
Zusammenfassung
In der Nase wird die Atemluft: - gereinigt (Filterwirkung der Nasenhaare, Partikelniederschlag auf dem Nasenschleim, rachenwärts gerichtete Zilienbewegung des Epithels) - erwärmt (fast bis auf Körpertemperatur) - befeuchtet (nahezu vollständige Sättigung mit Wasserdampf) Gestörte Nasenatmung kann daher Reizung und Austrocknung der Bronchialschleimhaut bewirken. Dem ist bei künstlicher Beatmung (Trachealkatheter, Trachealkanüle) Rechnung zu tragen.
You have full access to this open access chapter, Download chapter PDF
2.1 Die normale Atmung
2.1.1 Funktionen der oberen Luftwege
2.1.1.1 Bedeutung der Nasenatmung
In der Nase wird die Atemluft:
-
gereinigt (Filterwirkung der Nasenhaare, Partikelniederschlag auf dem Nasenschleim, rachenwärts gerichtete Zilienbewegung des Epithels)
-
erwärmt (fast bis auf Körpertemperatur)
-
befeuchtet (nahezu vollständige Sättigung mit Wasserdampf)
Gestörte Nasenatmung kann daher Reizung und Austrocknung der Bronchialschleimhaut bewirken. Dem ist bei künstlicher Beatmung (Trachealkatheter, Trachealkanüle) Rechnung zu tragen.
2.1.1.2 Selbstreinigung des Tracheobronchialsystem s
Erfolgt durch den schleimhaltigen Schutzfilm der Trachea und der Bronchien in Verbindung mit der rachenwärts gerichteten Zilienbewegung des Epithels (Transportgeschwindigkeit 10–15 cm/h).
Bestandteile des Zigarettenrauchs hemmen die Ziliaraktivität!
Husten und Niesen sind zusätzliche reflektorisch ausgelöste Reinigungsmechanismen.
Abschwächung oder Ausschaltung des Hustenreflexes kann lebensgefährlich sein, z. B. Aspiration bei Narkosen oder im Koma oder mangelhaftes Abhusten bei fortgeschrittener obstruktiver Lungenerkrankung.
2.1.2 Funktionen der Lunge
2.1.2.1 Ventilation
Die Ventilation umfasst die Einatmung mit Belüftung des Alveolarraumes und die Ausatmung. Die Einatmung erfolgt aktiv durch Kontraktion der Atemmuskeln, die Ausatmung überwiegend passiv durch elastische Rückstellkräfte der Lungen und des Brustkorbes. Bei erschwerter Ausatmung treten die exspiratorischen Muskeln (Bauchpresse) verstärkt in Aktion. Bei erschwerter oder verstärkter Einatmung dienen Skalenus, Sternokleidomastoideus, und beim Aufstützen der Arme die Mm. pectoralis minor et major als Atemhilfsmuskeln. Das Volumen eines normalen Atemzuges beträgt etwa 500 ml. Davon entfallen auf die Luftwege (Totraum) 150 ml, auf das Kompartiment der Alveolen 350 ml. Bei 15 Atemzügen pro Minute ergibt sich eine alveoläre Ventilation von etwa 5 l/min. Die Sauerstoffaufnahme beträgt dabei 300 ml, die CO2-Abgabe 250 ml/min. Unter Belastung kann die Sauerstoffaufnahme auf 4–5 l/min steigen und das Atemminutenvolumen auf das 15fache.
2.1.2.2 Gasaustausch
Durch Diffusion werden zwischen Alveolarluft und Lungenkapillarblut die Atemgase ausgetauscht. Die treibende Kraft dabei ist die Differenz der Partialdrücke zwischen dem Alveolarraum und dem Kapillarblut (◘ Abb. 2.1).


Gasaustausch in der Lunge
Der Gasaustausch erfolgt durch die respiratorische Membran, deren Diffusionskapazität ist abhängig von:
-
Membrandicke (Schichten: Flüssigkeitsfilm in den Alveolen, Alveolarepithel, Interstitium, Basalmembran der Kapillaren, Kapillarendothel)
-
Membranoberfläche (wird bei körperlicher Belastung durch Alveolendehnung vergrößert)
-
Diffusionskoeffizienten (abhängig vom Molekulargewicht des Gases und seiner Löslichkeit in der Membran; für CO2 zwanzigmal größer als für O2).
Unter Ruhebedingungen ist die Sauerstoffsättigung des Blutes nach weniger als einem Drittel der Passagezeit durch die Lungenkapillaren beendet. Beschleunigter Blutumlauf bei Belastung beeinträchtigt daher die O2-Sättigung nicht. Nur beim Zusammentreffen von Belastung und herabgesetztem atmosphärischem Sauerstoffdruck (Höhenluft) sinkt die arterielle O2-Sättigung ab, weil der verkleinerte O2-Druckgradient die Diffusionsgeschwindigkeit vermindert. Der CO2-Austausch zwischen Alveolarluft und Kapillarblut erfolgt wegen der hohen Diffusionsrate dieses Gases sehr schnell und wird durch die respiratorische Membran niemals behindert.
2.1.2.3 Perfusion
Der pulmonale Blutfluss hat die gleiche Größe wie das Herzminutenvolumen und beträgt etwa 5 l/min. Da auch die alveoläre Ventilation 5 l/min beträgt, ergibt sich global ein Ventilation-Perfusion-Verhältnis von annähernd 1. Ein kleiner Teil des Herzminutenvolumens (1–2 %) passiert die Lunge unter Umgehung der Alveolen. Diese venöse Zumischung zum arterialisierten Lungenvenenblut lässt den pO2 des arteriellen Blutes (paO2) auf 95 mm Hg absinken.
2.1.2.4 Verteilung
Ventilation und Perfusion erreichen in verschiedenen Lungenabschnitten ein unterschiedliches Ausmaß (◘ Abb. 2.2). Die Perfusion pro Volumeneinheit nimmt in aufrechter Körperhaltung von der Lungenbasis zur Spitze kontinuierlich ab. Das gleiche gilt für die Ventilation, doch ist der Gradient nicht so steil. Folglich nimmt das Ventilation-Perfusion-Verhältnis von der Basis zur Spitze zu. Den Alveolen der Spitzenregion wird wegen des geringen Kapillardurchflusses relativ wenig O2 entzogen und wenig CO2 zugeführt. Das Gegenteil ist in den stark durchbluteten Alveolen der Lungenbasis der Fall. Trotz der ungleichen Verteilung von Ventilation und Perfusion ist die globale Sauerstoffsättigung des Lungenblutes normalerweise gewährleistet. Doch treten Störungen der arteriellen O2-Sättigung auf, wenn sich bei Lungenerkrankungen das Ventilation-Perfusion-Verhältnis erheblich verändert. Im Extremfall wird eine belüftete Alveole überhaupt nicht perfundiert (Gefäßverschluss) bzw. eine perfundierte Alveole nicht belüftet (Atelektase). Kompensatorische Reflexmechanismen sorgen dafür, dass die Durchblutung schlecht belüfteter Alveolen gedrosselt wird bzw. dass die Belüftung schlecht durchbluteter Alveolen abnimmt.


Regionale Differenzen der Ventilation (VA), Blutperfusion (Q) und des Ventilation-Perfusion-Quotienten (VA/Q) in der normalen Lunge bei aufrechter Haltung (nach J.B. West)
2.1.3 Regulation der Atmung
2.1.3.1 Atemzentrum
2.1.3.1.1 Lokalisation
Neuronengruppen bilateral in der Medulla oblongata und im Pons:
-
Dorsale respiratorische Gruppe: Erzeugt durch spontane inspiratorische Impulse den Grundrhythmus der Atmung, der ohne stimulierte Exspiration auskommt. Empfängt und beantwortet afferente Signale von den peripheren Chemorezeptoren und verschiedenen Rezeptoren der Lunge. Wird bei Belastung durch übergeordnete Zentren stimuliert. Die willkürliche Atmung läuft über somatische Nerven von der Hirnrinde zur Atemmuskulatur.
-
Pneumotaktisches Zentrum: Lokalisiert im oberen Pons. Kontrolliert die Dauer des inspiratorischen Signals und damit Frequenz und Tiefe der Atmung. Das normale inspiratorische Signal erstreckt sich über 2 Sekunden, damit sich das Zwerchfell wie eine Hebebühne senken und einen ruhigen Einstrom der Atemluft bewirken kann.
-
Ventrale respiratorische Gruppe: Dicht vor und seitlich der dorsalen respiratorischen Gruppe gelegen und bei normaler Ruheatmung inaktiv. Erhält bei gesteigerter Atmung Impulse der dorsalen respiratorischen Gruppe und stimuliert dann die Exspiration und verstärkt mit einigen Neuronen auch die Inspiration.
2.1.3.1.2 Funktion
Die alveolare Ventilation wird so geregelt, dass der Gasaustausch den metabolischen Bedürfnissen des Organismus entspricht und paO2 und paCO2 auch unter Belastung annähernd normal bleiben. Die Stimulation des Atemzentrums bewirkt Zunahme der Atemfrequenz und der Atemtiefe.
2.1.3.2 Regulation des Atemzentrums durch Dehnungsrezeptoren der Lunge
Die Muskelschicht in der Wand von Bronchien und Bronchiolen ist mit Dehnungsrezeptoren ausgestattet, die bei Überdehnung die Inspiration verkürzen. Die Atemtiefe nimmt ab, die Atemfrequenz zu. Dieser als Hering-Breuer-Reflex bezeichnete Vorgang ist beim Menschen nicht an der normalen Kontrolle der Ventilation beteiligt, sondern schützt lediglich vor extremer Lungenblähung.
2.1.3.3 Humorale Regulation des Atemzentrums
2.1.3.3.1 Direkte Kontrolle
Sie erfolgt durch CO2 (Messgröße paCO2) und Wasserstoffionen (Messgröße pH). Das Atemzentrum wird über eine benachbarte chemosensitive Region stimuliert, die auf Wasserstoffionen anspricht. CO2 reagiert im Liquor und in der interstitiellen Flüssigkeit des Gehirns mit H2O zu H2CO3, das in H+ und HCO3 - dissoziiert. Das geschieht am wirksamsten im Liquor, der kaum Säurepuffer enthält. Auf die H-Ionen des Blutes reagiert das Atemzentrum viel unempfindlicher, weil die Blut-Hirn- und die Blut-Liquor-Schranke, die CO2 schnell passieren kann, für H+ weitgehend undurchlässig sind.
2.1.3.3.2 Indirekte Kontrolle
Exzitation des Atemzentrums über die Chemorezeptoren im Karotissinus und Aortenbogen, die hauptsächlich durch O2-Mangel im arteriellen Blut (paO2 <60 mmHg) stimuliert werden. Viel schwächer sprechen die Chemorezeptoren auch auf CO2 und H+ an. Bei Blutdruckabfall steigern sie via Sympathikus Herzfrequenz und Vasomotorentonus.
2.1.3.3.3 Quantitative Aspekte
Wichtigste Regelgröße ist der paCO2. Ein pCO2-Anstieg von 40 auf 63 mmHg in den Alveolen bewirkt eine 10-fache Ventilationssteigerung. Zum einen wirkt der paCO2-Anstieg im Blut direkt, zum anderen indirekt über die respiratorische Azidose atmungsstimulierend.
Ein pH-Abfall durch Vermehrung der fixen Säuren bewirkt geringere Ventilationssteigerung, da paCO2 infolge kompensatorischer Hyperventilation sinkt. Im pH-Bereich 7,3–7,5 ist der Effekt auf die Ventilation 10-mal geringer als bei paCO2-Drücken zwischen 35 und 60 mmHg.
Die O2-Regulation der Atmung über die Chemorezeptoren ist normalerweise unbedeutend, da ein geringer Abfall des alveolären pO2 die Sauerstoffsättigung des Blutes nicht wesentlich beeinträchtigt und nicht zur Stimulation der Chemorezeptoren führt. Außerdem wird eine durch O2-Mangel induzierte Hyperventilation durch den mit der Ventilationssteigerung verbundenen Abfall des paCO2 und Anstieg des pH gebremst. Bei behinderter Ventilation (obstruktives Emphysem) jedoch entfällt die Bremsung der O2-Mangel-Stimulation, da sich das Atemzentrum an erhöhte CO2-Drucke adaptiert. In diesen Situationen ist der O2-Mangel maßgebendes Stimulans für die Respirationssteigerung, was man daran erkennt, dass Sauerstoffbeatmung die Atmung abschwächt und die CO2-Konzentration im Blut steigen lässt. Es drohen respiratorische Azidose und Koma.
Auch bei niedrigen atmosphärischen O2-Drücken im Höhenklima wird die Atmung von den Chemorezeptoren stimuliert. Nach 2- bis 3-tägiger Akklimatisation verliert das Atemzen-trum 80 % seiner Sensitivität gegen Änderungen des paCO2. Folglich wird eine anstrengungsbedingte Hyperventilation (z. B. beim Bergsteigen) durch Absinken des paCO2 nicht mehr gebremst.
2.1.3.3.4 Pufferfunktion der Atmung
Die Atmung wirkt pH-Verschiebungen im arteriellen Blut durch Änderungen des paCO2 entgegen. Eine metabolische Azidose kann durch Hyperventilation (Abatmen von CO2) teilweise kompensiert werden, eine metabolische Alkalose durch Hypoventilation (Retention von CO2). Auf der anderen Seite können respiratorische Azidose und Alkalose metabolisch teilweise kompensiert werden. Eine ausführliche Darstellung erfolgt im Abschnitt über den Säure-Basen-Haushalt (▶ Kap. 3).
2.1.3.4 Zentrale Atemstimulation bei körperlicher Arbeit
Bei gesunden Athleten können O2-Verbrauch und CO2-Produktion bis zum 20fachen ansteigen. Bei körperlicher Anstrengung passt sich die alveolare Ventilation sofort der Stoffwechselsteigerung an. Offenbar gehen vom ZNS mit den Impulsen zur Skelettmuskulatur kollaterale Impulse zum Atemzentrum. Dieses Ansprechen der Atmung kann durch Training gesteigert werden. Die Feinregulation der Atmung unter Belastung erfolgt durch die Blutgase.
2.2 Lungenfunktionsprüfungen
Lungenfunktionsprüfungen
-
Ventilation
-
Lungenvolumina
-
Messmethoden
-
Spirometrie
-
Fremdgasverdünnungsmethode
-
Dynamische Ventilationsgrößen
-
-
Atemmechanik
-
Statische Compliance
-
Bronchialer Strömungswiderstand (Resistance
-
-
Verteilung
-
Nachweis ventilatorischer Verteilungsstörungen
-
-
Diffusion
-
Messung der Diffusionskapazität
-
Single-Breath-Methode
-
Steady-State-Methode
-
-
Atemgase und pH-Wert
-
Sauerstoffmessungen
-
Messung von paCO2- und pH-Wert
-
Blutgasanalyse bei dosierter Belastung
-
Spiroergometrie
-
2.2.1 Ventilation
2.2.1.1 Lungenvolumin a
Definition der Volumina und Kapazitäten (◘ Tab. 2.1):
-
Atemzugvolumen (VT): Volumen, das bei jedem normalen Atemzug ein- bzw. ausgeatmet wird (ca. 500 ml).
-
Inspiratorisches Reservevolumen (IRV): Extravolumen, das über das normale Atemzugvolumen hinaus eingeatmet werden kann (ca. 3000 ml).
-
Exspiratorisches Reservevolumen (ERV): Extravolumen, das am Ende einer normalen Ausatmung noch maximal ausgeatmet werden kann (ca. 1100 ml).
-
Residualvolumen (RV): Volumen, das nach extremer Ausatmung in der Lunge verbleibt (ca. 1200 ml).
-
Funktionelle Residualkapazität (FRC): Residualvolumen plus exspiratorisches Reservevolumen (ca. 2300 ml). Die FRC ist das Lungenvolumen am Ende einer normalen Ausatmung.
-
Vitalkapazität (VC): Die Summe von Atemzugvolumen, exspiratorischem und inspiratorischem Reservevolumen. Abhängig von Alter, Größe und Geschlecht (ca. 4600 ml).
-
Inspiratorische Vitalkapazität (IVC): Volumen, das nach langsamer maximaler Exspiration maximal eingeatmet werden kann.
-
Exspiratorische Vitalkapazität (EVC): Volumen, das nach maximaler Inspiration maximal ausgeatmet werden kann (ca. 5 % kleiner als die inspiratorische Vitalkapazität).
-
Forcierte exspiratorische Vitalkapazität (FVC): Volumen, das nach maximaler Einatmung mit größtmöglicher Geschwindigkeit ausgeatmet werden kann. Wichtig zur Erfassung der Obstruktion der Atemwege.
-
Totale Lungenkapazität (TC): Das maximale Lungenvolumen (ca. 5800 ml), erreicht bei tiefster Einatmung (Atemzugvolumen + inspiratorisches + exspiratorisches Reservevolumen + Residualvolumen).
2.2.1.2 Messmethoden
2.2.1.2.1 Spirometrie
Das klassische Spirometer besteht aus einer Trommel, die umgekehrt in ein Wasserbad eintaucht und durch ein Gegengewicht gehalten wird. In der Trommel ist Luft, und ein Rohr verbindet die Gaskammer mit dem Mund. Das atemsynchrone Heben und Senken der Trommel wird durch einen Zeiger auf einer rotierenden Trommel gegen die Zeit aufgezeichnet. In ◘ Abb. 2.3 ist das Diagramm der respiratorischen Atemexkursionen schematisch dargestellt.


Schematisches Spirogramm (FEV1 = Einsekundenausatmungskapazität)
2.2.1.2.2 Fremdgasverdünnungsmethode
Dient zur Bestimmung der funktionellen Residualkapazität (FRS), die durch einfache Spirometrie nicht zu erfassen ist. Ein Spirometer von bekanntem Volumen wird mit Luft gefüllt, der in bekannter Konzentration Helium beigemischt ist. Am Ende einer normalen Exspiration wird der Patient an dieses Spirometer angeschlossen und atmet zunächst forciert weiter aus. Dabei wird das exspiratorische Reservevolumen registriert. Es schließt sich eine minutenlange Ruheatmung an, bei der das Helium im Volumen der FRC verdünnt wird. Der Konzentrationsausgleich ist erreicht, wenn die Heliumkonzentration im Spirometer nicht weiter absinkt. Aus dem Grad der Heliumverdünnung im Spirometer und Spirometervolumen lässt sich die FRC berechnen. Die Methode wird heute nur noch selten angewandt.
2.2.1.3 Dynamische Ventilationsgrößen
Die Messungen erfolgen mit Spirographen oder integrierenden Pneumotachographen. Letztere messen während der Atemphasen die Strömungsgeschwindigkeit der Luft (l/s) und stellen sie graphisch dar. Der Pneumotachograph besteht aus einer offenen Röhre, in der sich zahlreiche parallele Lamellen befinden, um einen laminaren Luftstrom zu gewährleisten. Am Anfang und Ende der Röhre befinden sich Druckrezeptoren, die den Druckgradienten im Rohr während der Strömung messen. Bei gegebenem Rohrdurchmesser ergeben sich aus den Druckgradienten die Flussgeschwindigkeiten. Mit Hilfe eines Integrators wird aus der Strömung das Volumen bestimmt.
Messwerte sind:
-
Atemfrequenz: Ruhewert 12–18/min, je nach Atemtiefe.
-
Atemminutenvolumen (AMV): Der Ruhewert beträgt ca. 6–8 l/min. Sollwertberechnung: Grundumsatz × 4,73.
-
Atemgrenzwert (AGW): Maximale willkürliche Ventilation (MVV). Praktische Durchführung: Hyperventilation für 10–15 s, Umrechnung auf l/min. Normalwerte: 80–150 l/min.
-
Forciertes exspiratorisches Einsekundenvolumen (FEV 1 ): Das nach maximaler Einatmung bei forcierter Ausatmung in der ersten Sekunde exspirierte Volumen (◘ Abb. 2.3). Absolutwerte abhängig von Körpergröße und Alter. Das FEV1 wird in Prozent der exspiratorischen forcierten Vitalkapazität angegeben: FEV1/FVC %. Es ist dem exspiratorischen Strömungswiderstand umgekehrt proportional.
-
Fluss-Volumen-Kurve: Mit dem Pneumotachographen werden die Flussgeschwindigkeiten (l/s) unter Messung des Flussvolumens während der Atemphasen, insbesondere der forcierten maximalen Exspiration gemessen. Auf der Ordinate ist die Flussgeschwindigkeit (l/s), auf der Abszisse das Atemvolumen aufgetragen. Es lassen sich dann neben der maximalen die Flussgeschwindigkeiten bei 25, 50 und 75 % der forcierten exspiratorischen Vitalkapazität (FVC) ablesen (◘ Abb. 2.4).
Abb. 2.4 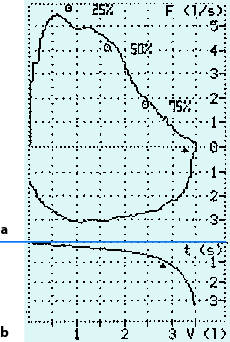
Pneumotachographie (Normalbefund). a Atemstromstärke bei forcierter Exspiration, b forciertes exspiratorisches Volumen


