
Abstract
Maintaining fetal patency and facilitating neonatal closure of the ductus arteriosus are complex processes that involve nuanced developmental programing and a precise balance of perinatal vasodilators and constrictors. Work over the past 40 years has identified “master regulators” of ductus tone, namely oxygen, prostaglandins, and nitric oxide signaling. Yet, prolonged neonatal patency of the ductus remains a significant clinical problem with limited therapeutic options. Understanding the molecular and genetic underpinnings of normal ductus function and PDA pathophysiology will facilitate the development of new therapeutic strategies to regulate ductus tone.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
The ductus arteriosus (DA) is an essential fetal vessel connecting the main pulmonary artery and aorta. In utero, high pulmonary vascular resistance coupled with relatively low systemic resistance promotes right-to-left blood flow through the DA, allowing blood oxygenated by the placenta to bypass the developing lungs [1, 2]. Soon after birth, the DA must close in order to facilitate proper perfusion of the newly inflated lungs, a process normally complete by 12–48 h in full-term neonates.
In some cases, the DA fails to close after birth, a condition termed patent ductus arteriosus (PDA). PDA is a common congenital heart defect, occurring in up to 80% of premature infants weighing <1000 g [3]. Several comorbidities including neurodevelopmental impairment, intraventricular hemorrhage, pulmonary hemorrhage, respiratory distress syndrome, bronchopulmonary dysplasia, necrotizing enterocolitis, and spontaneous intestinal perforation have been attributed to or associated with prolonged patency of the DA [4]. In contrast, keeping the DA open to preserve pulmonary or systemic circulation is essential in certain cardiovascular conditions where blood flow to the lungs or body is disrupted [5]. Unfortunately, there are surprisingly few therapies currently available to promote DA closure or maintain vessel patency. Pharmacology-based therapeutics are non-specific and relatively inefficient. While surgical ligation, catheter-based closure, and stent implantation are effective alternatives, these mechanical approaches come with their own risks and are sometimes limited by the anatomical size constraints of extremely premature newborns [6].
2 Molecular Regulation of the DA
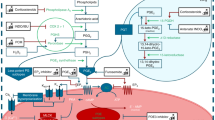
The fetal DA has intrinsic tone and requires factors to maintain its patency in utero (summarized in Table 34.1) [2, 7, 8]. While nitric oxide (NO) and prostaglandin E2 (PGE2) are typically considered the primary mediators of DA dilation, other factors clearly play a role. Potassium (K+) channels are well-characterized DA dilators. Voltage-gated K+ channels, large-conductance voltage-dependent and calcium-activated K+ (BKCa) channels, and ATP-gated K+ (KATP) channels are all specifically enriched in the DA compared to other vascular beds [9]. KATP channels are hetero-octameric complexes of pore-forming inward rectifier K+ channel subunits (Kir6.1 or Kir6.2) and regulatory sulfonylurea receptor subunits (SUR1, SUR2a, or SUR2b) [10]. Different combinations of these subunits are expressed in a tissue-specific manner and exhibit different pharmacological properties, thereby making them attractive targets for DA-specific therapies. This notion is supported by animal studies demonstrating that activating or inhibiting KATP channels directly modulates DA tone [11, 12].
While nitric oxide (NO) and prostaglandin E2 (PGE2) are typically considered the primary mediators of DA dilation, other factors clearly play a role. Potassium (K+) channels are well-characterized DA dilators. Voltage-gated K+ channels, large-conductance voltage-dependent and calcium-activated K+ (BKCa) channels, and ATP-gated K+ (KATP) channels are all specifically enriched in the DA compared to other vascular beds [9]. KATP channels are hetero-octameric complexes of pore-forming inward rectifier K+ channel subunits (Kir6.1 or Kir6.2) and regulatory sulfonylurea receptor subunits (SUR1, SUR2a, or SUR2b) [10]. Different combinations of these subunits are expressed in a tissue specific manner and exhibit different pharmacological properties, thereby making them attractive targets for DA-specific therapies. This notion is supported by animal studies demonstrating that activating or inhibiting KATP channels directly modulates DA tone [11, 12].
