–190
Smoller JW (2016) The genetics of stress-related disorders: PTSD, depression, and anxiety disorders. Neuropsychopharmacology 41:297–319
Snijders C, de Nijs L, Baker DG, Hauger RL, van den Hove D, Kenis G, Nievergelt CM, Boks MP, Vermetten E, Gage FH, Rutten BPF (2018) MicroRNAs in post-traumatic stress disorder. Curr Top Behav Neurosci 38:23–46
Spadaro PA, Bredy TW (2012) Emerging role of non-coding RNA in neural plasticity, cognitive function, and neuropsychiatric disorders. Front Genet 3:132
Stein A, Pearson RM, Goodman SH, Rapa E, Rahman A, McCallum M, Howard LM, Pariante CM (2014) Effects of perinatal mental disorders on the fetus and child. Lancet 384:1800–1819
Sun Y, Lu W, Du K, Wang JH (2019) microRNA and mRNA profiles in the amygdala are relevant to fear memory induced by physical or psychological stress. J Neurophysiol. https://doi.org/10.1152/jn.00215.2019
Taguchi YH (2013) MicroRNA-mediated regulation of target genes in several brain regions is correlated to both microRNA-targeting-specific promoter methylation and differential microRNA expression. BioData Min 6:11
Tardito D, Mallei A, Popoli M (2013) Lost in translation. New unexplored avenues for neuropsychopharmacology: epigenetics and microRNAs. Expert Opin Investig Drugs 22:217–233
Tolin DF (2010) Is cognitive-behavioral therapy more effective than other therapies? A meta-analytic review. Clin Psychol Rev 30:710–720
Tovote P, Fadok JP, Luthi A (2015) Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16:317–331
Vaisvaser S, Modai S, Farberov L, Lin T, Sharon H, Gilam A, Volk N, Admon R, Edry L, Fruchter E, Wald I, Bar-Haim Y, Tarrasch R, Chen A, Shomron N, Hendler T (2016) Neuro-epigenetic indications of acute stress response in humans: the case of MicroRNA-29c. PLoS One 11:e0146236
van der Ree MH, van der Meer AJ, de Bruijne J, Maan R, van Vliet A, Welzel TM, Zeuzem S, Lawitz EJ, Rodriguez-Torres M, Kupcova V, Wiercinska-Drapalo A, Hodges MR, Janssen HLA, Reesink HW (2014) Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antivir Res 111:53–59
Vidigal JA, Ventura A (2015) The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol 25:137–147
Vieland VJ, Goodman DW, Chapman T, Fyer AJ (1996) New segregation analysis of panic disorder. Am J Med Genet 67:147–153
Volk N, Pape JC, Engel M, Zannas AS, Cattane N, Cattaneo A, Binder EB, Chen A (2016) Amygdalar MicroRNA-15a is essential for coping with chronic stress. Cell Rep 17:1882–1891
Wang Y, Li X, Hu H (2011) Transcriptional regulation of co-expressed microRNA target genes. Genomics 98:445–452
Wang F, Pereira A (2016) Neuromodulation, emotional feelings and affective disorders. Mens Sana Monogr 14:5–29
Wang W, Kwon EJ, Tsai LH (2012) MicroRNAs in learning, memory, and neurological diseases. Learn Mem 19:359–368
Wang X, Sundquist K, Hedelius A, Palmer K, Memon AA, Sundquist J (2015) Circulating microRNA-144-5p is associated with depressive disorders. Clin Epigenetics 7:69
Wessa M, Flor H (2007) Failure of extinction of fear responses in posttraumatic stress disorder: evidence from second-order conditioning. Am J Psychiatry 164:1684–1692
Whittle N, Schmuckermair C, Gunduz Cinar O, Hauschild M, Ferraguti F, Holmes A, Singewald N (2013) Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model. Neuropharmacology 64:414–423
WHO (1993) The ICD-10 classification of mental and behavioral disorders: diagnostic criteria for research. World Health Organization, Geneva