2.2.2 Atemmechanik
2.2.2.1 Statische Compliance
Die statische Compliance ist definiert als Volumenänderung der Lunge pro Einheit der Druckänderung (ml/cmH2O bzw. l/kPa) und somit ein Maß für die Dehnbarkeit der Lunge. Für die Lungendehnung ist der intrapleurale Druck maßgebend. Das ist der negative Druck (Sog), welcher nötig ist, um einen Kollaps der Lunge zu verhindern. Bei angehaltenem Atem besteht zwischen der Retraktionskraft der Lunge und dem an der Lungenoberfläche angreifenden negativen intrapleuralen Druck ein Gleichgewicht. Wird der negative intrapleurale Druck bei der Inspiration vergrößert, ist der Volumenzuwachs der Lunge pro Zentimeter Wassersäule (oder kPa) (negativer) Druckzunahme der Dehnbarkeit der Lunge proportional. Methodisch wird die Änderung des intrapleuralen Druckes während der Inspiration statt im Pleuraspalt mittels Ballonsonde im unteren Ösophagus gemessen (Ösophagusdruckmethode).
Die Compliance ist der Quotient aus eingeatmetem Volumen und der Druckdifferenz zwischen Beginn und Ende der Einatmung. Normalwerte: Männer 0,21 l/cmH2O (kPa) und Frauen: 0,17 l/cmH2O (kPa).
Die Compliance ist den elastischen Rückstellkräften der Lunge umgekehrt proportional. Diese setzen sich zu einem Drittel aus der elastischen Kraft des Lungengewebes, zu zwei Dritteln aus der elastischen Kraft zusammen, die von der Oberflächenspannung des Flüssigkeitsfilms in den Alveolen ausgeht. Durch einen Lipidfilm (Surfactant) wird die Oberflächenspannung herabgesetzt und ein Kollaps der Alveolen verhindert.
2.2.2.1.1 Klinische Bedeutung
Eine pathologische Abnahme der Compliance führt zu einer Zunahme der Atemarbeit, da mehr (negativer) Druck aufgewandt werden muss, um die steife Lunge mit demselben Volumen zu füllen. Sie findet sich häufig bei restriktiven Lungenerkrankungen, kann aber auch bei akuten Veränderungen wie Ödem, Lungenentzündung und ARDS auftreten.
Bei Lungenfibrosen und Frühgeborenen mit Surfactantmangel ist die Compliance herabgesetzt, beim Lungenemphysem erhöht.
2.2.2.2 Bronchialer Strömungswiderstand (Resistance )
Die Luftbewegung in den Atemwegen wird durch einen Druckgradienten zwischen Außenluft und Alveolarraum erzeugt. Am Ende der normalen Exspiration ist der Alveolardruck gleich dem atmosphärischen Druck der Außenluft, da keine Luft strömt. Mit Beginn der Inspiration wird der Alveolardruck durch die Thoraxdehnung seitens der Atemmuskeln negativ. Es entsteht ein Druckgradient von außen nach innen, der am Ende der Inspiration wieder verschwindet. Mit Beginn der Exspiration steigt der Alveolardruck infolge Kompression der Alveolen über den atmosphärischen Druck der Außenluft; es entsteht ein Druckgradient von innen nach außen. Die Atemstromstärke (V) in l/s ist dem Druckgradienten (ΔP) zwischen Außenluft und Alveolarraum proportional und dem endobronchialen Strömungswiderstand (R) umgekehrt proportional:
 und
und 
Die Resistance ist definiert als die Druckdifferenz zwischen Alveolarluft und Außenluft, die erforderlich ist, um 1 Liter Luft/Sekunde im Bronchialsystem strömen zu lassen. Sie kann mit verschiedenen Methoden bestimmt werden.
2.2.2.2.1 Unterbrechermethode
Während der Spontanatmung am Pneumotachographen werden die Atemwege am Mund 2–5×/s für weniger als 0,1 s verschlossen. In der Verschlussphase gleicht der Munddruck dem Alveolardruck. Die Atemstromstärke wird während der Öffnungsphase gemessen. Etwas aufwendiger ist die Oszillometrie, bei der am Mundstück in den Luftstrom eine schnelle kleine Wechselströmung eingeleitet wird. Aus dem Verhältnis Wechseldruck/Wechselströmung wird der Widerstand der Atemwege bestimmt.
2.2.2.2.2 Ganzkörperplethysmographie
Der Patient wird in den Ganzkörperplethysmographen, eine luftdichte Kammer, eingeschlossen (◘ Abb. 2.5). Während er darin atmet, werden die Änderungen des Kammerdrucks und mittels eines Pneumotachographen die Atemstromstärke gemessen und beide auf einem XY-Schreiber registriert. Wenn sich der Brustkorb bei der Inspiration erweitert, entsteht in den Alveolen ein Unterdruck (Sog) und in der Kammer ein korrespondierender Überdruck, die beide bis zum Ende der Inspiration verschwinden, weil dann die verdrängte Kammerluft in die Lunge geströmt ist. Bei der Exspiration wird umgekehrt ein Überdruck in den Alveolen und ein Unterdruck in der Kammer erzeugt. Je größer der Strömungswiderstand, desto stärker weichen die Drucke in der Kammer bzw. in den Alveolen von der Ausgangslage ab. Das während eines Atemzyklus vom Schreiber aufgezeichnete Druckströmungsdiagramm zeigt eine Schleifenform (Resistanceschleife) mit der Inspiration im rechten oberen und der Exspiration im linken unteren Quadranten (◘ Abb. 2.6a). Je steiler die Achse der Schleife steht, desto kleiner die Resistance. Für die Bestimmung des absoluten Wertes der Resistance muss bekannt sein, welche Alveolardruckänderung einer definierten Änderung des Kammerdrucks entspricht. Um diese Relation zu ermitteln, wird der Kammerdruck gegen den Munddruck registriert, nachdem man das Atemrohr verschlossen hat, weil dann Munddruck und Alveolardruck übereinstimmen (◘ Abb. 2.6b). Der Patient hechelt bei diesen Messungen (Atemexkursionen ohne Luftströmung). Bei Erwachsenen sind Resistancewerte über 3,0 cm H2O (= 0,3 kPa/l/s) als pathologisch anzusehen (◘ Abb. 2.7). Aus den Druckänderungen bei der Hechelatmung lässt sich auch das thorakale Gasvolumen errechnen, weil aus den Druckänderungen in der Kammer die Volumenänderungen abzuleiten sind.


Ganzkörperplethysmograph. a Kammer. b Schema der Arbeitsweise (Pn = Pneumotachograph, V/t = Druckmanometer für Stromstärke, ∆PK = Druckmanometer für die Messung des Kammerdrucks


Druckströmungsdiagramm. a während eines Atemzyklus, b Mundverschlussdruckkurve (V = Atemfluss, pk = Kammerdruck, pmo = Munddruck, VT = Atemzugvolumen


Ganzkörperplethysmographische Resistenzkurven. Links Normalbefund, daneben Kurven von Patienten mit Atemwegsobstruktion
2.2.3 Verteilung
2.2.3.1 Nachweis ventilatorischer Verteilungsstörungen
Bei ventilatorischen Verteilungsstörungen wird die Lunge nicht gleichförmig beatmet. Es besteht ein Nebeneinander von hyper- und hypoventilierten Alveolen. Eine der Nachweismethoden ist die exspiratorische CO2-Druckkurve (◘ Abb. 2.8).


Exspiratorische CO2-Druckkurve. Nach tiefer Einatmung wird die Ausatmungsluft durch einen Infrarotabsorptionsschreiber geleitet, der den CO2-Druck fortlaufend registriert. In der Druckkurve sind 3 Abschnitte zu unterscheiden: absoluter Totraum (A), Mischluftanteil (B) und Alveolarluftanteil (C). Wenn der paCO2 während der Exspiration kein Plateau erreicht, sondern kontinuierlich ansteigt, muss die endinspiratorische CO2-Konzentration in verschiedenen Lungenbezirken unterschiedlich sein (Zeichen eines gestörten Ventilation-Perfusion-Verhältnisses). Außerdem muss sich die Lunge asynchron entleeren (Obstruktion). Denn bei gleichmäßiger Ventilation (synchroner Exspiration) würden sich die Alveolarluftanteile mit niedriger und hoher CO2-Konzentration während der Ausatmung wie im Normalfall in konstantem Verhältnis mischen
2.2.4 Diffusion
Die Diffusionskapazität , auch als Transferfaktor bezeichnet, ist definiert als das Gasvolumen, das bei einem Druckgradienten von 1 mmHg in jeder Minute von den Alveolen ins Blut bzw. in die Erythrozyten übertritt. Es muss dabei die alveolokapilläre Membran (Gewebeschranke) und das Blut mit seinen flüssigen und festen Bestandteilen (Blutschranke) passieren. Der Normalwert für die O2-Diffusionskapazität bei ruhiger Atmung beträgt 21 ml O2/min/Torr. Bei Diffusionsstörungen infolge Verdickung der respiratorischen Membran (Lungenfibrose, Stauungslunge) und bei Verkleinerung der Gesamtdiffusionsfläche (Emphysem, Lungenresektionen) ist dieser Wert herabgesetzt. Methodisch ist die Bestimmung der O2-Diffusionskapazität für die Praxis zu aufwendig und störanfällig. Deshalb bestimmt man die Diffusionskapazität für CO, dessen Partialdruck im Lungenkapillarblut wegen der starken Affinität des CO zum Hämoglobin gleich Null gesetzt werden kann und dessen alveolärer Partialdruck folglich dem Druckgradienten an der respiratorischen Membran gleichzusetzen ist.
2.2.4.1 Single-Breath-Methode
Die Einatemzugtechnik stellt gegenwärtig das Standardverfahren dar. Sie basiert auf der CO-Diffusion während einer Apnoezeit von 10 Sekunden. Dazu wird ein CO-haltiges Mischgas nach maximaler Exspiration tief inspiriert (bis zur Totalkapazität). Nach der Apnoe enthält die Exspirationsluft weniger CO als die Inspirationsluft. Die Differenz ist ein Maß für den CO-Transfer. Die Berechnung erfordert die Kenntnis des Alveolarvolumens zur Apnoezeit und die inspiratorische Verdünnung des Kohlenmonoxids. Dazu wird dem Mischgas in geringer Konzentration Helium zugesetzt dessen Transfer zu vernachlässigen ist.
2.2.4.2 Steady-State-Methode
Bei dieser Gleichgewichtsmethode atmet der Patient für einige Minuten ein Luftgemisch mit 0,1 % Kohlenmonoxid ein. Die Bestimmung der CO-Konzentration in der Atemluft erfolgt mit einem Gaschromatographen, z. B. CO-Uras.
2.2.5 Atemgase und pH-Wert im Blut
2.2.5.1 Diagnostische Bedeutung
Blutgasanalyse und pH-Bestimmung des Blutes liefern die wichtigsten Daten zu Beurteilung der globalen Lungenfunktion. Ihre Ergebnisse hängen allerdings auch vom Zustand des Herz-Kreislauf-Systems (z. B. Shunt-Vitien) und vom Säure-Basen-Haushalt (Azidose, Alkalose) ab.
2.2.5.1.1 Messmethode
Moderne automatisierte, computergesteuerte Mikro-pH/Blutgas-Analysatoren messen nach Eingabe einer Blutprobe aus dem arterialisierten Ohrläppchen Blutgase und pH-Wert mit Spezialelektroden. Sie liefern folgende Parameter: paO2 (arterieller O2-Partialdruck), paCO2 (arterieller CO2-Partialdruck), pH, Plasmabikarbonat, Basenüberschuss, O2-Sättigung und O2-Gehalt des Blutes.
2.2.5.2 Sauerstoffmessung en
2.2.5.2.1 paO2
Normalwerte für Männer und Frauen 75–100 mmHg. Im Liegen sind die Mittel- und Grenzwerte etwa 5 mmHg niedriger als im Stehen.
2.2.5.2.2 SaO2 (O2-Sättigung )
Prozentuale Sättigung des Hämoglobins im arteriellen Blut mit Sauerstoff. Normalwerte für beide Geschlechter 95–98 %. Im steilen Teil der Dissoziationskurve führt ein relativ kleiner Anstieg von paO2 zu einer starken Zunahme der O2-Sättigung, ein Effekt, der bei hypoxischen Patienten den Nutzen der O2-Inhalation erklärt.
2.2.5.2.3 O2-Kapazität
Die bei Vollsättigung mit Sauerstoff an das Hämoglobin gebundene O2-Menge in Vol.% (ml O2/100 ml Blut). Wird aus dem Hämoglobingehalt berechnet: 1 g Hb bindet 1,34 ml O2. Bei einem Hb-Gehalt von 16 g/100 ml Blut ergibt sich eine O2-Kapazität von 21,4 Vol%. Der physikalisch gelöste Sauerstoff erreicht bei einem pO2 der Atemluft von 95 mmHg nur 0,29 Vol%.
2.2.5.2.4 Hypoxie
Erniedrigter paO2-Partialdruck im Blut. Ursachen: O2-Mangel in der Atemluft (Höhenklima), Ventilationsstörungen, Störungen des Ventilation-Perfusion-Verhältnisses, Diffusionsstörungen, Rechts-Links-Shunt.
2.2.5.2.5 Hypoxämie
Verminderte O2-Menge pro Volumeneinheit Blut, verglichen mit dem O2-Gehalt des Blutes einer Normalperson unter denselben atmosphärischen Bedingungen. Ursachen: Wie bei Hypoxie, außerdem bei Anämien und CO-Intoxikation. Kann bei herabgesetztem pO2 durch sekundäre (kompensatorische) Polyzythämie teilweise ausgeglichen werden.
2.2.5.3 Messung von paCO2 und pH-Wert mit Berechnung der Basen
-
paCO2: Normalwerte für Männer 35–45 mmHg, für Frauen 32–42 mmHg.
-
Hypokapnie: Erniedrigter paCO2, stets durch Hyperventilation bedingt.
-
Hyperkapnie : Erhöhter paCO2-Partialdruck, stets durch alveoläre Hypoventilation bedingt.
-
pH-Normalwerte: Männer: 7,34–7,44; Frauen 7,35–7,45. Die Variationsbreite ist beim Individuum kleiner als im Kollektiv.
Bei pH-Werten <7,34 liegt eine Azidose, bei pH-Werten >7,44 eine Alkalose vor.
-
Plasmabikarbonat : Aktuelle Konzentration von Bikarbonat im Plasma.
-
Normalwerte: Für Männer 22–26, für Frauen 20–24 mmol/l.
-
Erhöhte Werte:
-
bei primärer metabolischer Alkalose (pH >7,45, paCO2 durch kompensatorische Hypoventilation erhöht)
-
kompensatorisch bei respiratorischer Azidose (pH <7,34, paCO2 durch primäre Hypoventilation erhöht)
-
-
Erniedrigte Werte:
-
bei metabolischer Azidose (pH <7,34, paCO2 durch kompensatorische Hyperventilation herabgesetzt)
-
kompensatorisch bei respiratorischer Alkalose (pH >7,45, paCO2 durch primäre Hyperventilation herabgesetzt)
-
-
Standardbikarbonat: Konzentration von Bikarbonat im Blutplasma, äquilibriert mit einem Gas von 40 mmHg pCO2 und 100 mmHg pO2 bei 37 °C. Normalwerte: Für Männer und Frauen 22–26 mmol/l.
-
Weitere Ausführungen über den Säure-Basen-Haushalt im ▶ Kap. 3.
2.2.5.4 Ergometrie : Blutgasanalyse bei dosierter Belastung
2.2.5.4.1 Methode
Gemessen werden paO2, paCO2 und pH-Wert bei stufenförmiger Belastung am Fahrradergometer oder Laufband.
2.2.5.4.2 Normalbefunde
Bei aerober dynamischer Arbeit bis etwa 50 % der maximalen Sauerstoffaufnahmekapazität (VO2max) steigt paO2 leicht an, weil zusätzliche Alveolen ventiliert werden (Vergrößerung des Ventilation-Perfusion-Quotienten). Dabei bleiben paCO2 und pH-Wert konstant, weil das vermehrt gebildete CO2 abgeatmet wird.
Bei weiter steigender Belastung wird zur aeroben auch anaerobe Energie benötigt, bei deren Gewinnung Laktat entsteht. Während paO2 noch etwas ansteigt, senkt das Laktat den Bikarbonatspiegel. Um den pH-Abfall zu bremsen, wird durch Ventilationssteigerung vermehrt CO2 abgeatmet, was zum Absinken von paCO2 führt. Wenn der pH-Wert auf etwa 7,24 abgesunken ist, erfolgt durch die Azidose eine Hemmung der Muskelkontraktion, und die absolute Belastungsgrenze ist erreicht.
2.2.5.5 Pathologische Befunde
2.2.5.5.1 Diffusionsstörung
In Ruhe ist der paO2 herabgesetzt und der paCO2 durch kompensatorische Hyperventilation ebenfalls erniedrigt. Unter Belastung sinkt der paO2 weiter ab, während der paCO2 durch vermehrten Anfall von CO2 auf normale Werte ansteigt.
2.2.5.5.2 Verteilungsstörung
In Ruhe erniedrigter paO2, der paCO2 normal oder herabgesetzt. Unter Belastung Anstieg von paO2 durch Verbesserung des Ventilation-Perfusion-Quotienten; Absinken von paCO2 durch Laktatazidose.
2.2.5.5.3 Hypoventilation
In Ruhe herabgesetzter paO2 und erhöhter paCO2 (da CO2 ungenügend abgeatmet wird). Unter Belastung kann sich paO2 normalisieren, paCO2 bleibt erhöht.
2.2.5.6 Spiroergometrie
2.2.5.6.1 Methode
Messung der O2-Aufnahme (VO2) und CO2-Abgabe (VCO2) unter pH-Kontrolle bei steigender Belastung am Fahrradergometer oder Laufband. Dazu werden mit einem Pneumotachographen das Atemminutenvolumen und mit Sensoren die Gaskonzentrationen in der Ein- und Ausatemluft bestimmt.
2.2.5.6.2 Parameter
-
Anaerobe Schwelle: VO2 und VCO2 steigen in der aeroben Phase im Verhältnis 1:1 an. Der respiratorische Quotient (RQ) ist 1. Die anaerobe Schwelle ist erreicht, sobald die VCO2 wegen Laktatbildung stärker anzusteigen beginnt als VO2. Der RQ (VCO2/VO2) wird >1.
-
Dauerleistungsgrenze (Arbeitskapazität): Sie ist erreicht, wenn die Laktatazidose nicht mehr ventilatorisch kompensiert wird und der pH-Wert zu sinken beginnt. Zu diesem Zeitpunkt geht die VCO2 leicht zurück, während das Atemminutenvolumen exponentiell ansteigt.
-
Maximale Saueraufnahmekapazität: Definiert als die O2-Aufnahme bei einem pH-Wert von 7,25 bzw. vorzeitiger Erschöpfung. Bei Lungenkrankheiten und Herzinsuffizienz ist die maximale O2-Aufnahme deutlich herabgesetzt. Untrainierte junge Männer erreichen 3.600 ml/min, trainierte Athleten 4.000 ml/min, Marathonläufer 5.100 ml/min. Die maximale Sauerstoffaufnahmekapazität steigt mit der Zahl der Mitochondrien in der Skelettmuskulatur, denn dort finden die Oxidationsprozesse statt.
2.3 Störungen der Atmung
Störungen der Atmung
-
Allgemeine Störungen
-
Dyspnoe
-
Hyperventilation
-
Hypoventilation
-
periodische Atmung
-
Zyanose
-
-
Obstruktion
-
Restriktion
-
Verteilungsstörungen
-
Diffusionsstörungen
-
Respiratorische Insuffizienz
-
ARDS (adult/acute respiratory distress syndrome)
-
Schlafapnoe
2.3.1 Allgemeine Symptome
2.3.1.1 Dyspnoe
2.3.1.1.1 Definition
Unangenehme Empfindung einer inadäquaten Anstrengung bei der Atmung. Vom Patienten beschrieben als Lufthunger, Kurzatmigkeit, Atemlosigkeit, Atembeklemmung, schweres Atmen, Atemnot und im Extremfall als Erstickungsgefühl.
2.3.1.1.2 Pathophysiologie
Die Empfindung Dyspnoe kann durch mindestens 3 Mechanismen hervorgerufen werden:
-
Gesteigerte Atemarbeit: Durch erhöhten Kraftaufwand für eine adäquate Atmung bei obstruktiven und restriktiven Ventilationsstörungen, insbesondere Lungenstauung (herabgesetzte Compliance infolge Blutüberfüllung bzw. Ödembildung in der Lunge). Vermittelt durch afferente, über den N. vagus geleitete Impulse von Gelenk-, Sehnen- und Muskelrezeptoren der Brustwand. Die Atemnot während und nach erschöpfender körperlicher Anstrengung wird auch empfunden, aber als ein normales Phänomen ohne Beunruhigung.
-
Abnorme Konstellation der Blutgase: Stimulation des Atemzentrums durch arterielle Hypoxie (Höhenklima, Diffusionsstörungen), Hyperkapnie (Hypoventilation bei Atemmuskelparese und Lungenaffektionen) und Azidose (diabetisches Koma).
-
Psychogene Faktoren: Emotional bedingte Stimulation des Atemzentrums mit einer die metabolischen Bedürfnisse übersteigenden Ventilation, die zur Hyperventilationstetanie führen kann. Meistens sind es Patienten mit Angstneurosen.
2.3.1.1.3 Klinik
Das Symptom Dyspnoe lässt sich nach folgenden Kriterien weiter differenzieren:
-
Qualität: Asthmatiker geben Giemen und exspiratorische, aber auch inspiratorische Dyspnoe an. Erstickungsgefühl bei Lungenödem, massiven Pleuraergüssen und Atemmuskellähmung.
-
Zeitliche Entwicklung: Zu unterscheiden sind folgende Kategorien:
-
plötzlich und dramatisch (in Minuten): Pneumothorax, große Lungenembolie, akutes Lungenödem, Aspiration
-
akut (in Stunden): Pneumonie, akute Lungeninfiltrationen (allergische Alveolitis), Asthma, Linksinsuffizienz
-
subakut (über Tage): Pleuraerguss, Bronchialkarzinom, subakute Lungeninfiltrationen (z. B. Sarkoidose)
-
chronisch (über Monate oder Jahre): chronische Obstruktion der Atemwege, diffuse Lungenfibrose, nicht pulmonale Ursachen (Anämie, Hyperthyreose)
-
intermittierend: Bronchialasthma, Linksinsuffizienz, rezidivierende Lungenembolien
-
2.3.1.1.4 Differenzialdiagnosen
Unterscheidung zwischen pulmonaler und kardialer Dyspnoe nach dem körperlichen Untersuchungsbefund, dem Röntgenbild des Thorax und der Spirometrie. Kardiale Ursachen sind durch EKG (akuter Infarkt), Echokardiographie (Linksinsuffizienz) und einen Anstieg des BNP bzw. NT-ProBNP im Serum zu erfassen.
2.3.1.1.5 Therapie
Kausal nach zugrunde liegender Erkrankung. Symptomatisch: Sauerstoffzufuhr durch die Nasensonde, bei Lungenstauung i.v. Schleifendiuretika.
2.3.1.2 Hyperventilation
2.3.1.2.1 Definition
Im Verhältnis zum Energiestoffwechsel gesteigerte Gesamtventilation.
2.3.1.2.2 Klassifikation
-
Primäre Hyperventilation: Gesteigerte Ventilation bei psychogener Stimulation des Atemzentrums (Angst, Schmerz, Erregung). Führt zur Hypokapnie und respiratorischen Alkalose.
-
Kompensatorische Hyperventilation:
-
Bei Hypoxie durch Diffusionsstörungen. Das Atemzentrum wird über die peripheren Chemorezeptoren stimuliert.
-
Bei diabetischer Azidose zur Bremsung des pH-Abfalls.
-
Die gesteigerte Ventilation bei körperlicher Arbeit ist keine Hyperventilation.
2.3.1.3 Hypoventilation
2.3.1.3.1 Definition
Im Verhältnis zum Energiestoffwechsel herabgesetzte Gesamtventilation.
2.3.1.3.2 Klassifikation
-
Primäre Hypoventilation: Bei respiratorischer Insuffizienz mit erhöhtem paCO2 und respiratorischer Azidose.
-
Kompensatorische Hypoventilation: Zur CO2-Retention bei metabolischer Alkalose (pH-Anstieg hemmt das Atemzentrum).
2.3.1.4 Periodische Atmung
2.3.1.4.1 Cheyne-Stokes-Atmung
Alternierende Perioden von Hyperpnoe (mit Zu- und Abnahme der Atemtiefe bei normaler Atemfrequenz) und Apnoe (◘ Abb. 2.9a).