At birth, the DA constricts in response to a sharp increase in oxygen (O2) tension coupled with a loss of vasodilators. Several mechanisms have been proposed to explain the DA’s unique ability to sense and respond to O2. These include cytochrome P450-mediated induction of endothelin 1 and production of “constrictor” isoprostanes (8-iso-PGF2α), and mitochondrial-mediated reactive oxygen species inhibition of Kv1.5 and Kv2.1 [8]. O2 can also inhibit KATP channels resulting in membrane depolarization, activation of voltage-dependent calcium (Ca2+) channels, and increased intracellular Ca2+ accumulation [12]. Other factors known to regulate DA contraction are listed in Table 34.1 [2, 6,7,8]. While it is clear that multiple mechanisms are involved, all pathways eventually converge on Ca2+-mediated phosphorylation of myosin light chain, leading to actin/myosin interaction and ultimately DA smooth muscle cell contraction [2, 8].
3 Genetic Regulation of the DA
While occurring most often in the setting of prematurity, twin studies gave the first indication that PDA has a genetic component [6]. Several mouse models of PDA have been created and are summarized in Table 34.2 [6,7,8]. Not surprisingly, many of these models result from disruption of genes involved in smooth muscle function or prostaglandin signaling.
Syndromes featuring PDA (Table 34.2) can also be informative regarding genetic regulation of DA development and function [6, 13]. For instance, clinically significant PDA occurs in 50% of patients with Cantu syndrome, a condition caused by gain-of-function mutations in KATP channel genes, KCNJ8 and ABCC9, which encode the vascular-specific KATP channel subtype Kir6.1/SUR2B. In these cases, PDA is resistant to indomethacin therapy and often requires surgical ligation to achieve closure [14].
While only 10% of PDA cases are associated with chromosomal abnormalities [7], identifying single-nucleotide polymorphisms (SNPs) associated with non-syndromic PDA may be more informative regarding the more common sporadic cases of PDA (Table 34.2). Variants in TFAP2β, TRAF1 (TNF receptor-associated factor 1), and AGTR1 (Angiotensin II type 1 receptor) were associated with an increased risk of PDA while SNPs in PTGIS (prostaglandin I2 synthase), ESR1 (estrogen receptor-α), and IFN-γ (interferon-γ) were found to be protective [6]. Unfortunately, replicating many of these findings in other cohorts has proved difficult to this point [15].
4 Pharmacological Regulation of the DA
Only three drugs are currently available to treat PDA (indomethacin, ibuprofen, and acetaminophen). All are non-selective cyclooxygenase inhibitors that suppress prostaglandin signaling. Alternately, infusion of prostaglandin E1 (PGE1) is the only pharmacologic option used to maintain DA patency in cases of ductus-dependent congenital heart defects. None of these therapies specifically target the DA, leading to off-target effects on other vascular beds. Furthermore, indomethacin and ibuprofen have been associated with spontaneous intestinal perforation and necrotizing enterocolitis while PGE1 has been associated with apnea, fever, and other physiologic disturbances [16, 17].
The fetal DA can also be affected by drugs administered to pregnant women (summarized in Table 34.3). Use of tocolytics, ACE inhibitors, antihistamines, anticonvulsants, amphetamines, and alcohol during pregnancy have all been associated with PDA [18]. In contrast, maternal use of NSAID pain-relievers and antidepressants can cause fetal DA constriction [18, 19].
Furthermore, drugs commonly administered to neonates often have vasoactive properties and may inadvertently affect postnatal DA closure [18]. Several drugs have been shown to specifically dilate the DA or cause resistance to indomethacin therapy (Table 34.3). Of note, DA reopening has been reported after exposure to diazoxide, a KATP channel activator used to treat neonatal hyperinsulinism [20].