Wibrand K, Panja D, Tiron A, Ofte ML, Skaftnesmo KO, Lee CS, Pena JT, Tuschl T, Bramham CR (2010) Differential regulation of mature and precursor microRNA expression by NMDA and metabotropic glutamate receptor activation during LTP in the adult dentate gyrus in vivo. Eur J Neurosci 31:636–645
Wingo AP, Almli LM, Stevens JS, Klengel T, Uddin M, Li Y, Bustamante AC, Lori A, Koen N, Stein DJ, Smith AK, Aiello AE, Koenen KC, Wildman DE, Galea S, Bradley B, Binder EB, Jin P, Gibson G, Ressler KJ (2015) DICER1 and microRNA regulation in post-traumatic stress disorder with comorbid depression. Nat Commun 6:10106
Wingo AP, Almli LM, Stevens JS, Jovanovic T, Wingo TS, Tharp G, Li Y, Lori A, Briscione M, Jin P, Binder EB, Bradley B, Gibson G, Ressler KJ (2017) Genome-wide association study of positive emotion identifies a genetic variant and a role for microRNAs. Mol Psychiatry 22:774–783
Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jonsson B, Olesen J, Allgulander C, Alonso J, Faravelli C, Fratiglioni L, Jennum P, Lieb R, Maercker A, van Os J, Preisig M, Salvador-Carulla L, Simon R, Steinhausen HC (2011) The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol 21:655–679
Yang YL, Lu KT (2005) Facilitation of conditioned fear extinction by d-cycloserine is mediated by mitogen-activated protein kinase and phosphatidylinositol 3-kinase cascades and requires de novo protein synthesis in basolateral nucleus of amygdala. Neuroscience 134:247–260
Yehuda R, Bierer LM (2009) The relevance of epigenetics to PTSD: implications for the DSM-V. J Trauma Stress 22:427–434
You HJ, Park JH, Pareja-Galeano H, Lucia A, Shin JI (2016) Targeting MicroRNAs involved in the BDNF signaling impairment in neurodegenerative diseases. NeuroMolecular Med 18:540
Zhang Y, Wang Y, Wang L, Bai M, Zhang X, Zhu X (2015) Dopamine receptor D2 and associated microRNAs are involved in stress susceptibility and resistance to escitalopram treatment. Int J Neuropsychopharmacol 18:pyv025
Zhou R, Yuan P, Wang Y, Hunsberger JG, Elkahloun A, Wei Y, Damschroder-Williams P, Du J, Chen G, Manji HK (2009) Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology 34:1395–1405
Zhou J, Nagarkatti P, Zhong Y, Ginsberg JP, Singh NP, Zhang J, Nagarkatti M (2014) Dysregulation in microRNA expression is associated with alterations in immune functions in combat veterans with post-traumatic stress disorder. PLoS One 9:e94075
Zhu J, Chen Z, Tian J, Meng Z, Ju M, Wu G, Tian Z (2017) miR-34b attenuates trauma-induced anxiety-like behavior by targeting CRHR1. Int J Mol Med 40:90–100
Acknowledgments
Research of the authors’ work related to this book chapter is supported by the Austrian Science Fund (FWF) (Grant No, SFB F4410 (to NS)), Signal Processing in Neurons (SPIN) (FWF Grant No, DK W-1206 (to NS)), and the European Molecular Biology Organization (ASTF Grant No. 459-2015 (to CPM)). The authors report no financial interests or conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this chapter

Cite this chapter
Murphy, C.P., Singewald, N. (2019). Role of MicroRNAs in Anxiety and Anxiety-Related Disorders. In: Binder, E., Klengel, T. (eds) Behavioral Neurogenomics. Current Topics in Behavioral Neurosciences, vol 42. Springer, Cham. https://doi.org/10.1007/7854_2019_109
Download citation
DOI: https://doi.org/10.1007/7854_2019_109
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-31264-0
Online ISBN: 978-3-030-31265-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)