Periodische Atmung. a Cheyne-Stokes-Atmung, b Biot-Atmung
2.3.1.4.1.1 Pathogenese
Verzögerter Bluttransport von der Lunge zum Atemzentrum im Hirnstamm. Infolgedessen wird noch weiter geatmet, obwohl der pCO2 im Lungenvenenblut bereits stärker abgesunken ist. Erreicht das arterielle Blut mit niedrigem paCO2 das Atemzentrum, kommt es zur Apnoe. Ursachen der Strömungsverzögerung: Herzinsuffizienz mit Vergrößerung des linken Ventrikels, Vitien, Kreislaufschock. In den apnoischen Phasen sinkt der paO2, während der paCO2 steigt. Die anschließende Ventilationssteigerung kommt durch den paCO2-Effekt auf das Atemzentrum und den paO2-Effekt auf die Chemorezeptoren zustande. Durch O2-Beatmung kann letztere Komponente ausgeschaltet werden. Auch direkte Läsionen des Atemzentrums (Hirndruck etc.) können zur Cheyne-Stokes-Atmung führen.
2.3.1.4.2 Biot-Atmung
Auf mehrere Atemzüge von gleichem Abstand und gleicher Amplitude folgt eine apnoische Pause. Die Dauer der Zyklen ist variabel (◘ Abb. 2.9b).
2.3.1.4.2.1 Pathogenese
Direkte Schädigung des Atemzentrums durch Traumen, Kompression, Ischämie etc.
2.3.1.5 Zyanose
2.3.1.5.1 Definition
Blaufärbung der Haut und der sichtbaren Schleimhäute durch erhöhten Gehalt des Kapillarblutes an reduziertem Hämoglobin.
2.3.1.5.2 Pathogenese
Reduziertes Hämoglobin (Hb red.) hat eine intensiv blaue Eigenfarbe, die viel kräftiger ist als die rote Farbe des Oxyhämoglobins. Maßgebend ist nicht die relative, sondern die absolute Konzentration des reduzierten Hämoglobins:
-
leichte Zyanose: Hb red. 3–4 g/100 ml Blut
-
starke Zyanose: Hb red. 5 g/100 ml Blut und mehr
Bei Polyzythämie genügt ein relativ geringer Abfall der O2-Sättigung des Blutes, um eine Zyanose entstehen zu lassen, bei Anämien muss die O2-Untersättigung stärker sein als im normalen Blut. Selten kommt eine Zyanose durch Methämoglobinbildung vor. Nach dem Entstehungsmechanismus unterscheidet man 2 Formen der Zyanose:
-
Zentrale Zyanose: Resultiert aus einer Herabsetzung der arteriellen O2-Sättigung, z. B. bei respiratorischer Insuffizienz. Sie betrifft Haut (Lippen, Wangen, Nagelbett) und Schleimhäute (Zunge). Bei kardialem und pulmonalem Rechts-Links-Shunt nimmt sie unter körperlicher Belastung zu.
-
Periphere Zyanose: Entsteht durch gesteigerte O2-Ausschöpfung des Kapillarblutes der Haut bei verminderter arterieller Durchblutung und langsamem Blutfluss. Ursachen: Vasokonstriktion durch Kälteeinwirkung, Arterienspasmen (Morbus Raynaud), arterielle Embolien (Hautblässe mit zyanotischer Komponente), Herz- und Kreislaufinsuffizienz mit kompensatorischer kutaner Vasokonstriktion und bei venöser Stauung.
2.3.2 Obstruktion
2.3.2.1 Definition
Obstruktion bedeutet Verengung der Atemwege.
Unterschieden werden 2 Formen:
-
Endobronchiale Obstruktion: Lumeneinengung durch Schleimhautschwellung, Sekret und Spasmen der Bronchialmuskulatur (durch β-Rezeptorenblocker begünstigt). Kommt bei Asthma und chronischer obstruktiver Bronchitis vor.
-
Exobronchiale Obstruktion:
-
Lumeneinengung infolge Herabsetzung der die Bronchien offenhaltenden elastischen Zugkräfte des Lungengewebes. Vorkommen: beim Emphysem.
-
Abnorme Kollapsbereitschaft der Bronchien durch chronische Bronchitis.
-
Kompression von außen durch Malignome.
-
2.3.2.2 Pathophysiologie
Die Obstruktion beeinträchtigt Atemmechanik, Ventilation und Lungenkreislauf. Sie kann dadurch zu schweren Funktionsstörungen der Lunge und des rechten Herzens führen.
2.3.2.3 Veränderungen der Ventilation und Atemmechanik
Die obstruktionsbedingte Erhöhung des bronchialen Strömungswiderstandes steigert die Atemarbeit. Da die Exspiration auch bei schwerer Obstruktion in Ruhe passiv (durch die Retraktionskraft der Lunge) erfolgt, muss die Lunge zur Vergrößerung der Retraktionskraft inspiratorisch stärker vorgedehnt werden. Bei schwerer Obstruktion ist die Exspiration so verlangsamt, dass sie von der Inspiration vor Erreichen des normalen Ruhevolumens unterbrochen wird. Es kommt zur Hyperinflation der Lunge mit Erhöhung der funktionellen Reservekapazität, die bei geschwächter Lungenstruktur eine Emphysembildung begünstigt. Mit sekundärem Elastizitätsverlust der Lunge nimmt das Residualvolumen und folglich auch die Totalkapazität zu. Während die Inspiration bei der Obstruktion durch die an den Bronchien radiär angreifende Retraktionskraft des Lungengewebes erleichtert wird, kommt es bei forcierter Exspiration, also bei körperlicher Belastung, durch Kompression der kleinen Bronchien zu einer erheblichen Steigerung des Strömungswiderstandes. Durch den kompensatorischen Einsatz der exspiratorischen Atemmuskeln werden die endexspiratorischen Intrapleuraldrucke bei Arbeitsbelastung positiv, während sie beim Gesunden unter dem Atmosphärendruck bleiben. Die Atemfrequenz nimmt auf Kosten der Atemtiefe zu. Es resultiert eine Belastungsdyspnoe in schweren Fällen eine Ruhedyspnoe.
2.3.2.4 Veränderungen der Verteilung
Die Atemwegsobstruktion führt zur regionalen Minderbelüftung der Alveolen mit Absinken des Ventilation-Perfusion-Quotienten. Zunächst fällt dadurch der paO2 ab, in schweren Fällen mit globaler alveolärer Hypoventilation kommt es außerdem zum Anstieg des paCO2 und zur respiratorischen Azidose.
2.3.2.5 Steigerung des pulmonalen Strömungswiderstandes
Die mit der Obstruktion verbundene Verteilungsstörung bewirkt in den minderbelüfteten Lungenbezirken eine reflektorische Vasokonstriktion, die eine pulmonale Hypertonie mit Rechtsinsuffizienz zur Folge haben kann.
2.3.2.6 Diagnostische Kriterien
-
Auskultation: Überwiegend exspiratorisches Giemen und Brummen über allen Lungenabschnitten.
-
Spirometrie: Herabsetzung des forcierten exspiratorischen Einsekundenvolumens (FEV1) und des Quotienten FEV1/FVC(%). In schweren Fällen ist auch die FVC herabgesetzt. Abnahme der maximalen exspiratorischen Atemstromstärke. Abnorme Fluss-Volumen-Kurve.
-
Resistance-Messung: In Ruhe erhöhte Resistance-Werte bei der Ganzkörperplethysmographie und mit der Unterbrecher- oder Oszillationsmethode.
-
Volumenmessungen: Erhöhung des Residualvolumens (RV) und der funktionellen Residualkapazität (FRC). Das führt zur Vergrößerung der Totalkapazität.
-
Blutgasbestimmung: Nachweis der partiellen oder globalen respiratorischen Insuffizienz als Obstruktionsfolge (▶ unten).
2.3.3 Restriktion
2.3.3.1 Definition
Restriktion bedeutet verminderte Dehnbarkeit der Lunge oder der Brustwand.
2.3.3.2 Vorkommen
Bei schweren Thoraxdeformitäten, massiver Fettsucht, Pneumothorax, Pleuraergüssen, Lungenstauung, Lungenödem, Atelektasen, massiven Pneumonien und diffusen Fibrosierungsprozessen (Tuberkulose, Sarkoidose, Pneumokoniosen, zystischer Fibrose, Strahlenfibrose, idiopathischer Lungenfibrose).
2.3.3.3 Pathophysiologie
Steigerung der Atemarbeit durch Erschwerung der Inspiration. Verminderung der Lungenbelüftung durch Begrenzung der Inspiration. Verteilungsstörungen infolge Inhomogenität der Belüftung, in schweren Fällen respiratorische Insuffizienz.
2.3.3.4 Diagnostische Kriterien
-
Volumenmessungen: Verkleinerung der inspiratorischen Vitalkapazität, des Residualvolumens und der Totalkapazität. Der absolute Wert des forcierten exspiratorischen Einsekundenvolumens (FEV1) ist zwar verkleinert, der Quotient FEV1/FVC% jedoch normal, da keine Obstruktion vorliegt.
-
Compliance-Bestimmung: Herabgesetzte Werte, bei pulmonalen Ursachen mit erhöhtem, bei pleuralen und thorakalen Ursachen der Restriktion mit normalem transpulmonalem Druck.
-
Blutgasbestimmung: Nachweis der sekundären respiratorischen Insuffizienz (▶ unten).
2.3.4 Verteilungsstörungen
2.3.4.1 Definition
Störungen des Ventilation-Perfusion-Verhältnisses, d. h. des Gleichgewichtes zwischen Ventilation und Perfusion der Alveolen mit Beeinträchtigung des Gasaustausches.
2.3.4.2 Ventilatorische Verteilungsstörungen
Primäre Störung der Ventilation mit erniedrigtem Ventilation-Perfusion-Quotienten (obstruktive und restriktive Ventilationsstörungen aller Art) oder Wegfall der Ventilation (Atelektasen, Pneumothorax, Kompression durch Ergüsse).
2.3.4.3 Zirkulatorische Verteilungsstörungen
Primäre Störung der Lungenperfusion aus verschiedenen Ursachen (z. B. durch Lungenembolien) und bei intrapulmonalen Shunts.
2.3.4.4 Pathophysiologie
Das schon in der normalen Lunge vorhandene, funktionell aber belanglose Ungleichgewicht zwischen Ventilation und Perfusion wird durch die meisten generalisierten Lungenerkrankungen und Okklusionen der Lungengefäße erheblich verstärkt. Sowohl O2-Aufnahme als auch CO2-Ausscheidung werden erschwert. Letztere kann durch kompensatorische Hyperventilation normal belüfteter und perfundierter Lungenbezirke ausgeglichen werden. Da die O2-Sättigung des Kapillarblutes in normal belüfteten Alveolen schon fast maximal ist, kann sie durch Hyperventilation nur begrenzt gesteigert werden. Es resultiert eine Hypoxämie, die in schweren Fällen nur durch Erhöhung des Sauerstoffpartialdrucks der Atemluft, also durch Sauerstoffzufuhr per Nasensonde gebessert werden kann.
2.3.4.5 Diagnostische Kriterien
Eine Verteilungsstörung liegt vor, wenn der paO2 bei normalem oder herabgesetztem paCO2 erniedrigt ist. In leichteren Fällen normalisiert sich der paO2 während körperlicher Belastung durch Zunahme der Ventilation und Perfusion unter Ausgleich von Inhomogenitäten. Auf Verteilungsstörungen weist auch eine abnorme exspiratorische CO2-Druckkurve hin (◘ Abb. 2.8). Direkt zu erfassen sind Verteilungsstörungen (besonders nach Lungenembolie) durch kombinierte Anwendung der Ventilationsszintigraphie mit 133Xenon und der Perfusionsszintigraphie mit 99mTc-Mikrosphären.
2.3.5 Diffusionsstörungen
2.3.5.1 Definition
Störungen der Atmung infolge Verkleinerung der O2-Diffusionskapazität der Lunge.
2.3.5.2 Ätiologie
Nach den Determinanten der O2-Diffusionskapazität ergibt sich die folgende Einteilung:
-
Alveokapillärer Block: Verlängerung der Diffusionsstrecke zwischen Alveolarwand und Erythrozyten. Kommt bei interstitiellen Lungenfibrosen, interstitieller Pneumonie und Lungenstauung vor. Für eine Hypoxie weniger bedeutsam als die oft gleichzeitig bestehende Verteilungsstörung.
-
Verkleinerung der Gasaustauschfläche: Bei Lungenemphysem, Alveolarzellkarzinom und als Pneumektomiefolge.
-
Anämie: Reduziert die O2-Aufnahmekapazität des Blutes. Bei CO-Intoxikation ist sie blockiert.
2.3.5.3 Pathophysiologie
Diffusionsstörungen sind meistens mit Verteilungsstörungen kombiniert. Sie betreffen nur die O2-Diffusion, da CO2 sehr leicht diffundiert. Die Hypoxämie löst eine kompensatorische Hyperventilation aus.
2.3.5.4 Diagnostische Kriterien
Bestimmung des CO-Transferfaktors, der herabgesetzt ist (▶ oben). Bei Emphysem ist wegen des Elastizitätsverlustes gleichzeitig die Compliance vergrößert.
2.3.6 Respiratorische Insuffizienz
2.3.6.1 Definition
Kritische Hypoxämie mit einem paO2 <60 mmHg bei normalem, erniedrigtem oder erhöhtem paCO2. Erst unterhalb eines paO2 von 60 mmHg sinkt die arterielle O2-Sättigung unter 90 %. In jedem Fall geht von der Hypoxämie die wesentliche Bedrohung des Organismus aus.
2.3.6.2 Klassifikation
2.3.6.3 Typ I: Akute hypoxämische respiratorische Insuffizienz
Primär ist die arterielle Sauerstoffaufnahme in der Lunge herabgesetzt (paO2 <60 mmHg), während CO2 normal oder gesteigert abgeatmet wird. Erst wenn die kompensatorische Ventilationssteigerung zur Erschöpfung der Atemmuskulatur führt, resultiert eine Hypoventilation mit Anstieg des paCO2. Kommt bei Überflutung der Alveolen mit Flüssigkeit aus folgenden Ursachen vor:
-
Lungenödem durch Stauung im kleinen Kreislauf bei Herzinsuffizienz
-
Lungenödem ohne Drucksteigerung im linken Vorhof bei ARDS (Acute Respiratory Distress Syndrome), dem eine primäre Lungenschädigung durch Sepsis, Pneumonie, Magensaftaspiration, multiple Bluttransfusionen und Pankreatitis zugrunde liegen kann.
2.3.6.4 Typ II: Ventilatorischer Typ
Das Primäre ist eine alveoläre Hypoventilation , die zum Absinken des paO2 und zum Anstieg des paCO2 führt, also zu einer globalen respiratorischen Insuffizienz. Mit dem Anstieg des paCO2 auf >50 mmHg ist ein gleichhoher Anstieg des pCO2 in der Alveolarluft verbunden, der gemäß der Alveolargasformel den pO2 in der Alveolarluft herabsetzt. Die Sauerstoffaufnahme wird dadurch zusätzlich beeinträchtigt. Mechanismen der ventilatorischen Insuffizienz sind:
-
Störungen der zentralen Atemstimulation durch überdosierte Sedativa, Hirnstammschädigung, Schlafapnoe-Erkrankungen, Hypothyreose
-
Schwäche oder Lähmung der Atemmuskulatur bei Myasthenia gravis, amyotrophischer Lateralsklerose, Guillain-Barré-Syndrom, Phrenikusläsion, Myopathie, Hypokaliämie, Botulismus
-
Erschöpfung der Atemmuskulatur durch gesteigerte Atemarbeit bei herabgesetzter Compliance der Lunge (alveolares Ödem, Atelektasen) oder der Brustwand (extreme Kyphoskoliose, ◘ Abb. 2.10), Pneumothorax, Pleuraergüsse, Zwerchfellhochstand und bei erhöhtem bronchialen Strömungswiderstand (COPD)
Abb. 2.10 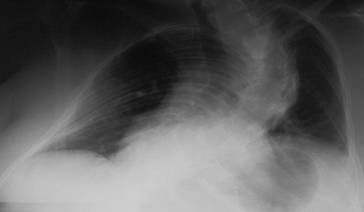
72-jähriger Patient mit schwerer Kyphoskoliose. Im Verlauf Ausbildung einer schweren restriktiven Ventilationsstörung mit respiratorischer Insuffizienz. Stabilisierung unter nichtinvasiver Maskenbeatmung