5 Clinical Implications and Future Directions
PDA remains a significant problem that is inefficiently managed with currently available therapies. Therefore, a greater emphasis must be placed on identifying other factors that can be targeted to regulate DA tone.
The identification of drug targets outside of the prostaglandin pathway is informed by the molecular findings, mouse models of PDA, and human genetic studies mentioned above. While these provide several pathways that could be targeted therapeutically, one with a high likelihood of success is KATP channels. The vascular form of KATP channels (i.e., Kir6.1/SUR2B) is enriched in smooth muscle cells of the DA [9, 21] making it a promising target for developing DA-preferring dilators and constrictors. Advances in molecular target-based high-throughput screening of large compound libraries have enabled the ability to discover small molecules that target specific KATP channel subtypes [22]. We recently performed a thallium flux-based high-throughput screen of compounds for modulators of Kir6.1/SUR2 and identified several novel-scaffold inhibitors of this channel (Denton, unpublished data). The most potent inhibitor, termed VU278, exhibited preferential activity toward the vascular form of KATP channels (Kir6.1/SUR2) over the pancreatic beta/neuronal cell form (i.e. Kir6.2/SUR1). Using isolated vessel pressure myography assays, we evaluated the specificity and vasoactive potential of these drugs (Fig. 34.1). Fetal mouse DAs were exposed to increasing concentrations of glibenclamide (non-specific sulfonylurea receptor inhibitor), VU063 (pancreatic/neuronal Kir6.2/SUR1 activator) [22], or VU278 (vascular Kir6.1/SUR2 inhibitor). As expected, glibenclamide constricted the DA in a dose-dependent manner. Importantly, VU063 had no effect on the DA while VU278 induced constriction.

Term-gestation mouse DAs were treated with increasing concentrations of glibenclamide, VU063, or VU278. Changes in lumen diameter were measured and plotted as a percent change from the initial lumen diameter reading under baseline (bl) conditions. Glibenclamide and VU279 caused DA constriction while VU063 had no effect
In conclusion, an emerging body of genetic, physiological, and pharmacological evidence paints a bright future for the treatment of PDA. Developing a comprehensive understanding of the molecular mechanisms and signaling pathways that regulate DA patency, coupled with focused efforts to develop specific pharmacological modulators of these new targets, is creating unprecedented opportunities for improving PDA outcomes. Among several potential drug targets emerging from these studies, vascular-specific Kir6.1/SUR2 channels hold significant promise due to their enriched expression in the DA and unique pharmacological properties. Ongoing efforts by our laboratories will explore the therapeutic potential of Kir6.1/SUR2B channels in regulating DA patency.
References
Drayton MR, Skidmore R. Ductus arteriosus blood flow during first 48 hours of life. Arch Dis Child. 1987;62(10):1030–4.
Smith GC. The pharmacology of the ductus arteriosus. Pharmacol Rev. 1998;50(1):35–58.
Koch J, Hensley G, Roy L, Brown S, Ramaciotti C, Rosenfeld CR. Prevalence of spontaneous closure of the ductus arteriosus in neonates at a birth weight of 1000 grams or less. Pediatrics. 2006;117(4):1113–21.
Reese J, Shelton EL, Slaughter JC, McNamara PJ. Prophylactic indomethacin revisited. J Pediatr. 2017;186:11–14.e1.
Yun SW. Congenital heart disease in the newborn requiring early intervention. Korean J Pediatr. 2011;54(5):183–91.
Lewis TR, Shelton EL, Van Driest SL, Kannankeril PJ, Reese J. Genetics of the patent ductus arteriosus (PDA) and pharmacogenetics of PDA treatment. Semin Fetal Neonatal Med. 2018;23:232.
Bokenkamp R, DeRuiter MC, van Munsteren C, Gittenberger-de Groot AC. Insights into the pathogenesis and genetic background of patency of the ductus arteriosus. Neonatology. 2010;98(1):6–17.