2.3.6.5 Typ III
Respiratorische Insuffizienz durch ausgedehnte, meist perioperativ auftretende Atelektasen mit Hypoxämie und gesteigerter Atemarbeit. Entstehungsmechanismus: Abnahme des Residualvolumens unter der Allgemeinnarkose. Dadurch Kollaps der abhängigen Lungenpartien.
2.3.6.6 Typ IV
Respiratorische Insuffizienz im Schock aufgrund einer Hypoperfusion der Atemmuskulatur, die unter respiratorischem Distress bis zu 40 % des Herzminutenvolumens benötigt und in ein O2-Defizit gerät.
2.3.6.7 Klinik
2.3.6.8 Symptome der Hypoxie
Benommenheit, Desorientiertheit, Verwirrtheit, Unruhe, Aggressivität, Störungen der Intelligenz sowie klinisch-physikalische Befunde wie Tachypnoe oder Hypopnoe, Tachykardie, leichte Hypertonie, Zyanose, periphere Vasokonstriktion, seltener Bradykardie und Hypotonie.
2.3.6.9 Symptome der Hyperkapnie
Konzentrationsschwäche, Müdigkeit, Muskelschwäche, respiratorische Azidose.
2.3.6.10 Kausale Therapie
Behandlung der Grundkrankheit soweit möglich.
2.3.6.11 Symptomatische Therapie
2.3.6.12 Sauerstoffinhalation
Zur Beseitigung der Hypoxämie von vitaler Bedeutung. Darf nicht aus Furcht vor CO2-Narkose und respiratorischer Azidose unterlassen werden. Applikation über Gesichtsmaske oder Nasensonde. Mit der nasalen Sonde ist bei einem O2-Fluss von 1–6 l/min eine Sauerstoff-Fraktion in der inspiratorischen Atemluft (FiO2) von 0,24–0,35 zu erreichen. Erst oberhalb einer FiO2 von 0,6 beginnen Intoxikationserscheinungen (Tracheobronchitis, Atelektasen, respiratorische Insuffizienz). Der paO2 soll auf >60 mmHg gebracht werden.
2.3.6.13 Nichtinvasive Atemhilfe
Teilweise oder komplette maschinelle Ventilation ohne endotracheale Intubation über Nasen- oder Gesichtsmaske. Indiziert bei Hypoventilation mit Hyperkapnie infolge Schwäche der Atemmuskulatur. Die Inspiration wird mit Druckluft herbeigeführt, die Exspiration erfolgt spontan. Getriggert werden die Atemzüge durch die Atemanstrengung des Patienten oder den Ventilator entsprechend der eingestellten Frequenz. Das Atemzugvolumen kann volumen- oder druckgesteuert appliziert werden. Es handelt sich um assistierte Beatmung, bei der die Spontanatmung erhalten bleibt. Volumengesteuert ist die IPPV (Intermittend Positive Pressure Ventilation ), druckgesteuert die BIPAP (Biphasic Positive Airway Pressure Ventilation ).
2.3.6.14 Invasive Atemhilfe
Maschinell assistierte oder kontrollierte Ventilation über Endotrachealtubus.
-
Zugangswege: Orotracheal, nasotracheal, Tracheostoma (nach Dilatationstracheotomie)
-
Indikationen: Atemstillstand, schwere respiratorische Insuffizienz
-
Ventilatortypen:
-
Druckgesteuerter Ventilator: Beatmung bis zu einem vorgegebenen Atemwegsdruck, danach passive Exspiration. Das Atemzugvolumen ist von der thorakopulmonalen Compliance und dem Atemwegswiderstand abhängig.
-
Volumengesteuerter Ventilator: Das Atemzugvolumen und der maximal zugelassene Spitzendruck werden vorgewählt. Der Atemwegsdruck passt sich innerhalb der Grenzen den Volumenbedürfnissen an.
-
2.3.6.15 Lungentransplantation
Lungentransplantationen kommen im Endstadium der meisten nichtmalignen Lungenerkrankungen als letzte therapeutische Option in Betracht. Im Jahr 2010 wurden in Deutschland 298 Lungentransplantationen durchgeführt, verteilt auf 13 Zentren. Die Transplantationsfrequenz wird weltweit durch den Mangel an Organspendern begrenzt.
2.3.6.16 Indikationen
Am häufigsten bei folgenden Grundkrankheiten:
-
chronische obstruktive Lungenerkrankung (39 %)
-
Lungenfibrose (17 %)
-
zystische Fibrose (10 %)
-
α1-Proteinasen-Inhibitor-Mangel (9 %)
-
idiopathische pulmonale Hypertonie (4 %)
-
Bronchiektasen (2,5 %)
2.3.6.17 Operationsverfahren
-
Einseitige Lungentransplantation (SLTx): Nur ein Lungenflügel wird transplantiert. Der zweite Lungenflügel des Empfängers wird belassen.
-
Bilaterale oder doppelseitige Lungentransplantation (DLTx): Es werden beide Lungenflügel übertragen.
-
Herz-Lungen-Transplantation (HLTx): Herz und beide Lungenflügel des Spenders werden en bloc transplantiert.
Eine bilaterale Transplantation ist bei Bronchiektasen notwendig, weil die bronchiale Infektion auf das Transplantat überspringen könnte. Die Herz-Lungen-Transplantation ist beim Eisenmenger-Syndrom mit komplexen Herzanomalien obligatorisch und in Fällen wo Lungen- und Herzkrankheit im Endstadium sind. Ein Cor pulmonale muss nicht ersetzt werden, weil sich der rechte Ventrikel nach der Transplantation erholt. Bei den übrigen Lungenkrankheiten sind SLTx oder DLTx akzeptabel. Letztere erzielt bei COPD und α1-Antitrypsinmangel eine längere Überlebensdauer.
2.3.6.18 Empfängerauswahl
Voraussetzung für die Transplantation ist die Blutgruppenkompatibilität zwischen Spender und Empfänger gemäß den ABO-Kriterien. HLA-Kompatibilität bleibt aus Zeit- und Spendermangel unberücksichtigt. Patienten mit entsprechendem Schweregrad der Lungenerkrankung kommen zunächst auf eine Warteliste des Transplantationszentrums und werden dort bis zur Transplantation kontrolliert. Die Wartezeit beträgt etwa 2 Jahre. Als Alterslimit für eine Transplantation gilt das 65. Lebensjahr.
2.3.6.19 Kontraindikationen
-
Absolute Kontraindikationen: Floride Infektionen, maligne Tumorerkrankung, Suchtverhalten einschließlich Nikotinkonsum während der letzten 6 Monate.
-
Relative Kontraindikationen: Kachexie, schwere Adipositas, maschinelle Beatmung (ausgenommen intermittierende Selbstbeatmung), HIV-Infektion, Niereninsuffizienz, chronische Virushepatitis (B oder C), Leberzirrhose, Herzinsuffizienz (für Lungentransplantation), symptomatische Osteoporose, neurologische, neuromuskuläre und psychiatrische Krankheiten, Systemkrankheiten mit relevanter extrapulmonaler Manifestation, psychosoziale Probleme, schlechte Mitarbeit bei der bisherigen Therapie.
2.3.6.20 Immunsuppression
Im Vergleich zu anderen Organtransplantationen ist nach Lungentransplantationen eine besonders intensive Immunsuppression notwendig, die lebenslang fortgesetzt werden muss. Zum Einsatz kommt eine Dreierkombination aus einem Calcineurininhibitor (Ciclosporin, Tacrolimus), einem Inhibitor der T-Zellproliferation (Azathioprin, Mycophenolat, Sirolimus) und Prednisolon.
2.3.6.21 Komplikationen
-
Abstoßungsreaktionen: In akuter Form am häufigsten während der ersten 3 Monate nach der Transplantation. Erkennbar an Abgeschlagenheit, Fieber, Hypoxämie und sehr empfindlich am Abfall der Einsekundenkapazität (FEV1). Die chronische Abstoßung manifestiert sich als Bronchiolitis-obliterans-Syndrom . Therapie: Bei akuter Abstoßung Methylprednisolon (1-mal 500–1.000 g/ Tag für 3 Tage) und monoklonale Antikörper gegen T-Lymphozyten. Bei chronischer Abstoßung Intensivierung bzw. Umstellung der Immunsuppression, evtl. Retransplantation.
-
Infektionen: Meistens bakterielle Atemwegsinfektionen mit gramnegativen Erregern, Pneumokokken und Hämophilus. Am zweithäufigsten sind Zytomegalie-Virus-Infektionen.
-
Malignome: Erhöhtes Risiko für Plattenepithelkarzinome und Lymphome.
2.3.6.22 Ergebnisse
Durch eine erfolgreiche Transplantation werden die kardiopulmonale Funktion und damit auch die Lebensqualität unabhängig von der Vorerkrankung eindrucksvoll gebessert. Die ergospirometrische Belastbarkeit ist nach einseitiger und zweiseitiger Lungentransplantation nicht signifikant unterschiedlich. Die körperliche Leistungsfähigkeit bleibt aber reduziert, und weniger als die Hälfte der Empfänger nimmt eine Voll- oder Teilzeitbeschäftigung auf.
Die Überlebensraten variieren unter den Grundkrankheiten relativ wenig:
-
3 Monate: 79–89 %
-
1 Jahr: 66–79 %
-
3 Jahre: 50–61 %
-
5 Jahre: 38–52 %
Häufigste Todesursachen im ersten Jahr nach der Transplantation sind technische Probleme bei der Operation, Transplantatversagen infolge ischämischer Schädigung und Infektionen. Akute Abstoßungsreaktion und CMV-Infektionen kommen im ersten Jahr ziemlich oft vor, enden aber selten tödlich. Nach dem ersten Jahr sind die meisten Todesfälle auf chronische Abstoßung und Infektionen zurückzuführen.
2.3.7 Acute respiratory distress syndrome (ARDS )
2.3.7.1 Definition
Schnell einsetzende schwere Dyspnoe mit Hypoxämie und diffusen Lungeninfiltraten, die zur akuten respiratorischen Insuffizienz, auch als akutes Lungenversagen bezeichnet, führen.
2.3.7.2 Ätiologie
2.3.7.3 Direkte Schädigung des Alveolarepithels
Durch Pneumonie, Aspiration von Mageninhalt, Lungenkontusion, Beinaheertrinken oder Toxininhalation.
2.3.7.4 Indirekte Lungenschädigung
Sepsis, schwere Verletzungen (multiple Knochenbrüche, Kopftraumen, Verbrennungen), multiple Transfusionen, Pankreatitis, Darminfarkt, Operation mit Herzlungenmaschine. Hohes Risiko bei Traumen plus Sepsis.
2.3.7.5 Pathogenese
2.3.7.6 Exsudative Phase
Permeabilitätssteigerung in den Alveolen durch entzündliche Schädigung der Endothelzellen der alveolären Kapillaren und der alveolären Epithelzellen. Dadurch Ansammlung eiweißreicher Flüssigkeit in den Alveolen und im Interstitium. Auslösend sind proinflammatorische Zytokine (Tumornekrosefaktor-α, Interleukine 1 und 8) und Leukotriene. Es werden Effektorzellen rekrutiert und aktiviert, vor allem Neutrophile, die in der Lunge sequestriert werden und toxische O2-Metaboliten freisetzen. In den Alveolen bilden sich Niederschläge aus aggregierten Proteinen und Detritus, in den Kapillaren Mikrothromben. Durch Zerstörung des Surfactant entstehen Atelektasen.
Dyspnoe und Tachypnoe beginnen 12–36 h nach dem auslösenden Ereignis, selten wenige Tage später. Die exsudative Phase dauert etwa 7 Tage. Es entwickelt sich eine zunehmende Hypoxämie (paO2 <60 mmHg) mit normalem oder erniedrigtem CO2. Der Quotient paO2/FiO2 sinkt auf <200, wobei FiO2 die O2-Fraktion der inspirierten Luft ist, die bei der Intensivtherapie durch O2-Atmung erhöht wird. Um einen paO2 von 90 mmHg zu erreichen, muss die Sauerstoff-Fraktion >0,45 betragen. Das Röntgenbild der Lunge zeigt Trübungen in den unteren Zweidritteln der Lungen sowie Atelektasen.
2.3.7.7 Proliferationsphase
Sie dauert vom 7.–21. Tag. Es gehen die neutrophilen in überwiegend mononukleäre Infiltrate über. Im günstigen Fall bilden sich neue Alveolarzellen, die wieder Surfactant produzieren und die Lösung der Infiltrate einleiten, was letztlich zur vollen Ausheilung führen kann. Die Sekretion von alveolärem Typ-III-Kollagen bedeutet dagegen den Übergang zur Fibrosierung.
2.3.7.8 Fibrotische Phase
Gekennzeichnet durch extensive duktale und interstitielle Fibrosierung mit Umbau der Lungenarchitektur zu emphysemartigen bullösen Strukturen und entsprechende Abnahme der Compliance. Durch Intimaproliferation in den kleinen Lungengefäßen entwickelt sich eine schwere pulmonale Hypertonie, häufig mit letalem Ausgang.
2.3.7.9 Therapie
Unter Intensivpflege intensive Behandlung der auslösenden Krankheit. Meistens wird eine mechanische Beatmung zur Entlastung der Atemmuskulatur und zur Beseitigung der Hypoxämie notwendig. Um eine Überdehnung der ventilierten oberen Lungenbezirke zu vermeiden, sollte das Atemvolumen 6 ml/kg nicht übersteigen. Zur besseren Lungenentfaltung wird mit positivem endexspiratorischem Druck (PEEP) wechselnd in Bauch- und Rückenlage beatmet. Die FiO2 sollte möglichst <0,6 betragen.
2.3.7.10 Prognose
Die Letalität des ARDS beträgt 41–65 %. Die meisten Patienten sterben am Grundleiden oder an finalem Multiorganversagen.
2.3.8 Schlafapnoe-Syndrom
2.3.8.1 Definition
Als Schlafapnoe wird die intermittierende Unterbrechung des Atemstromes an Nase und Mund für mindestens 10 Sekunden bezeichnet. Die Dauer der Unterbrechung beträgt meisten 20–30 s und kann 2–3 min erreichen. Wenn es zu Krankheitserscheinungen kommt, spricht man vom Schlafapnoe-Syndrom.
2.3.8.2 Häufigkeit
Betroffen sind rund 4 % der Männer und 2 % der Frauen mittleren Lebensalters.
2.3.8.3 Klassifikation
Zu unterscheiden sind zwei pathogenetisch ganz unterschiedliche Formen der Schlafapnoe:
-
Obstruktive Schlafapnoe: Die weitaus häufigste Form, bei der es in der Apnoephase unter Atembewegungen des Thorax zum Verschluss der Atemwege im Pharynxbereich kommt.
-
Zentrale Schlafapnoe: Bei der zentralen Apnoe setzt der zentrale Atemantrieb aus.
2.3.8.4 Obstruktives Schlafapnoe-Syndrom
2.3.8.4.1 Pathogenese
Kollaps der oberen Atemwege im Schlaf durch Herabsetzung der elektromyographischen Aktivität der oberen Atemwegsmuskeln. Sitz der Obstruktion ist meistens der Pharynx. Andere Mechanismen: Kollaps der Seitenwand des Oropharynx, Zurücksinken der Zunge gegen den weichen Gaumen und die Rachenhinterwand. Die meisten Patienten sind mittel- bis hochgradig übergewichtig. Bei ihnen wird die Schlafapnoe wahrscheinlich durch ein Zurücksinken des schweren Unterkiefers begünstigt. Ein zusätzlicher Risikofaktor ist starker Alkoholkonsum. Disponiert sind auch Patienten mit starker Hypertrophie der Tonsillen, mit Mikrognathie und Retrognathie. Vielfach wurde computertomographisch bei Patienten ohne auffallende Anomalien eine Enge des oropharyngealen Lumens nachgewiesen.
In der Apnoephase sinkt der pO2 in der Alveolarluft und im arteriellen Blut, während der paCO2 ansteigt. Schließlich geht von den Chemorezeptoren ein Weckreiz aus, der zwar nicht zu vollem Bewusstsein, aber zur Normalisierung der Ventilation führt. Die Patienten registrieren diesen halbwachen Zustand nicht und berichten am Morgen, dass sie gut geschlafen hätten. In Wirklichkeit war ihr Schlaf mehr oder weniger stark fraktioniert und deshalb nicht regeneratorisch. Pro Nacht können bis zu 500 Apnoephasen auftreten.
Wichtigste Konsequenz der Schlafapnoe ist ein intermittierendes O2-Defizit. Es kann durch kontinuierliche Messung der arteriellen Sauerstoffsättigung (SaO2) mittels Ohroximetrie gemessen werden. In langen Apnoephasen kann die SaO2 unter 60 % sinken, entsprechend einem paO2 <40 mmHg. Der Mittelwert der während des Schlafes gemessenen SaO2 ist ein Maß für den Schweregrad der obstruktiven Schlafapnoe. In einem größeren Patientenkollektiv hat man Werte zwischen 62 und 96 % registriert. Die wichtigsten Variablen für den Abfall der SaO2 sind der paO2 im Wachzustand, das Lungenvolumen und die prozentuale Schlafzeit in der Apnoe.
Kardiovaskuläre Reaktionen sind vagusinduzierte Sinusbradykardie, nächtliche Hypertonie, Zunahme der diastolischen Ventrikelfüllung und damit Steigerung des Afterloads durch Unterdruck im Thorax, vor allem aber eine pulmonale Hypertonie infolge hypoxischer Vasokonstriktion im Lungenkreislauf. In schweren Fällen resultiert ein Cor pulmonale mit Polyglobulie und Rechtsinsuffizienz. Extremer Vagustonus kann zu Asystolien von bis zu 13 Sekunden führen. Durch myokardialen O2-Mangel entstehen in schweren Fällen tachykarde Rhythmusstörungen bis hin zum tödlichen Kammerflimmern.
Neuropsychiatrische Störungen kommen durch den Mangel an regeneratorischem Schlaf und durch intermittierende zerebrale Hypoxie zustande.
2.3.8.4.2 Klinik
Fast alle Patienten sind Schnarcher, aber längst nicht alle Schnarcher haben eine obstruktive Schlafapnoe. Leitsymptom ist eine ausgeprägte Tagesschläfrigkeit bei Patienten, die nicht über Schlaflosigkeit klagen. Der Ehegatte sollte nach Atempausen befragt werden. In leichteren Fällen wird nur über unerklärliche Müdigkeit geklagt. Hinzu kommen Konzentrationsschwäche, Intelligenzabbau, Persönlichkeitsänderungen und Verhaltensstörungen. In schweren Fällen resultieren Zyanose durch Polyglobulie, Leberschwellung und Ödeme. Zu achten ist auf Tonsillengröße und Kieferanomalien. Mit dem Langzeit-EKG ist nach nächtlichen Rhythmusstörungen zu fahnden.
2.3.8.4.3 Diagnostik
2.3.8.4.4 Screening mit nächtlicher Ohroximetrie
In den meisten Fällen wird ein intermittierender Abfall der SaO2 gefunden. Bei großem Lungenvolumen und kürzeren Apnoephasen kann der Befund annähernd normal sein.
2.3.8.4.5 Polysomnographie
Erlaubt eine sichere Diagnose, die Beurteilung des Schweregrades und die Abgrenzung von der zentralen Schlafapnoe (◘ Abb. 2.11). Sollte in jedem Verdachtsfall durchgeführt werden. Im Schlaflabor werden fortlaufend gemessen: EEG, Elektrookulogramm und Kinn-EMG, außerdem an Mund- und Nasenöffnung der Atemstrom (z. B. mit Termosistor), die thorakoabdominale Atemaktivität mittels Induktionsplethysmographie und die SaO2 mittels Ohroximetrie. Mit einer Ballonsonde im Ösophagus kann auch der inspiratorische Druckabfall im Thorax registriert werden.


Polysomnographie bei Schlafapnoe (schematisch)
2.3.8.4.6 Konservative Therapie
2.3.8.4.7 Allgemeine Maßnahmen
Gewichtsreduktion, Verzicht auf Alkohol, schlafen mit erhöhtem Oberkörper, Nasenatmung optimieren. In leichten bis mittelschweren Fällen erreicht man so deutliche Linderung.
2.3.8.4.8 Technische Hilfen
Dentale Applikatoren, die Unterkiefer und Zunge nach vorn verlagern.
2.3.8.4.9 Nasale CPAP (Continous Positive Airway Presssure )
Durch einen Druck von 4,5–14 cm H2O wird der intraluminale Druck in den oberen Atemwegen während der Inspiration offengehalten. Die Anpassung des Gerätes und die Einstellung des richtigen Druckes erfolgt im Schlaflabor. Für alle schweren Formen die optimale Therapie. Die Tagesschläfrigkeit hört sofort auf, die übrigen Symptome bilden sich umgehend zurück. Komplikationen: Trockenheit und Reizung der Nasenschleimhaut, Bindehautreizungen. Nicht alle Patienten behalten die Methode bei. Manche wenden sie nur alternierend an.
2.3.8.4.10 Operative Therapie
2.3.8.4.11 Tracheotomie
Bleibt schwersten Fällen vorbehalten, bei denen die CPAP versagt. Subjektive Belastung und sekundäre Morbidität durch Infektionen schränken die Methode ein.
2.3.8.4.12 Uvulopalatopharyngeoplastik (UPPP)
Reseziert werden Uvula, Teile des Gaumensegels, Tonsillen und Falten der Rachenhinterwand. Indikation in schweren Fällen bei Versagen der Überdruckbeatmung. Erfolgsquote ca. 30–50 %. Vorher im Einzelfall schwierig abzuschätzen.
2.3.8.5 Zentrales Schlafapnoe-Syndrom
2.3.8.5.1 Pathogenese
Auf diese Form entfallen nicht mehr als 10 % der Schlafapnoefälle. Anders als bei der obstruktiven Form ist die Atemmuskulatur während der Apnoephase mangels zentraler Stimulation inaktiv. Es kommen aber Mischformen vor, bei denen die zentrale in eine obstruktive Apnoe übergeht. Im Schlaf wird die Atmung primär durch den arteriellen pCO2 gesteuert, der über die Chemorezeptoren am Atemzentrum angreift. Zur Apnoe kann es durch Fehlen der ventilatorischen Chemosensitivität kommen, wie beim Fettsucht-Hypoventilations-Syndrom (Pickwick) und bei der zentralen alveolären Hypoventilation. Ferner werden nächtliche Apnoephasen durch dysrhythmische Atmung mit periodischer Hypokapnie ausgelöst, was bei Herzinsuffizienz zu beobachten ist. In vielen Fällen gibt es für die intermittierende nächtliche Apnoe keine Erklärung.
2.3.8.5.2 Klinik
Patienten mit reiner zentraler Schlafapnoe sind im Gegensatz zu den mit der obstruktiven nicht übergewichtig. Sie klagen selten über Tagesschläfrigkeit, sondern über Schlaflosigkeit, Schlafunterbrechung bzw. häufiges Erwachen während der Nacht. Nicht selten kommt es zu Depressionen und Libidoverlust.
2.3.8.5.3 Diagnostik
Die Diagnose ist nur mittels Polysomnographie zu stellen (◘ Abb. 2.11). Diese zeigt in den Apnoephasen keine Aktivität der Atemmuskulatur und keinen Druckabfall im Ösophagus. Der Schweregrad ergibt sich aus dem Mittelwert der SaO2 während der Nacht.
2.3.8.5.4 Therapie
Patienten mit Hypoxie profitieren von nächtlicher O2-Zufuhr per Nasensonde. Manche reagieren auf Ansäuerung mit Acetazolamid. Bei Kombination mit obstruktiven Phasen zeigt die nasale CPAP gute Erfolge. Bei Herzinsuffizienz hat sich die adaptive Servo-Ventilation bewährt. In schweren Fällen mit Polyglobulie und Cor pulmonale sind assistierte Beatmung oder Diaphragma-Pacing mittels elektrischer Phrenikusstimulation indiziert.
2.4 Diagnostische Methoden
Diagnostische Methoden
-
Körperliche Untersuchung
-
Sputumuntersuchung
-
Makroskopische Beurteilung
-
Mikroskopische Untersuchung
-
Bakteriologische Untersuchung
-
-
Radiologische Untersuchungsmethoden
-
Röntgenuntersuchung des Thorax
-
Computertomographie (CT)
-
Magnetresonanztomographie (MRT)
-
-
Nuklearmedizinische Untersuchungsmethoden
-
Perfusionsszintigraphie
-
Radiospirometrie
-
67Gallium-Szintigraphie und Emmissionstomographie
-
-
Thoraxsonographie
-
Bronchoskopie, Biopsie und Lavage
2.4.1 Körperliche Untersuchung
2.4.1.1 Methoden
Mit den einfachen Mitteln der Inspektion, Auskultation, Perkussion und der Prüfung des Stimmfremitus lassen sich nach wie vor wichtige diagnostische Aufschlüsse gewinnen. Die typischen Befundkonstellationen bei Erkrankungen der Bronchien und der Lunge sind in der ◘ Tab. 2.2 zusammengestellt.
2.4.2 Sputumuntersuchung
2.4.2.1 Makroskopische Beurteilung
2.4.2.1.1 Sammeln
Durch spontane Expektoration oder nach Inhalation eines Expektorans (z. B. Salbutamol), in besonderen Fällen durch transtracheale Aspiration (zur Ausschaltung der Mundflora). Größere Mengen sollten in Glasgefäßen gesammelt werden.
2.4.2.1.2 Inspektion
-
Menge: Viel Auswurf bei Bronchiektasen (1 Liter/Tag und mehr), Lungenzysten und Abszessen, Gangrän und Empyemdurchbruch
-
Farbe und Konsistenz:
-
weißlich-schleimig: normal
-
gelbgrün-geballt: eitrig
-
serös-schaumig: Lungenödem
-
weiß und zäh-schleimig (Curschmann-Spiralen): Asthma bronchiale
-
rostbraun: Pneumonie, Stauungslunge
-
hellrot-schaumig: Hämoptoe
-
-
Geruch: Unauffällig (meistens), fötide (Gangrän, Abszess)
2.4.2.2 Mikroskopische Untersuchung
-
Eosinophile Leukozyten: Asthma bronchiale.
-
Herzfehlerzellen: Lungenstauung (Alveolarzellen mit Eisenspeicherung).
-
Elastische Fasern: Lungengewebezerfall.
-
Zytodiagnostik: Untersuchung auf Tumorzellen.
2.4.2.3 Bakteriologische Untersuchung
Erregernachweis: Eitererreger, Tuberkelbazillen etc.
Resistenzbestimmung: Testung der antibiotischen Empfindlichkeit.
2.4.3 Radiologische Untersuchungsmethoden
2.4.3.1 Röntgenuntersuchung des Thorax
2.4.3.1.1 Standardaufnahmen
Anterior-posteriore (a. p.) Übersichtsaufnahme (◘ Abb. 2.12) und Seitenaufnahme (rechts oder links anliegend) in tiefer Inspirationslage. Erfasst werden umschriebene und diffuse Krankheitsprozesse im Lungenparenchym (Herdnachweis ab 5 mm Durchmesser) und im Pleuraraum (Ergüsse), Hilusvergrößerungen und weniger genau Prozesse im Mediastinum. Erkannt werden auch Größen- und Formänderungen des Herzens, Aortenaneurysmen und abnorme Befunde am Brustkorbskelett.


Thoraxübersichtsaufnahme (Normalbefund)
2.4.3.1.2 Thoraxdurchleuchtung
Ermöglicht Beurteilung der Zwerchfellbewegung, Abgrenzung von Pleuraergüssen gegen Schwarten, Lokalisation von Lungenprozessen, Nachweis abnormer Pulsationen des Hilus und intrapulmonaler Gebilde.
2.4.3.2 Computertomographie (CT)
2.4.3.2.1 Konventionelle CT
Erweitert die diagnostischen Möglichkeiten der Röntgenuntersuchung in folgenden Punkten:
-
Genaue Lokalisation von Lungenherden, Einschmelzungen und Emphysemblasen (◘ Abb. 2.13).
Abb. 2.13 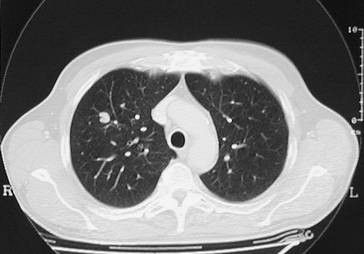
CT-Bild mit Rundherd im rechten Oberlappen. Nachweis eines Adenokarzinoms, das mittels Oberlappenektomie entfernt wurde
-
Erfassung von Prozessen mit geringen Dichteunterschieden zum normalen Lungengewebe.
-
Nachweis hilärer und mediastinaler Prozesse (z. B. Lymphknotenbefall bei Bronchialkarzinomen).
-
Calciumnachweis in Rundherden.


2.4.3.2.2 Hochauflösendes CT (HRCT)
Die Schichtdicke der Bilder beträgt 1–2 mm statt 10 mm. Dadurch gelingt die subtile Darstellung der Atemwege und des Parenchyms. Liefert spezifische Muster bei den interstitiellen Lungenerkrankungen.
2.4.3.2.3 Spiral-CT
Erlaubt die Sammlung von kontinuierlichen Daten über ein größeres Lungenvolumen während eines Atemanhalte-Manövers. Kombiniert mit Kontrastmittelinjektion können Emboli in den Lungenarterien erkannt werden.
2.4.3.3 Magnetresonanztomographie (MRT)
Gegenüber der CT geringere räumliche Auflösung im Lungenparenchym. Vaskuläre können besser von nichtvaskulären Strukturen unterschieden werden. Eingesetzt zur ergänzenden Darstellung mediastinaler Raumforderungen und von Prozessen an der Knochen-Weichteil-Lungengrenze.
2.4.4 Nuklearmedizinische Untersuchungsmethoden
2.4.4.1 Perfusionsszintigraphie
Nachweis von Perfusionsstörungen im Lungenkreislauf durch Injektion radioaktiv markierter Partikel. Zum Nachweis von Lungenembolien (▶ Kap. 1).
2.4.4.2 Radiospirometrie
Quantitative Messung der Radioaktivität über beiden Lungen nach:
-
intravenöser Injektion von 133Xenon (Perfusionsverteilungsmessung) und
-
Inhalation von 133Xenon (Ventilationsverteilungsmessung).
Damit kann festgestellt werden, wie sich verschiedene Volumina (Residualvolumen, inspiratorische Vitalkapazität) über beide Lungen aufteilen. Außerdem werden Verteilungsstörungen erfasst und die Halbwertszeiten bei der Abatmung des radioaktiven Gases. Indiziert zur präoperativen regionalen Lungenfunktionsdiagnostik.
2.4.4.3 67Gallium-Szintigraphie und Emissionstomographie
Ermöglicht Markierung von neoplastischem und entzündlichem Gewebe, insbesondere bei Sarkoidose und Lymphomen. Nachweis von entzündlicher Aktivität bei idiopathischer pulmonaler Fibrose.
2.4.5 Thoraxsonographie
2.4.5.1 Methodische Besonderheiten
An der gesunden Lunge wird der Ultraschall durch den Luftgehalt fast völlig reflektiert. Pulmonale Prozesse können mit Ultraschall abgebildet werden, wenn konsolidiertes Lungengewebe an die viszerale Pleura reicht und durch ein Schallfenster eine pathologische Schallemission ermöglicht. Störende Schallschatten, die von den Rippen ausgehen, können durch Atemmanöver überbrückt werden. Transjugulär ist das vordere obere Mediastinum bis zum aortopulmonalen Fenster gut einsehbar.
2.4.5.2 Indikationen
Häufig eingesetzte Standardtechnik bei pleuranahen Lungenveränderungen und pleuralen Prozessen aller Art. Methode der Wahl zum Nachweis auch kleiner Pleuraergüsse. Als Ergänzungen bieten sich die transösophageale und endobronchiale Sonographie an, die allerdings nach Aufwand und Handhabung invasive Untersuchungsmethoden sind (◘ Abb. 2.14).


Bild eines endobronchialen Ultraschalls. Die Sonde ist im Bronchus intermedius (BI) platziert. Parabronchial stellt sich ein vergrößerter Lymphknoten (LN) dar
2.4.6 Bronchoskopie , Biopsie und Lavage
2.4.6.1 Diagnostische Indikationen
2.4.6.1.1 Inspektion
Bei ungeklärter Bronchitis, Hämoptoe, Verdacht auf Bronchialkarzinom (◘ Abb. 2.15), Verdacht auf Bronchusstenosen bzw. Atelektasen, bei unspezifischer Pneumonie, Tuberkuloseverdacht (zur Keimgewinnung) und Kontrolle nach Operationen.


Endoskopisches Bild eines Carcinoma in situ im Oberlappen links
2.4.6.1.2 Biopsie
Bei allen tumorösen Veränderungen und den interstitiellen Lungenerkrankungen.
2.4.6.1.3 Bronchoalveoläre Lavage (BAL)
Spülung durch das Bronchoskop in Portionen von 20 ml physiologischer Kochsalzlösung und zytologische bzw. bakteriologische Untersuchung der Spülflüssigkeit. Anwendung zur Abgrenzung der granulozytären (idiopathische Fibrose) von der lymphozytären Alveolitis (Sarkoidose) und zur Differenzierung der T-Lymphozyten bei Asthma bronchiale (keine Routinemethode).
2.4.6.2 Interventionelle Bronchoskopie
Darunter versteht man die Durchführung therapeutischer Eingriffe in Trachea und Bronchien mittels minimalinvasiver bronchoskopischer Techniken. Es handelt sich um ein Spezialgebiet der Pneumologie, das sich in den letzten Jahren stark weiterentwickelt hat. Die Eingriffe werden in günstigen Fällen ambulant durchgeführt, im Allgemeinen aber während eines kurzen stationären Aufenthaltes. Welches Verfahren im Einzelfall angewendet wird, hängt von der Art der Läsion und im Falle einer malignen Stenose von der anatomischen Obstruktionsform ab.
Techniken der interventionellen Bronchoskopie
-
Starre Bronchoskopie
-
Ballondilatation
-
Laser
-
Elektro- und Argon-Plasma-Koagulation
-
Stentimplantation
-
Endobronchiale Brachytherapie
-
Kryotherapie
-
Photodynamische Therapie
2.4.6.3 Mediastinoskopie
2.4.6.3.1 Indikationen
Beurteilung der Operabilität maligner Lungentumoren, Klärung von Hilusverbreiterungen unbekannter Ursache.
2.4.6.3.2 Kontraindikationen
Akut entzündliche mediastinale und Pleuraprozesse.
2.4.6.3.3 Technik
Operativer Eingriff in Intubationsnarkose, bei dem nach kollarer Mediastinotomie das Mediastinoskop entlang der Vorderwand der Trachea bis zur Bifurkation vorgeschoben wird. Entnahme möglichst ganzer Lymphknoten zur histologischen Untersuchung (◘ Abb. 2.16)