Stoller JZ, Demauro SB, Dagle JM, Reese J. Current perspectives on pathobiology of the ductus arteriosus. J Clin Exp Cardiolog. 2012;8(1):S8-001.
Shelton EL, Ector G, Galindo CL, et al. Transcriptional profiling reveals ductus arteriosus-specific genes that regulate vascular tone. Physiol Genomics. 2014;46(13):457–66.
Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–6.
Toyoshima K, Momma K, Ishii T, Nakanishi T. Dilatation of the ductus arteriosus by diazoxide in fetal and neonatal rats. Pediatr Int. 2017;59(12):1246–51.
Waleh N, Reese J, Kajino H, et al. Oxygen-induced tension in the sheep ductus arteriosus: effects of gestation on potassium and calcium channel regulation. Pediatr Res. 2009;65(3):285–90.
Hajj H, Dagle JM. Genetics of patent ductus arteriosus susceptibility and treatment. Semin Perinatol. 2012;36(2):98–104.
Grange DK, Nichols CG, Singh GK. Cantu syndrome and related disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle, WA: University of Washington; 1993.
Kawase K, Sugiura T, Nagaya Y, et al. Single nucleotide polymorphisms in AGTR1, TFAP2B, and TRAF1 are not associated with the incidence of patent ductus arteriosus in Japanese preterm infants. Pediatr Int. 2016;58(6):461–6.
Christmann V, Liem KD, Semmekrot BA, van de Bor M. Changes in cerebral, renal and mesenteric blood flow velocity during continuous and bolus infusion of indomethacin. Acta Paediatr. 2002;91(4):440–6.
Lewis AB, Freed MD, Heymann MA, Roehl SL, Kensey RC. Side effects of therapy with prostaglandin E1 in infants with critical congenital heart disease. Circulation. 1981;64(5):893–8.
Reese J, Veldman A, Shah L, Vucovich M, Cotton RB. Inadvertent relaxation of the ductus arteriosus by pharmacologic agents that are commonly used in the neonatal period. Semin Perinatol. 2010;34(3):222–30.
Hooper CW, Delaney C, Streeter T, et al. Selective serotonin reuptake inhibitor exposure constricts the mouse ductus arteriosus in utero. Am J Physiol Heart Circ Physiol. 2016;311(3):H572–81.
Demirel F, Unal S, Cetin II, Esen I, Arasli A. Pulmonary hypertension and reopening of the ductus arteriosus in an infant treated with diazoxide. J Pediatr Endocrinol Metab. 2011;24(7–8):603–5.
Bokenkamp R, van Brempt R, van Munsteren JC, et al. Dlx1 and Rgs5 in the ductus arteriosus: vessel-specific genes identified by transcriptional profiling of laser-capture microdissected endothelial and smooth muscle cells. PLoS One. 2014;9(1):e86892.
Raphemot R, Swale DR, Dadi PK, et al. Direct activation of beta-cell KATP channels with a novel xanthine derivative. Mol Pharmacol. 2014;85(6):858–65.
Acknowledgments
This work was supported by R21HL132805 and AHA15SDG25280015 awarded to E.L.S. and by NIHR01DK082884 awarded to J.S.D.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Shelton, E.L., Denton, J.S. (2020). Molecular, Genetic, and Pharmacological Modulation of the Ductus Arteriosus: KATP Channels as Novel Drug Targets. In: Nakanishi, T., Baldwin, H., Fineman, J., Yamagishi, H. (eds) Molecular Mechanism of Congenital Heart Disease and Pulmonary Hypertension. Springer, Singapore. https://doi.org/10.1007/978-981-15-1185-1_34
Download citation
DOI: https://doi.org/10.1007/978-981-15-1185-1_34
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-1184-4
Online ISBN: 978-981-15-1185-1
eBook Packages: MedicineMedicine (R0)