Mediastinoskopie. a Schematische Darstellung. b Verdacht auf ein mediastinales Lymphom bei einer jungen Frau. Sicherung der Diagnose eines Non-Hodgkin-Lymphoms mittels Mediastinoskopie
2.4.6.3.4 Komplikationen
Blutungen, Pneumothorax, Phrenikusparesen, Mediastinitis. Letalität 0,1–0,7 %.
2.4.6.4 Präskalenische Lymphknotenbiopsie (nach Daniels)
Chirurgische Lymphknotenexstirpation in der Lücke zwischen M. scalenus anterior und M. sternocleidomastoideus. Seitenwahl nach Lokalisation des verdächtigen Lungenbefundes. Trefferquote bei Verdacht auf Bronchialkarzinom 45 %, bei Sarkoidose 65 %, bei Morbus Hodgkin 80 %. Die diagnostische Ausbeute ist geringer als bei der Mediastinoskopie.
2.4.6.5 Transkutane Pleura - und Lungenbiopsie
Die Pleurabiopsie ist bei unklarer Pleuritis (Tbc, Tumor etc.) zur bakteriologischen und molekularbiologischen Untersuchung des Punktats indiziert, nicht beim Emphysem. Die Lungenbiopsie wird als Punktionsbiopsie mit Hohl- oder Spreiznadel bei Lungenherden in Außenzonen eingesetzt. Punktionsaspiration bei schweren Pneumonien mit unklarem Erreger (immunsupprimierte Patienten) zur bakteriologischen und molekularbiologischen Untersuchung.
2.4.6.6 Thorakoskopie und Biopsie
2.4.6.6.1 Indikationen
Umschriebene oder diffuse primäre Pleuraerkrankungen, unklarer Pleuraerguss, diffuse Lungenparenchymerkrankungen, Pneumothorax, Tumoren des hinteren Mediastinums.
2.4.6.6.2 Technik
Anlage eines Pneumothorax. Unter Lokalanästhesie oder Intubationsnarkose Einführung starrer oder flexibler Thorakoskope. Biopsie mit Zange, nötigenfalls Koagulation mit einer Elektrode. Durch eine Minithorakotomie können die offene Thoraxbiopsie und Thorakoskopie kombiniert werden.
2.4.6.7 Diagnostische Thorakotomie
Indiziert bei peripheren Rundherden und umschriebenen Mediastinaltumoren, die bei dem Eingriff in toto entfernt werden. Diagnostik und Therapie erfolgen somit gleichzeitig.
2.5 Erkrankungen der Trachea und Bronchien
Erkrankungen der Trachea und Bronchien
-
Tracheitis
-
Akute Bronchitis
-
Chronische Bronchitis und Emphysem
-
Zystische Fibrose (Mukoviszidose)
-
Asthma bronchiale
-
Bronchiektasen
-
Bronchialkarzinom
-
Nichtkleinzelliges Bronchialkarzinom
-
kleinzelliges Bronchialkarzinom
-
-
Bronchoalveoläres Karzinom
-
Lungenmetastasen
-
Semimaligne und benigne Bronchialtumoren
-
Bronchialadenome
-
Benigne Bronchialtumoren
-
2.5.1 Tracheitis
2.5.1.1 Akute Tracheitis
2.5.1.1.1 Definition
Akute Entzündung der Trachea.
2.5.1.1.2 Ätiologie
Meistens als Folge einer Virusinfektion der oberen Luftwege (Rhinoviren, Adenoviren, Influenza A und B, Parainfluenza-Viren, Coronaviren u. a.), seltener durch Infektion mit Mykoplasmen und Chlamydien oder durch Reizgase ausgelöst. Häufig Mitbefall des Kehlkopfes (Laryngotracheitis).
2.5.1.1.3 Klinik
Husten, Heiserkeit, Brennen hinter dem Brustbein, bei Kindern wegen des kleinen Trachealumens auch akute Obstruktion mit in- und exspiratorischem Stridor und Dyspnoe. Abgrenzung gegen Fremdkörperaspiration durch Tracheoskopie.
2.5.1.1.4 Therapie
Ausschaltung inhalativer Noxen (Rauchverbot). Inhalationen mit Emser Salz oder Kamillendämpfen, Antitussiva. Bei Verdacht auf bakterielle Infektion Makrolid-Antibiotika (Azithromycin). Bei Allergie Kortikoide als Aerosol.
2.5.1.2 Chronische Tracheitis
2.5.1.2.1 Definition
Chronische Entzündung der Trachea, die länger als 3 Monate persistiert, nicht selten im Anschluss an eine akute Entzündung und mit einer chronischen Bronchitis kombiniert.
2.5.1.2.2 Ätiologie
Verursacht durch chronische Inhalationsschäden (Raucher) und Tracheastenosen (Kompression von außen durch Strumen oder Lymphknoten, interne Obstruktion durch Tumore, Intubationsverletzungen, Tracheomalazie).
2.5.1.2.3 Klinik
Chronischer Hustenreiz mit etwas Auswurf, Heiserkeit, bei Stenosen Globusgefühl und Atemnot.
2.5.1.2.4 Diagnostik
Stenosen führen zur in- und exspiratorischen Widerstandserhöhung im Ganzkörperplethysmogramm. Bei jeder chronischen Tracheitis ist die Bronchoskopie angezeigt.
2.5.1.2.5 Therapie
Symptomatisch wie bei akuter Tracheitis. Behandlung der Stenose nach der Ursache.
2.5.2 Akute Bronchitis
2.5.2.1 Ätiologie
2.5.2.2 Virusinfekte
Rhinoviren, Myxoviren (Influenza A und B), REO-Viren, Adenoviren u. a. Primäre Virusinfektionen können die Anfälligkeit gegenüber bakteriellen Infektionen steigern und ihnen den Weg bahnen: Influenza-Viren → Streptococcus pneumoniae, Staphylococcus aureus, Haemophilus influencae, E. coli, Neisseria meningitidis, Aspergillus fumigatus. Respiratory syncytial virus → S. pneumoniae und H. influenzae als Otitis-media-Erreger. Zytomegalievirus bei Immunsuppression → Pseudomonas aerogenes, E. coli, Klebsiella, Listeria, Aspergillus, Candida albicans, Pneumocystis carinii. Die Mechanismen dieses Synergismus sind noch weitgehend ungeklärt. Pertussis ▶ Kap. 10.
2.5.2.3 Mycoplasma pneumoniae
Infektionen hauptsächlich bei Schulkindern, von denen die Familie angesteckt wird. Verursacht auch atypische Pneumonien. Antibiotikaempfindlich.
2.5.2.4 Bakterielle Infekte
Selten ohne vorausgehende Virusinfekte oder Vorschädigung der Bronchialschleimhaut. Scharlach, Typhus und Diphtherie beginnen oft mit Bronchitis. In der Praxis dominieren Pneumokokken und H. influenzae, in der Klinik S. aureus, Enterokokken, Kolibakterien, Proteus, Klebsiellen und Pseudomonas.
2.5.2.5 Inhalationsnoxen
Smog (SO2-haltiger Kohlenstaub und Nebel) ergibt einen die Atemwege reizenden schwefelsäurehaltigen Nebel. Weitere Reizstoffe: Nitrosegase (bei Sprengungen mit Dynamit), Ozon, Fluorkohlenstoffpolymere u. a.
2.5.2.6 Klinik
Husten (zuerst trocken, dann schleimig bis eitrig), Kopfschmerz, Abgeschlagenheit, auch leichtes Fieber.
2.5.2.7 Diagnostik
Über den Lungen manchmal Nebengeräusche. Starkes Giemen spricht für Exazerbation einer chronischen obstruktiven Bronchitis. Thoraxaufnahme ohne pathologischen Befund.
2.5.2.8 Therapie
Bei Fieber Bettruhe in feuchtwarmer Zimmerluft. Brust warm halten, keine Zugluft. Gegen Hals- und Kopfschmerz leichte Analgetika. Sekretolytische und spasmolytische Mittel meistens entbehrlich. Codeinhaltige Antitussiva bei starkem Hustenreiz. Vitaminreiche Kost, Zitrusfrüchte, Vitamin C. Antibiotika: Bei Mykoplasmeninfektion Tetrazykline (spezifisch), bei eitrigem Sputum Trimethoprim/Sulfamethoxazol, Makrolide (Azithromycin) oder Moxifloxacin.
2.5.3 Chronische Bronchitis und Emphysem
2.5.3.1 Definition
2.5.3.2 Chronische Bronchitis
Klinisch definiert als Husten mit Auswurf für wenigstens 3 Monate im Jahr während einer 2-Jahresperiode. Bei der einfachen chronischen Bronchitis ist das Sputum schleimig, bei der chronischen mukopurulenten Bronchitis durch persistierende oder rezidivierende Purulenz des Sputums gekennzeichnet.
2.5.3.3 Chronische obstruktive Bronchitis
Chronische Bronchitis mit permanenter, überwiegend irreversibler Obstruktion der Bronchien. Infekte und inhalierte Noxen können zu einer reversiblen Exazerbation der Obstruktion führen.
2.5.3.4 Emphysem
Pathologisch anatomisch definiert als Erweiterung der Lufträume distal der terminalen Bronchiole mit Zerstörung der Alveolarsepten. Durch Abnahme der elastischen Zugkräfte des Lungenparenchyms kommt es dabei obligatorisch zu einer irreversiblen exobronchialen Obstruktion der kleinen Bronchien.
2.5.3.5 Chronische obstruktive Lungenkrankheit (COPD : chronic obstructive pulmonary disease)
Die in der Regel vorliegende, meistens durch Zigarettenrauchen verursachte, Kombination von chronischer obstruktiver Bronchitis und Emphysem mit progredienter Atemflusslimitierung.
2.5.3.6 Pathogenese
2.5.3.7 Chronische Bronchitis
Hyperplasie und Hypertrophie der submukösen Schleimdrüsen in den großen (knorpelhaltigen) Bronchien. In den kleinen (knorpelfreien) Bronchien Hyperplasie der Becherzellen, entzündliche Zellinfiltrate und Ödem der Mukosa und Submukosa, obturierende Schleimpfröpfe und peribronchiale Fibrose. Vermehrung der glatten Muskelzellen. Die das Krankheitsbild beherrschende Obstruktion ist vor der Entwicklung des Emphysems hauptsächlich durch die Verengung und das Verschwinden der kleinen Atemwege (Durchmesser <2 mm) bedingt.
2.5.3.8 Emphysem
Zu unterscheiden sind 2 Formen, die häufig nebeneinander vorkommen:
-
Zentroazinäres Emphysem: Dilatation und Destruktion hauptsächlich der respiratorischen Bronchiolen und Alveolargänge. Zusammengedrängte kleine Alveolen in der Azinusperipherie.
-
Panazinäres Emphysem: Destruktion und Dilatation des ganzen Azinus mit Verkleinerung der Gasaustauschfläche.
2.5.3.9 Ätiologie
2.5.3.10 Chronische Bronchitis
Die Entstehung wird auf ein Zusammenwirken exogener und endogener Faktoren zurückgeführt:
-
Zigarettenrauchen: Wichtigster Kausalfaktor. Hemmt Zilienbewegung des Flimmerepithels bei gleichzeitiger Staubbelastung (150 Milliarden Staubteilchen pro Zigarette). Chemische Reizung (Ammoniak, Ameisensäure, Essigsäure, kanzerogene Teerbestandteile), Hemmung der alveolären Makrophagen, Induktion von Hyperplasie und Hypertrophie der submukösen Schleimdrüsen. Auslösung von Bronchospasmus (via N. vagus) durch Reizung submuköser Rezeptoren. Da nur 15–20 % der Raucher eine chronische obstruktive Bronchitis bekommen, wird bei den Erkrankten eine Hyperreaktivität der Bronchien auf konstriktorische Reize angenommen.
-
Luftverunreinigung: Größere Erkrankungshäufigkeit in stark industrialisierten städtischen Regionen (SO2).
-
Berufliche Gas- und Staubexposition: NO2, Asphalt, Teerstaub u. a.
-
Rezidivierende Infekte der Atemwege: Bedeutung für Pathogenese und Progression bisher nicht endgültig geklärt, scheint aber bei Rauchern gegeben zu sein.
2.5.3.11 Emphysem
Ein panazinäres Emphysem lässt sich tierexperimentell durch Inhalation oder Instillation Elastin-spaltender Fermentes (Papain, Elastase) aus Pankreas, segmentkernigen Leukozyten oder alveolären Makrophagen erzeugen. Die Elastase wird durch das in die Lunge übertretende α1-Antitrypsin des Plasmas gehemmt. Bei genetisch bedingtem Mangel an α1-Antitrypsin entwickelt sich in der Mehrzahl der Fälle ein panazinäres Emphysem. Zahlenmäßig fällt dieser genetische Defekt als Emphysemursache nicht ins Gewicht. Er zeigt aber, dass dem Emphysem ganz allgemein eine Elastindestruktion zugrunde liegt. Da in der Bronchiallavage bei chronischen Rauchern vermehrt segmentkernige Leukozyten und alveoläre Makrophagen nachzuweisen sind, könnte die von diesen Zellen gebildete Elastase für das bei Rauchern dominierende Emphysem der maßgebende pathogenetische Faktor sein.
2.5.3.12 Pathophysiologie
Bei der COPD greifen 2 pathophysiologische Komponenten ineinander:
-
Emphysem: Neben einer Verteilungsstörung bedingt es eine Verkleinerung der Gasaustauschfläche mit globaler Diffusionsstörung. Hinzu kommt eine nur exspiratorische exobronchiale Obstruktion durch den Elastizitätsverlust der Lunge. Einem Anstieg von paCO2 und einem Abfall von pO2 wird durch Ventilationssteigerung begegnet, so dass die Blutgase in Ruhe normal bleiben. Hypoxie und Hyperkapnie treten erst bei körperlicher Belastung auf. Polyglobulie und pulmonale Hypertonie entwickeln sich erst spät. Patienten vom Emphysemtyp sind gewöhnlich untergewichtige, hyperventilierende Astheniker mit blassrotem Hautkolorit („Pink Puffer“).
-
Chronische obstruktive Bronchitis: Vorherrschend ist eine endobronchiale Obstruktion (in- und exspiratorisch wirksam) mit erheblicher Verteilungsstörung. Die damit verbundene Hypoxämie stimuliert die Ventilation kaum. Sie führt durch Vasokonstriktion zur pulmonalen Hypertonie und zur reaktiven Polyglobulie. Im Stadium der respiratorischen Insuffizienz werden Hypoxie und Hyperkapnie ohne wesentliche Ventilationssteigerung akzeptiert. Dafür scheint eine verminderte Ansprechbarkeit der Chemorezeptoren, der afferenten Wege oder des Atemzentrums verantwortlich zu sein. Die Patienten vom bronchitischen Typ sind überwiegend hustende, zyanotische Pykniker mit Übergewicht und geblähtem Thorax („Blue Bloater “).
-
Mischtypen: In der Praxis kommen sie als COPD am häufigsten vor.
2.5.3.13 Epidemiologie
Die COPD ist eine verbreitete Krankheit, an der in Amerika nicht weniger als 16 Millionen Personen leiden. Männer sind häufiger betroffen als Frauen. Am höchsten ist dort die Prävalenz in den Bevölkerungsschichten mit niedrigem sozioökonomischem Status. Mehr als 90 % der Patienten sind Raucher oder haben früher geraucht.
2.5.3.14 Klassifikation
Nach Gold siehe Zusammenfassung in ◘ Tab. 2.3.
2.5.3.15 Klinik
Die COPD wird meistens zu Beginn des fünften Lebensjahrzehnts mit einer Herabsetzung der FEV1 klinisch manifest. Als einfacher Raucherhusten wird sie oft längere Zeit unterschätzt und ignoriert. Im Vordergrund stehen chronischer Husten mit Auswurf und zunehmende Belastungsdyspnoe. Die Obstruktion ist irreversibel und progredient. Durch Infekte der oberen Luftwege kommt es zu vorübergehenden Exazerbationen.
2.5.3.16 Diagnostik
2.5.3.17 Körperlicher Untersuchungsbefund
In leichten Fällen nur etwas Giemen bei forcierter Exspiration, im Verlauf starkes in- und exspiratorisches Giemen, das sich im Spätstadium wegen Hyperinflation der Lunge erheblich abschwächt. Forcierte Exspiration löst Husten aus. Auf ein Emphysem weisen ein abgeschwächtes Atemgeräusch und ein hypersonorer Klopfschall hin. Bezüglich körperlichem Habitus und die Hautfarbe siehe Pathophysiologie. Das Cor pulmonale führt zu tastbaren epigastrischen Pulsationen.
2.5.3.18 Lungenfunktion
Leitsymptom im Frühstadium ist eine herabgesetzte FEV1. Der Quotient FEV1/FVC liegt unter 70 %. Residualvolumen und Totalkapazität sind auf Kosten der Vitalkapazität und des inspiratorischen Reservevolumens vergrößert. Das pneumotachographisch registrierte Fluss-Volumen-Diagramm zeigt einen abrupten Flussabfall nach Erreichen des reduzierten Spitzenflusses („Emphysemknick“). Die Resistance ist erhöht. Ein Emphysem steigert die Ruheventilation.
Zur Differenzierung zwischen Obstruktion und Emphysem kann der Bronchospasmolysetest herangezogen werden (▶ Abschn. 2.5.5).
2.5.3.19 Blutgasbestimmung
paO2 zuerst nur unter Belastung später auch in Ruhe herabgesetzt; paCO2 steigt im fortgeschrittenen Stadium schon in Ruhe und führt zur respiratorischen Azidose. Im Finalstadium Ruhedyspnoe mit Einsatz der Atemhilfsmuskeln. Geringste Belastungen verursachen stärkste Atemnot. Oft Polyglobulie mit Zyanose und Symptome des dekompensierten Cor pulmonale.
2.5.3.20 Laboruntersuchungen
Blutbild zur Erfassung einer sekundären Polyglobulie. Sputumuntersuchung auf Eosinophile und Bestimmung des IgE im Serum zur Abgrenzung gegen ein allergisches Asthma bronchiale. Messung des α1-Antitrypsins im Serum bei frühmanifester COPD.
2.5.3.21 Röntgenaufnahmen der Lunge
Anfangs unauffällig. Bei fortgeschrittener COPD vermehrte Strahlendurchlässigkeit vor allem der Oberfelder mit rarefizierter Gefäßzeichnung durch Emphysem. Zwerchfelltiefstand infolge Hyperinflation der Lunge. Retrosternalraum erweitert (◘ Abb. 2.17). Zunächst kleiner Herzschatten, später durch pulmonale Hypertonie mit Cor pulmonale Vorwölbung der rechten Herzkontur und betonter Pulmonalisbogen.


Thoraxaufnahme in 2 Ebenen (a.p. und seitlich) bei ausgeprägtem Emphysem. Erhöhte Strahlentransparenz der Lunge, tiefstehendes Zwerchfell mit abgeflachten Kuppeln, erweiterter Retrosternalraum (Sammlung Prof. Jend)
2.5.3.22 Computertomographie
Zeigt die emphysematösen Veränderungen besonders deutlich (◘ Abb. 2.18).


Hochgradiges Emphysem bei einem 68-jährigen Raucher. Zusätzlich Raumforderung im linken Oberlappen (Adenokarzinom)
2.5.3.23 Therapie (◘ Tab. 2.3)
2.5.3.24 Prophylaktische Maßnahmen
Nikotinabstinenz, Fernhalten von Staub und industriellen Abgasen. Schutz vor Erkältung und Infektionen (Grippeschutzimpfung). Raucherscreening zur Früherfassung der chronischen Bronchitis und Obstruktion.
2.5.3.25 Nikotinentzug
Verbessert die Lebenserwartung und das FEV1, ohne die eingetretene Schädigung des Bronchialsystems zu beseitigen. Leider stellen 70–80 % der Patienten nur vorübergehend das Rauchen ein, und nicht mehr als 6 % bleiben dauerhaft abstinent. Zur Erklärung der Nikotinabhängigkeit hat man die Aktivierung von Endorphinen im Gehirn durch diese Substanz herangezogen. Maßnahmen zur Entwöhnung: Aufklärung über Risiken, Verhaltenstherapie, vorübergehend Nikotinpflaster, auch Clonidin und Antidepressiva haben sich manchmal bewährt. Das Rauchen sollte plötzlich beendet werden, weil die schrittweise Reduzierung keinen Erfolg verspricht. Vorbildfunktion des Arztes.
2.5.3.26 Medikamentöse Therapie
-
Bronchodilatatoren: Inhalative β2-Sympathikomimetika: Kurzwirkende (Fenoterol, Terbutalin, Salbuterol) sind 4–6 h, langwirkende (Salmeterol, Formoterol) sind mehr als 24+ h effektiv. Ebenso wirksam sind inhalative Anticholinergika, die als kurzwirkende (Ipatropiumbromid) und langwirkende (Tiotropium) verfügbar sind. Beide Substanzklassen sollten wegen eines deutlichen additiven Effektes kombiniert eingesetzt werden. Die Wirksamkeit kann mit dem Broncholysetest überprüft werden.
-
Theophyllin: Bei der COPD ist der bronchodilatorische Effekt anders als beim Bronchialasthma gering, weil es nur den Histamin-induzierten Bronchospasmus löst. Von begrenztem Nutzen ist Theophyllin für etwa 20 % der Patienten. In fortgeschrittenen Fällen kann es das Ansprechen des Atemzentrums auf Hyperkapnie und die Kontraktilität des Zwerchfells verbessern. Wegen der geringen therapeutischen Breite wird das Mittel kaum noch verwendet.
-
Glukokortikoide: Obwohl beim Asthma sehr wirksam, haben inhalative Steroide bei der COPD kaum einen therapeutischen Effekt. Im Sputum nehmen weder Entzündungszellen noch Zytokine ab. Die Steroidresistenz ist überwiegend auf den durch Rauchen und oxidativen Stress verursachten Aktivitätsabfall der Histondeacetylasen (HDACs) zurückzuführen. Diese Enzyme hemmen die Translation proinflammatorischer Gene (▶ Kap. 6.3.1).
-
PDE-4-Hemmer: Phosphodiesterasehemmer wirken der Anreicherung von cAMP in inflammatorischen Zellen entgegen und vermindern so deren Aktivität. Im Handel ist Roflumilast. Die regelmäßige Einnahme soll Exazerbationen verhindern und Entzündungen hemmen. Es gibt aber etliche Nebenwirkungen.
-
Antibiotika: Indiziert bei Exazerbationen mit eitrigem Sputum und Leukozytose. Am günstigsten wirken meistens die Makrolide (Azithromycin u. a.). Erregerzüchtung aus dem Sputum und Resistenzbestimmung verbessern die Erfolgsaussichten.
2.5.3.27 Physikalische Therapie
-
Atemübungen: Verlängerung der Exspiration bei geschürzten Lippen, um die exspiratorische Kompression der kleinen Bronchien einzuschränken. Betonung der Zwerchfellatmung zur Vergrößerung des Atemzugvolumens und Verkleinerung der funktionellen Residualkapazität. Lockerung und Training der Atemhilfsmuskulatur.
-
Körperliches Training: Mit Übungen, die der herabgesetzten Leistungsfähigkeit angepasst sind, lässt sich die Ausdauer verbessern. Sauerstoffgabe während des Trainings kann hilfreich sein.
2.5.3.28 Maßnahmen gegen die Hypoxämie
Bei respiratorischer Insuffizienz (paO2≤55 mmHg) Langzeit-O2-Heimtherapie mit Hilfe von O2-Separatoren. Beatmungsdauer 16 h pro Tag.
2.5.3.29 Operative Therapie
Resektion emphysematöser Lungenbezirke zur Verminderung der Totraumventilation und der Kompression gesunden Lungengewebes.
2.5.3.30 Lungentransplantation
Im präterminalen Stadium eine therapeutische Option. Überlebensraten nach einem Jahr 80–90 %, nach 5 Jahren über 60 %.
2.5.4 Zystische Fibrose (Mukoviszidose)
2.5.4.1 Definition
Autosomal rezessive Systemerkrankung mit sekretorischer oder resorptiver Dysfunktion des Epithels, die zu folgenden Manifestationen führt: Sekretverstopfung der Atemwege, Störungen der exokrinen Pankreassekretion, Herabsetzung der Flüssigkeitssekretion im proximalen und Steigerung der Flüssigkeitsresorption im distalen Intestinum, Obliteration des Vas deferens, abnorm hohe Chloridkonzentration im Schweiß.
2.5.4.2 Ätiologie
Der ursächliche genetische Defekt betrifft das am Chromosom 7 lokalisierte Gen für das CFTR-Protein (cystic fibrosis transmembrane regulator). Die häufigste Mutation des CF-Gens ist eine Deletion von 3 Basenpaaren an Position 508 (ΔF508) mit dem Ausfall eines Phenylalaninmoleküls. Sie bewirkt, dass CFTR in der Zelle abgebaut wird, also ganz fehlt. Nachgewiesen wurden noch >800 weitere Punktmutationen des CF-Gens, die zu funktionslosen CFTR-Molekülen oder solchen mit eingeschränkter Funktion führen. Etwa 95 % der Fälle von manifester zystischer Fibrose sind aber nur durch 22 der bekannten Genmutationen bedingt. Heterozygote Merkmalsträger erkranken nicht. Umstritten ist noch, ob sie bei manchen Mutationen zu Pankreatitiden disponiert sind.
2.5.4.3 Pathogenese
Das CFTR-Protein ist in der apikalen Epithelmembran lokalisiert und hat die Funktion eines cAMP-aktivierten Chloridkanals. Es reguliert aber auch andere Ionenkanäle. Seine Funktion variiert mit dem Standort des Epithels, woraus sich für Manifestationen der zystischen Fibrose unterschiedliche pathophysiologische Mechanismen ergeben.
2.5.4.4 Atemwege
Der CFTR-Defekt beschleunigt die Na+- und Wasserresorption des Epithels und hemmt die Cl--Sekretion in das Bronchialsekret, das dadurch viskös und klebrig wird. Die Entfernung des Schleims aus den kleinen Bronchien durch Ziliarbewegung und Husten reicht nicht mehr aus. Es kommt zur chronischen Obstruktion mit Sekretstau und sekundären Infektionen, die allmählich das Lungenparenchym zerstören.
2.5.4.5 Pankreas
Der Wegfall bzw. Defekt des cAMP-aktivierten Chloridkanals, den das CFTR-Protein repräsentiert, hemmt die Cl--Sekretion der Ductuli und damit auch deren Bikarbonat-Sekretion sowie die passive Sekretion von Na+ und Wasser. Das ins Lumen ausgeschiedene Cl- wird nämlich im Austausch gegen Bikarbonat wieder rückresorbiert. Es resultiert eine starke Eindickung des Pankreassekrets mit Okklusion der Ductuli, die zum Parenchymuntergang und zur exkretorischen Pankreasinsuffizienz führen.
2.5.4.6 Intestinaltrakt
Im oberen Dünndarm ist die Sekretion von Cl- und Wasser herabgesetzt, wodurch die Ausschwemmung von Muzinen und Makromolekülen aus den Krypten beeinträchtigt wird. Im distalen Dünndarm und im Kolon ist die Na+- und Wasserresorption abnorm gesteigert, was zu Eindickung des Kots und schon bei Neugeborenen zum Mekoniumileus führen kann.
2.5.4.7 Genitalsystem
95 % der männlichen Patienten haben eine Azoospermie infolge Obliteration des Vas deferens durch eingedicktes Sekret.
2.5.4.8 Epidemiologie
Die Prävalenz der zystischen Fibrose variiert mit dem ethnischen Ursprung der Population. In der weißen Bevölkerung Nordamerikas und Nordeuropas kommt ein Fall auf 3000 Lebendgeburten. In Afrika ist es ein Fall auf 17.000 und in Asien ein Fall auf 90.000 Lebendgeburten.
2.5.4.9 Klinik
Die meisten Patienten erkranken schon im Kindesalter, darunter 15 % an einem Mekoniumileus in den ersten 24 h. Bei etwa 7 % wird die Diagnose erst nach dem 18. Lebensjahr gestellt. Dank therapeutischer Fortschritte ist die Lebenserwartung der männlichen Patienten auf 32 Jahre gestiegen, die der weiblichen auf 29 Jahre.
2.5.4.10 Respirationstrakt
Die Patienten leiden an einer chronischen Sinusitis, oft mit Polypenbildung, chronischem Husten und einer zunehmenden obstruktiven Bronchitis. Das zunächst visköse Sputum wird purulent und nimmt schließlich eine grünliche Farbe an. Es kommt zu rezidivierenden bronchopneumonischen Schüben mit zunehmender Verschlechterung der Lungenfunktion. Unter den Sputumkeimen dominieren Haemophilus influenzae, S. aureus, Pseudomonas aeroginosa und Burkholderia cepacia. Im Röntgenbild und CT stark erweiterte und destruierte Bronchien (◘ Abb. 2.19). Komplikationen: Pneumothorax, Hämoptysen, Cor pulmonale. Das Krankheitsgeschehen mündet in eine finale respiratorische Insuffizienz, die mit einem dekompensierten Cor pulmonale einhergehen kann.


Mukoviszidose. Thoraxübersichtsaufnahme und CT. In der ganzen Lunge erweiterte und destruierte Bronchien. Das CT-Bild zeigt Sekretspiegel (Sammlung Prof. Jend)
2.5.4.11 Gastrointestinaltrakt
Bei 90 % der Patienten entwickelt sich eine exkretorische Pankreasinsuffizienz mit Malabsorption und Steatorrhö. Die initiale distale intestinale Obstruktion wird durch voluminöse Fettstühle abgelöst. Bei älteren Patienten kann ein insulinbedürftiger Diabetes mellitus in Erscheinung treten.
2.5.4.12 Genitalsystem
Männliche Patienten sind wegen Obstruktion der Samenwege infertil. Bei weiblichen Patienten ist die Menstruation durch die schwere Lungenkrankheit beeinträchtigt. Schwangerschaften werden jedoch meistens ausgetragen.
2.5.4.13 Diagnostik
2.5.4.14 Klinisches Bild
Bei Neugeborenen Unvermögen zu schreien, aufgetriebener Leib, ausbleibende Darmentleerung. Später ist die Kombination respiratorischer und gastrointestinale Symptome wegweisend.
2.5.4.15 Elektrolytbestimmung im Schweiß
Konzentrationen von Na und Cl >70 mval/l. Bei 1–25 % der Fälle von Mukoviszidose sind die Konzentrationen im Normbereich.
2.5.4.16 Genetische Analyse
Familienanamnese, DNA-Analyse.
2.5.4.17 Therapie
2.5.4.18 Lunge
Regelmäßige Atem- und Expektorationsübungen, Brustkorbperkussion. Zur Sekretverflüssigung Inhalation von Aerosolen mit hypertoner Salzlösung. Zur Schleimlösung sind Inhalationen mit rekombinanter humaner DNA-ase geeignet, da das Sputum konzentrierte DNA enthält. N-Acetylcystein ist als Mukolytikum unwirksam. Bei akuten entzündlichen Schüben ist die frühzeitige und prolongierte Antibiotikatherapie dringend erforderlich, möglichst gezielt nach Sputumkultur. Im Finalstadium kann nur eine Lungentransplantation helfen oder eine Herz-Lungen-Transplantation. Die 2-Jahresüberlebensraten liegen >50 %.
2.5.4.19 Gastrointestinaltrakt
Substitution der exokrinen Pankreasinsuffizienz mit konzentrierten Enzympräparaten zu den Mahlzeiten. Ausgleich entstandener Vitamindefizite. Gegen Darmobstruktion osmotische Abführmittel (Laktulose) und Einläufe.
2.5.4.20 Gentherapie
Versuche, das fehlende Gen in Körperzellen zu übertragen, sind seit Jahren im Gange. Gegenwärtig wird es in England und den USA mittels Liposomen erprobt.
2.5.5 Asthma bronchiale
2.5.5.1 Definition
Reversible Obstruktion der Atemwege bei Hyperreaktivität auf entzündlicher Basis.
2.5.5.2 Ätiologie und Pathogenese
Die Hyperreaktivität der Bronchien basiert auf einer persistierenden subakuten Entzündung der Bronchialschleimhaut mit Epithelabschilferung, Hyperplasie der Becherzellen, Verdickung der subepithelialen Basalmembran und subkutanen Infiltraten aus Eosinophilen, T-Lymphozyten und Neutrophilen. Auch im Intervall zwischen Asthmaanfällen ist die Entzündung vorhanden. Aus den zellulären Elementen und den Epithelzellen werden von Fall zu Fall Wirkstoffe freigesetzt, die durch Ödembildung, Steigerung der Schleimsekretion und Kontraktion der glatten Muskelzellen zur Obstruktion führen. Im Gegensatz zur COPD kommt es nicht zum destruktiven Ödem. Zwei Kategorien des Asthmas sind zu unterscheiden:
2.5.5.3 Allergisches Asthma
Es entsteht durch eine IgE-vermittelte immunologische Reaktion vom Soforttyp (mit Spätreaktion) gegen Antigene, die für gesunde Individuen nicht pathogen sind und deshalb als Allergene bezeichnet werden (▶ Kap. 8, Abschnitt Allergie). Die Allergene werden in der Schleimhaut von dendritischen Zellen aufgenommen und in den regionalen Lymphknoten von spezifischen T-Helferlymphozyten vom Typ TH2 präsentiert. Die aktivierten TH2-Zellen proliferieren und stimulieren passende B-Lymphozyten zur Produktion von Immunglobulin E. Mit diesem Immunglobulin werden die unter der Epithelschicht lokalisierten Gewebemastzellen bestückt. Sobald Allergene die Antigenbindungsstellen von zwei benachbarten IgE-Molekülen überbrückt haben, entleeren die Mastzellen den Inhalt ihrer Granula nach außen, die Histamin, Tryptase, Proteoglykane, Prostaglandin D2 und chemotaktische Peptide enthalten. Zusätzlich sezernieren aktivierte Makrophagen Leukotriene, die zusammen mit Histamin eine Bronchokonstriktion bewirken. Die in der Schleimhaut anwesenden TH2-Zellen stimulieren mittels IL-5 die Einwanderung von Eosinophilen, die mit ihren Inhaltsstoffen einen Entzündungsprozess in Gang setzen und unterhalten.
Ganz neu ist die Beobachtung, dass in der durch Lavage gewonnenen bronchoalveolären Flüsssigkeit von Asthmatikern unter den reichlich vorhandenen T-Lymphozyten natürliche Killer-T-Zellen dominieren (Anteil 60–80 %). Bei den übrigen T-Zellen handelt es sich um CD4+-TH2-Lymphozyten. Die NK-T-Zellen bilden wie die CD4+-T-Zellen IL-4 und IL-13, aber kein Interferon γ. Sie sind auch in den Schleimhautinfiltraten vorhanden. Nach Dauerbehandlung mit Glukokortikoiden waren sie nicht vermindert. Die NK-T-Zellen besitzen im Gegensatz zu den natürlichen Killerzellen T-Zellrezeptoren, deren Spezifizität aber nur wenig variiert. Sie erkennen Glykolipidantigene, die von antigenpräsentierten Zellen zusammen mit dem MHC-Klasse-I-Molekülen vom Typ CD1d exprimiert werden. Solche Glykolipide sind auch in Pollen enthalten.
Die Allergene lassen sich in nachstehende Gruppen gliedern:
-
tierische Produkte: Haare, Schuppen, Borsten, Federn, Duftstoffe, Insektenhämoglobine, Exkremente, Bakterienenzyme (in Waschmitteln)
-
pflanzliche Produkte: Getreide, Mehl, Kleie; Baumpollen (Frühlingstyp), Gräserpollen (Herbsttyp); ätherische Öle, Gewürze, Kolophonium, grüner Kaffee, Castorbohnen
-
chemische Stoffe: Penicillin, Cimetidin, Psyllium, Chloramine, Acrylate, Anhydride.
Die Hyperreaktivität der Bronchien hat zur Folge, dass auch unspezifische Reize wie Kaltluft, körperliche Anstrengung (Flüssigkeitsentzug der Schleimhaut bei Mundatmung und Hyperventilation), Luftverschmutzung und emotionaler Stress Asthmaanfälle auslösen können. In solchen Fällen kann die Erkennung der Atopie schwierig sein.
2.5.5.4 Nichtallergisches Asthma
Es ist nicht immunologisch bedingt, führt aber auch zur entzündlichen Reaktion der Bronchialschleimhaut. Man spricht auch vom idiosynkratischen Asthma, womit eine individuelle Disposition unterstellt wird. Die Pathogenese ist nicht einheitlich und nur teilweise geklärt. Irritanzien könnten durch Freisetzung von Neuropeptiden (SP, Neurokinin A) Bronchkonstriktion und Ödembildung bewirken. Bekannte auslösende Faktoren sind die folgenden:
-
Virusinfektionen der Atemwege: Unter den Pathogenen dominieren bei Kindern das respiratorische Synzytiumvirus und die Parainfluenzaviren, bei Erwachsenen Rhino- und Influenzaviren. Oft handelt sich um gewöhnliche Infekte der oberen Luftwege, denen im Abstand von Tagen oder einigen Wochen die Symptome der Obstruktion folgen. Nicht selten nimmt auch das allergische Asthma nach einem Infekt seinen Anfang. Offenbar zerstören die Erreger die Schleimhautbarriere und induzieren dadurch entzündliche Prozesse.
-
Aspirin und nichtsteroidale Antiphlogistika: Die meist erwachsenen Patienten präsentieren sich mit einer chronischen polypösen Rhinosinusitis und Asthmaanfällen. Nur durch die Vorgeschichte oder einen Expositionsversuch ist die Diagnose zu klären. Die Schleimhäute weisen eosinophile Infiltrate auf. Pathogenetischer Mechanismus: Durch Blockade der Cyclooxygenase wird die Arachidonsäure nur auf dem Lipoxygenaseweg metabolisiert. Dabei fallen vermehrt Leukotriene an, die bronchokonstriktorisch wirken und Gewebsmastzellen degranulieren.
-
Luftschadstoffe: Ozon, NO2 und SO2 wirken auf die Schleimhaut als Irritanzien und bei entsprechend starker Exposition asthmaauslösend.
-
Gewerbliches Asthma: Durch Azofarbstoffe (Tartrazin), Metalldämpfe (Platin, Nickel, Chrom u. a.), Halogene (Chlor, Fluor, Brom), Holzextrakte, Holzstaub, Pflanzenstaub (Baumwolle, Flachs, Hanf) und Insektizide, Formaldehyd. Pathogenetischer Mechanismus: Irritation der Schleimhaut mit Freisetzung entzündlicher und bronchokonstriktiver Mediatoren.
-
Psychogene Faktoren: Nicht als primäre Krankheitsursache, aber für die Intensität der Symptome im fördernden wie im hemmenden Sinne von Bedeutung. Emotionaler Stress und Angst können Asthmaanfälle auslösen, Suggestion kann sie abschwächen. Psychogene Einflüsse auf die Atemwege scheinen hauptsächlich über efferente Vagusfasern zu laufen. Stressbedingte Adrenalinausschüttung wirkt bronchodilatorisch.
2.5.5.5 Pathophysiologie
2.5.5.6 Ventilationsstörung
Die Obstruktion der Bronchien bewirkt primär eine exspiratorische Widerstandserhöhung für die Atemluft. Das hat folgende Konsequenzen:
-
Herabsetzung des forcierten exspiratorischen Volumens (FEV1), der forcieren exspiratorischen Vitalkapazität (FVC) und des Quotienten FEV1/FVC (in Prozenten).
-
Die maximale exspiratorische Flussgeschwindigkeit (PEF) ist verlangsamt und fällt während der Exspiration abrupt ab.
-
Die Lunge wird überbläht, weil die Exspiration vorzeitig zu Ende geht. Residualvolumen (RV) und funktionelle Residualkapazität (FVC) nehmen zu.
-
Die Atemarbeit steigt, weil die Atemfrequenz zunimmt und die Lunge inspiratorisch stärker gedehnt wird, um die elastischen Rückstellkräfte für die Exspiration zu erhöhen. In schweren Fällen muss die Exspiration muskulär unterstützt werden.
2.5.5.7 Verteilungsstörung
In minderbelüfteten Alveolen kommt es zur reflektorischen Vasokonstriktion. Daraus resultieren Veränderungen des Ventilations-Perfusions-Verhältnisses, die zur Hyoxämie mit kompensatorischer Hyperventilation und zur Hypokapnie führen. Außerdem kommt es zur pulmonalen Hypertonie mit Rechtsherzbelastung.
2.5.5.8 Epidemiologie
Das Bronchialasthma hat in den letzten Jahrzehnten deutlich zugenommen, besonders in den Industriestaaten. Heute schätzt man die Prävalenz auf 5 % der Bevölkerung. Bakterielle Infektionen im Kindesalter scheinen vor der Atopie zu schützen, weil sie die Immunantwort frühzeitig auf Th1-Helferzellen lenken, die keine IgE- sondern eine IgG-Produktion induzieren. Solche Infektionen werden aber durch gute Hygiene und freizügige Antibiotikaverordnung eingedämmt. Das allergische Asthma dominiert bei Kindern und jungen Erwachsenen. Die höchste Erkrankungsrate liegt vor dem 5. Lebensjahr. Etwa 50 % der betroffenen Kinder werden nach der Pubertät symptomfrei. Das nichtallergische Asthma hängt oft mit dem Gewerbe zusammen und ist im mittleren Lebensalter am häufigsten.
2.5.5.9 Klinik
2.5.5.10 Verlaufsformen
Die klinischen Manifestationen des Asthmas hängen davon ab, wie schnell sich die Obstruktion entwickelt, welchen Grad sie erreicht, wie lange sie andauert und wie weit sie sich zurückbildet. So kann der Asthmaanfall akut mit maximaler Intensität einsetzen (auch nachts) oder protrahiert (über Tage und Wochen) zum Höhepunkt gelangen. Schwere Anfälle von über 24-stündiger Dauer bezeichnet man als Status asthmaticus. Nicht selten ist die Rückbildung der Obstruktion unvollständig, auch bei symptomfreien Patienten, was sich durch die Messung der FEV1, feststellen lässt. Statt typischer Anfälle kann es zu längeren Phasen asthmatischer Dyspnoe kommen. Vor allem in den Frühstadien gewerblicher Asthmaformen beobachtet man oft eine chronische Dyspnoe („eosinophile asthmatische Bronchitis“). Hier entstehen Abgrenzungsschwierigkeiten gegen die chronische obstruktive Bronchitis.
2.5.5.11 Symptome
Der typische Asthmaanfall beginnt mit trockenem Husten und Engegefühl in der Brust. Danach folgen zunehmende Dyspnoe und Tachypnoe, in schweren Fällen mit Einsatz der auxillären Atemmuskeln und ein zuerst exspiratorisches, danach in beiden Atemphasen auf Distanz hörbares Giemen oder Pfeifen. Auf dem Höhepunkt des Anfalls lässt der Husten nach, und das Giemen wird leiser (Abnahme der Atemstromstärke). Eine Zyanose kann auch bei bedrohlicher Hypoxie fehlen. In der Abklingphase kommt es zum produktiven Husten mit schleimigem oder gelblichem Auswurf (kein Eiter, sondern Eosinophilie) und erneut zu stärkerem Giemen, das schließlich aufhört. Im Asthmaanfall ist die Exspiration stärker erschwert als die Inspiration, die vorzeitig einsetzt, so dass sich die Atemmittellage nach der inspiratorischen Seite verschiebt und der Thorax in Inspirationsstellung geht. Meistens besteht eine Tachykardie über 100/min, in bedrohlichen Situationen zugleich ein Pulsus paradoxus. Die Patienten sind erregt, haben Angst und ein Erstickungsgefühl. Die Anfälle enden spontan oder unter therapeutischen Maßnahmen. Leichtere, oft längere Zeit andauernde Grade der asthmatischen Obstruktion gehen mit Husten, Auswurf, Belastungsdyspnoe und dem Auskultationsbefund des Giemens und Brummens einher.
2.5.5.12 Diagnostik
2.5.5.13 Lungenfunktion
Inspiratorische Vitalkapazität normal, FEV1, FVC, Quotient FEV1/FVC herabgesetzt, RV erhöht, maximal auf 400 % der Norm, Totalkapazität normal oder etwas erhöht. Exspiratorischer Spitzenfluss um 20 % vermindert, konvexe exspiratorische Atemstromkurve. Resistance erhöht.
2.5.5.14 Bronchospasmolysetest
Nach Einatmen von 4 Hüben Salbutamol wird die prozentuale Reversibilität des FEV1-Wertes bestimmt: >15 % spricht für Asthma, <15 % weist auf Emphysem hin.
2.5.5.15 Blutgasanalyse
Die Normwerte für den paO2 sind abhängig von Geschlecht, Alter und Gewicht. Sie variieren von 70–100 mmHg. Im Asthmaanfall ist der paO2 herabgesetzt, hyperventilationsbedingt auch der paCO2. Schon ein normaler paCO2 ist ein bedrohliches Zeichen, ein erhöhter der Beweis für eine respiratorische Insuffizienz.
2.5.5.16 Röntgenaufnahme des Thorax
Im Anfall Überblähung der Lunge mit Tiefstand des Zwerchfells. Außerhalb der Asthmaanfälle ist das Röntgenbild der Lunge gewöhnlich normal (◘ Abb. 2.20).


Asthma bronchiale. a Im Anfall Überblähung der Lunge mit Tiefstand des Zwerchfells. b Nach dem Anfall Rückbildung der Überblähung (Sammlung Prof. Jend)
2.5.5.17 Laboruntersuchungen
Bestimmung des IgE im Serum. Im Blutbild ist auf Eosinophilie zu achten. Daneben Routineprogramm.
2.5.5.18 Identifizierung von Allergenen
In vitro mit dem Radio-Allergo-Sorbent-Test (RAST). In vivo durch Hautproben. Außerdem inhalative Provokationstests. Auch die Expositionsprophylaxe kann Aufschluss geben.
2.5.5.19 Identifizierung nichtallergischer Ursachen
Expositionsanamnese. Am Arbeitsplatz mit einem Flowmeter den exspiratorischen Spitzenfluss messen. Normalisieren sich pathologische Werte daheim, ist ein gewerbliches Asthma sehr wahrscheinlich. In anderen Fällen Provokationstests mit suspekten Stoffen.
2.5.5.20 Therapie
Zwei Zielrichtungen sind bei der Asthmabehandlung zu unterscheiden, die Ausschaltung des auslösenden Agens bzw. seine Blockierung und die symptomatische Behandlung von Schleimhautentzündung und Obstruktion.
2.5.5.21 Expositionsprophylaxe und Desensibilisierung
Dauerhafte Elimination des auslösenden Agens führt zur Heilung des Asthmas. Sie ist vor allem bei gewerblichem Asthma möglich (Berufs- oder Arbeitsplatzwechsel). Schwierigkeiten ergeben sich bei ubiquitären Allergenen. Nicht selten entfällt die Expositionsprophylaxe, weil das Allergen nicht bekannt ist oder weil eine Polyallergie besteht.
Durch spezifische Desensibilisierung lässt sich die Immunantwort so umstellen, dass gegen das Allergen überwiegend Antikörper vom Typ IgG gebildet werden, die das Allergen im Gewebe abfangen, bevor es mit noch vorhandenen IgE-besetzten Mastzellen reagieren kann. Das Verfahren setzt die genaue Identifizierung des Allergens voraus. Es besteht darin, das Allergen in langsam steigender Dosis über Wochen subkutan zu injizieren, wobei die Initialdosis keine positive Hautreaktion geben darf. Der Wirkungsmechanismus ist nicht genau bekannt. Im Erfolgsfall bleiben Asthmaanfälle jahrelang aus oder sie verlaufen stark abgeschwächt. Die Erfolge sind allerdings begrenzt. Außerdem besteht das Risiko des allergischen Schocks. Die Desensibilisierung hat deshalb keine weite Verbreitung gefunden. Neue Methoden der Downregulierung von antigenspezifischen Th2-vermittelten Immunantworten u. a. mit DNA-Vakzinen und rekombinantem Interleukin-4-Rezeptor sind in der Entwicklung. Man darf hoffen, dass sie zum Einsatz kommen und die klassische Methode der Desensibilisierung übertreffen.
2.5.5.22 Bronchodilatatoren
-
β-Sympathikomimetika: Die adrenerge Stimulation relaxiert über die β2-Rezeptoren die glatten Muskelzellen und hat mit Abstand den stärksten bronchodilatorischen Effekt. Am intensivsten wirken die nichtselektiven Adrenergika Adrenalin, gefolgt von den etwas schwächeren Isoprenalin (Aludrin®) und Orciprenalin (Alupent®). Sie stimulieren auch β1-Rezeptoren und haben deshalb erhebliche Nebenwirkungen (Tachykardie, Arrhythmie, Hypertonie, Unruhe, Schwitzen Schwindelgefühl). Außerdem ist ihre Wirkungsdauer kurz (90 min). Ihr Einsatz bleibt schwersten Fällen vorbehalten. Adrenalin kann nur injiziert werden und kommt bei stark herabgesetzter Atemstromstärke in Betracht (0,3–0,5 ml einer 1:1000 verdünnten Lösung s.c).
Therapeutischer Standard sind die inhalativen selektiven β2-Stimulatoren, von denen die kurzwirkenden (4–6 h) am häufigsten eingesetzt werden, z. B. Salbutamol (z. B. Sultanol®), Terbutalin (z. B. Bricanyl®) und Pirbuterol (Zeisin®). Man muss sie gewöhnlich mehrmals täglich per Dosieraerosol oder Inhalator applizieren. Für leichtere Fälle sind β2-Adrenergika mit 12-stündiger Wirkungsdauer verfügbar, z. B. Salmeterol (aeromax®) und Formoterol (Foradil®).
-
Anticholinergika: Beim Asthma ist ihre Bedeutung geringer als bei der COPD. Für manche Patienten, besonders solche mit psychogenen Exazerbation sind sie aber sehr nützlich. Verfügbar sind inhalative Präparate Ipatropiumbromid (Atrovent®) und Tiotropium, letzteres mit Langzeitwirkung.
-
Theophyllin: Die Substanz mit ihren verschiedenen Salzen ist ein mittelstarker Bronchodilatator, wirksam durch Steigerung des cAMP in den glatten Muskelzellen. Man setzt sie meistens in der Abklingphase des Asthmaanfalls ein. Die Applikation erfolgt per os. Die Dosis muss unter Kontrolle der Plasmaspiegel individuell erprobt werden, da die therapeutische Breite der Substanz klein und die Clearance sehr unterschiedlich ist. Therapeutische Plasmaspiegel liegen zwischen 5 und 15 µg/ml. Nebenwirkungen: Appetitmangel, Schlafstörungen, Tachykardie, Unruhe.
2.5.5.23 Entzündungshemmende Mittel
Ihnen obliegt es, den Asthmaattacken gewissermaßen den Boden zu entziehen.
-
Glukokortikoide: Sie wirken stark entzündungshemmend (▶ Kap. 6, Abschn. Glukokortikoidwirkung), supprimieren bei Inhalation die Mastzellendegranulation und potenzieren den Effekt der Katecholamine. Der Wirkungseintritt erfolgt aber erst nach 6 h h. Für die Basistherapie des Asthmas sind sie das wichtigste Mittel und müssen daher kontinuierlich angewendet werden. Gewöhnlich erfolgt die Applikation per Inhalation, in schweren Fällen zusätzlich per os, intramuskulär oder intravenös. Durch die topische Anwendung werden systemische Steroideffekte stark eingeschränkt, aber nicht ganz vermieden. Der Inhalation ist möglichst ein Bronchospasmolytikum vorzuschalten. Inhalative Glukokortikoide: Beclometason-dipropionat (Sanasthmyl® u. a.) Budenosid (Pulmicort® u. a.), Mometason furoat (Asmanex®) Fluticason-propionat (Flutide®). Beclometason-dipropionat wird erst in der Lunge wirksam (durch Esterasen), womit Einwirkungen auf Mund- und Rachenschleimhaut vermieden werden. Es gibt druckbetriebene ind Pulverinhalatoren. Letztere enthalten kein Treibmittel. Als Treibmittel wird statt Chlorfluorcarbon jetzt meistens Hydrofluoralkan verwendet, durch das etwa 60 % des Medikaments in die Lunge gelangt, mit Pulverinhalatoren bis zu 30 %, was bei der Dosierung zu berücksichtigen ist.
Zur systemischen Therapie verwendet man Prednison (oral); Prednisolon oder Triamcinolon (i.v. und in Kristallform i. m. als Depot). Falls eine längere systemische Therapie erforderlich ist, muss eine Osteoporoseprophylaxe (mit Vitamin D und Calcium) erfolgen.
Manche Asthmatiker sind steroidresistent. Die Gründe dafür sind ungenügend geklärt. Sicher hat aber das Zigarettenrauchen wie bei der COPD einen Wirkungsverlust zur Folge.
-
Cromoglycinsäure: Mastzellenstabilisator, der die antigeninduzierte und unspezifische Mediatorfreisetzung blockiert, aber nicht direkt bronchospasmolytisch und entzündungshemmend wirkt. Bewährtes Mittel zur Anfallsprophylaxe beim chronischen Asthma, im Asthmaanfall unwirksam. Zur Anwendung gelangt das Dinatriumsalz (Intal®), appliziert als Pulver oder Aerosol zur Inhalation (4×tgl. 1 Ampulle zu 20 mg). Hat die geringsten Nebenwirkungen aller Asthmamittel, deshalb für Kinder vorteilhaft.
2.5.5.24 Sekretolyse , Expektoration
Zur Entfernung des hochviskösen Sekrets tragen β-Adrenergika, Theophyllin und Kortikoide bei. Wichtig sind ausreichende Hydratisierung und mechanische Maßnahmen (Lagerung, Vibrationsmassage, Expektorationsgymnastik). Die zusätzliche Anwendung von Sekretolytika und Expektoranzien (Acetylcystein, Bromhexin, Detergenzien) in Form von Inhalationen ist hinsichtlich des therapeutischen Effektes umstritten. In schwersten Fällen Intubation und Bronchiallavage.
2.5.5.25 Strategien der Asthmabehandlung
-
Patientenunterweisung: Erklärung des Krankheitsbildes und der Wirkungsweise der Medikamente, deren Dosierung vom Patienten in Grenzen variiert werden kann. Wichtig ist die Früherkennung einer Verschlimmerung, am besten mit einem einfachen Flowmeter, damit vom Arzt rechtzeitig eingegriffen werden kann.
-
Asthmaanfall: Initial alle 20 Minuten 2–3 Sprühstöße mit einem β2-Adrenergikum, z. B. Salbutamol. Sofort 40–100 mg Prednisolon i.v., auch wenn der Effekt erst nach Stunden zu erwarten ist. In schwersten Fällen 0,5 mg Adrenalin subkutan. Nach der ersten Stunde zusätzlich Theophyllin und Anticholinergikum. Der Therapieeffekt ist mit dem Flowmeter zu verfolgen. Sehr bedrohliche Zeichen wie Pulsus paradoxus (inspiratorischer Blutdruckabfall ≥10 mmHg) und Atmung mit Hilfsmuskeln erfordern Einweisung auf eine Intensivstation. Dort kann intubiert, mechanisch beatmet und Sauerstoff appliziert werden.
-
Dauertherapie: Das Ziel ist, mit möglichst geringen Mitteln einen asymptomatischen Zustand mit guter Lungenfunktion zu erhalten. In leichten Fällen lässt man β2-Adrenergika nach Bedarf inhalieren. Bei häufigen Obstruktionsbeschwerden sind die Adrenergika und ein inhalatives Glukokortikoid für einige Zeit täglich anzuwenden. Um eine befriedigend Remission zu erreichen, kann vorübergehend die orale Gabe von Glukokortikoiden erforderlich werden. Dabei ist auf Soorbefall der Mundhöhle zu achten.
2.5.5.26 Prognose
Der Langzeitverlauf ist durch den Wechsel von Exazerbationen und Remissionen gekennzeichnet. Die Quote der Kinder, die 7–10 Jahre nach der Erstmanifestation noch Asthma haben, beträgt durchschnittlich 46 %. Patienten, die das Asthma im Erwachsenenalter erworben haben, erreichen in 20 % der Fälle eine Spontanremission, bei 40 % nimmt die Häufigkeit und Schwere der Anfälle ab. Irreversible Lungenschäden entstehen nur durch zusätzliche Einflüsse, vor allem durch Rauchen. Zur Sterblichkeit an Bronchialasthma gibt es keine genauen Daten. In den USA rechnet man mit etwas weniger als 6000 Todesfällen an Asthma bronchiale pro Jahr bei rund 10 Millionen Asthmapatienten. In Deutschland soll es jährlich etwa 1000 Asthmatote geben.
2.5.6 Bronchiektasen
2.5.6.1 Definition
Dauerhafte abnorme Erweiterung einzelner oder mehrerer Bronchialäste mit der Tendenz zu chronisch-eitriger Entzündung. Man unterscheidet folgende anatomische Formen (◘ Abb. 2.21):


Bronchiektasen. a Schematische Darstellung der verschiedenen Formen. b Operationspräparat
-
Zylindrische (spindelförmige) Bronchiektasen: Erstrecken sich über die dichotomen Bronchialbaumverzweigungen der 6.–10. Generation.
-
Variköse Bronchiektasen: Kombination von sack- und spindelförmigen Veränderungen, die ein varizenähnliches Bild ergeben.
-
Zystische (sackförmige) Bronchiektasen: Überwiegend in den großen proximalen Bronchien lokalisiert, die an der betroffenen Stelle (4. Bronchialverzweigung) in großen Säcken, Zysten oder traubenartigen Gebilden enden.
2.5.6.2 Ätiologie und Pathogenese
2.5.6.3 Hereditäre und kongenitale Faktoren
-
Kongenitale Bronchiektasie (Williams-Campbell-Syndrom): Sackförmige Bronchiektasenzystenbildung infolge eines Defektes der Knorpelbildung. Durch Sekretstau kommt es zu bakteriellen Infektionen und chronischer Entzündung.
-
Primäre ziliäre Dyskinesie: Rezessiv erbliches Syndrom mit variabler Penetranz. Frequenz bei der Geburt 1:15.000. Das für die Krümmung der Zilien zuständige Axonprotein ist durch eine Punktmutation defekt. Es kommt zum Sekretstau mit Infektion und Wandläsionen. Die Hälfte der Patienten hat ein Kartagener-Syndrom (Bronchiektasen, Sinusitis, Infertilität und partieller oder totaler Situs inversus).
-
α1-Antitrypsinmangel: Der Serin-Proteinase-Inhibitor wird überwiegend in der Leber gebildet, zirkuliert im Blut und supprimiert die Aktivität der Neutrophilen-Elastase in der Lunge. Der Erbgang des Enzymdefektes ist autosomal kodominant. Die Mutationen lassen Inhibitormoleküle mit unterschiedlichem Wirkungsverlust entstehen. Manche verbleiben in der Leber und verursachen eine Zirrhose. Klinisch relevant sind α1AT-Plasmaspiegel <11 µmol/l. In Europa beträgt die Prävalenz 1:2000 bis 1:7000. Die Erkrankung wird erst im mittleren Lebensalter manifest, überwiegend bei Rauchern (Kofaktor Nikotin). Symptome: Panazinäres Lungenemphysem, Bronchiektasen, chronische obstruktive Bronchitis (COPD). Nur wenige Nichtraucher werden symptomatisch.
-
Mukoviszidose (zystische Fibrose): Generalisierte erbliche Sekretionsanomalie der exokrinen Drüsen, die an den Bronchien auch ohne Pankreasbeteiligung manifest werden kann. Es resultiert ein abnorm visköser Bronchialschleim, der die mukoziliare Clearance beeinträchtigt. Folgen: Sekretretention, Verstopfung der Bronchien, sekundäre Infektion, Schädigung der Bronchialwand, Bronchiektasen.
-
Mounier-Kuhn-Syndrom: Seltene kongenitale Tracheobronchomegalie bei Kindern und jungen Erwachsenen mit Erweiterung und abnormer Dehnbarkeit von Trachea und Bronchien infolge Atrophie des elastischen Gewebes und der glatten Muskulatur. Erschwerte Expektoration durch Kollapsneigung der Atemwege und gehäufte Infekte.
-
Genetische Defekte des Immunsystems: Verschiedene Formen des Immunglobulinmangels, X-gebundene Agammaglobulinämie (Burton), IgA-Mangel bedingen eine Abwehrschwäche gegen pyogene Bakterien.
2.5.6.4 Erworbene Faktoren
-
Obstruktion: Lokal durch Fremdkörper, entzündliche Stenosen, endobronchiale Tumoren oder Kompression von außen. Diffus bei chronischer obstruktiver Bronchitis, Asthma und bronchopulmonaler Aspergillose. Die Wandschädigung entsteht erst durch bakterielle Infektion des gestauten Sekrets.
-
Nekrotisierende Entzündung: Bronchiektasenbildung bei vorher gesunden Personen durch Tuberkulose, Staphylokokken- und andere eitrige Pneumonien sowie nekrotisierende Pneumonien als Komplikation bei Masern, Pertussis, Adenovirusinfektionen und Influenza.
-
Begleiterkrankung: Bronchiektasen können einer rheumatoiden Arthritis vorausgehen und sind manchmal mit entzündlichen Darmerkrankungen assoziiert.
2.5.6.5 Klinik
Chronischer Husten mit voluminösem eitrigem Sputum (Dreischichtung), Hämoptysen und rezidivierende Pneumonien. Bei 75 % der Patienten Dyspnoe, bei 50 % pleuritische Schmerzen. Im Initialstadium rezidivierende, auf Antibiotika ansprechende Infekte der unteren Luftwege mit symptomfreien Intervallen. Später Übergang in chronischen Husten. Oft begleitende Sinusitis. Die Schwere des Krankheitsbildes ist von der Ausdehnung der Bronchiektasen abhängig.
2.5.6.6 Diagnostik
2.5.6.7 Auskultation
An umschriebenen Stellen inspiratorische feuchte Nebengeräusche, bei Obstruktion exspiratorisches Giemen.
2.5.6.8 Röntgenaufnahme des Thorax
In den meisten Fällen unspezifische Veränderungen: Lineare oder Plattenatelektasen, verstreute Trübungen, umschriebene pneumonische Herde (◘ Abb. 2.22a).


Bronchiektasen. a Röntgenbild mit doppelseitigen Bronchiektasen und bronchopulmonischen Infiltraten. b CT-Bild mit Bronchiektasen
2.5.6.9 CT
Erlaubt die sichere Erkennung und Klassifizierung von Bronchiektasen (◘ Abb. 2.22b). Das Spiral-CT erfasst zusätzlich subtile Veränderungen. Damit ist die Bronchographie mit Kontrastmittel überflüssig geworden.
2.5.6.10 Lungenfunktion
FVC normal oder etwas herabgesetzt, FEV1 vermindert, nicht immer durch inhalative Bronchodilatoren zu verbessern.
2.5.6.11 Laboruntersuchungen
Bestimmung der Immunglobuline im Serum. Blutbild (Eosinophilie?), BKS und Serumelektropherogramm zur Beurteilung der entzündlichen Aktivität.
2.5.6.12 Therapie
2.5.6.13 Exazerbationen
Symptome sind Zunahme der Sputummenge mit stärkerer Gelbfärbung sowie Zunahme von Dyspnoe und Husten, Fieber. Die häufigsten Keime sind H. influenzae, Pseudomonas aeruginosa, Branhamella catarrhalis. Antibiotika der Wahl sind die Fluorochinolone (Ciprofloxacin, Levofloxacin, Moxifloxacin). Sie müssen bis zu 10 Tagen gegeben werden. In zweiter Linie kommen Makrolide (Azithromycin) in Betracht.
2.5.6.14 Langzeitbehandlung
Die entzündliche Überaktivität kann durch inhalative Glukokortikoide gedämpft werden. Bei obstruktiver Symptomatik sind inhalative β-Adrenergika indiziert.
Bei Hämoptysen Seitenlagerung auf die vermutlich blutende Seite, evtl. bronchoskopische Versorgung. Weitere Maßnahmen: Drainage durch Hängelage und Krankengymnastik. Ausreichende Hydratation. Expektorantien von fraglichem Nutzen. In schweren Fällen fiberbronchoskopische Absaugung. Sauerstoffinhalation bei Dyspnoe. Grippeschutzimpfung.
2.5.6.15 Operative Maßnahmen
Segment- oder Lappenresektion bei lokalisierten Bronchiektasen, die auf medikamentöse Behandlung ungenügend ansprechen und oft zu Pneumonien und Blutungen führen. Ultima ratio Lungentransplantation.
2.5.7 Bronchialkarzinom
2.5.7.1 Definition
Maligne Tumoren der Lunge, die vom respiratorischen Epithel (Bronchien, Bronchiolen Alveolen) ausgehen.
2.5.7.2 Epidemiologie
Nach Auskunft des Statistischen Bundesamtes sind im Jahr 2000 in der Bundesrepublik 38955 Personen an bösartigen Neubildungen der Bronchien und der Lunge verstorben, darunter 29121 männlichen und 9834 weiblichen Geschlechts. Das sind 26,56 % aller männlichen und 9,73 % aller weiblichen Todesfälle an bösartigen Neubildungen. Der hohe Anteil des männlichen Geschlechts resultiert aus den unterschiedlichen Rauchgewohnheiten.
2.5.7.3 Ätiologie
Dominierender Risikofaktor ist das Zigarettenrauchen, wahrscheinlich durch Inhalation von Karzinogenen (z. B. Benzpyren). Zwischen konsumierter Zigarettenmenge und Lungenkrebssterblichkeit besteht eine quantitative Beziehung. Wer 20 Jahre lang täglich 40 Zigaretten raucht, hat gegenüber einem Nichtraucher ein 60- bis 70-faches Risiko, am Bronchialkarzinom zu sterben. Bei Exrauchern nimmt das Risiko kontinuierlich ab, ist aber nach 15 Jahren noch doppelt so hoch wie das der Nichtraucher.
Wesentlich seltenere Ursachen für Lungenkarzinome sind Expositionen mit Asbest, Radon, polyzyklischen Kohlenwasserstoffen, Nickel, Chromaten und anorganischen Arsenverbindungen.
2.5.7.4 Pathogenese
Bronchialkarzinome sind monoklonal, d. h. sie entstehen durch Proliferation einer einzelnen Tumorzelle, die durch mehrere somatische Mutationsschritte aus einer normalen Zelle hervorgegangen ist. Molekulargenetische Untersuchungen haben gezeigt, dass eine Tumorzelle im Genom ≤10 Läsionen aufweist. Es handelt sich um aktivierte dominante Onkogene oder inaktivierte Tumorsuppressorgene (rezessive Onkogene). Erstere sind wirksam, wenn nur ein Allel transformiert ist (durch Punktmutationen der K-ras-Genregion oder Amplifikation, Rearrangement und Verlust der transkriptionalen Kontrolle an den Onkogenen der myc-Familie (c-, N- und l-myc). Letztere erlangen erst Wirksamkeit, wenn die Inaktivierung oder ein Defekt an beiden Allelen aufgetreten ist (Allelverluste an mehreren Chromsomen, am häufigsten an 3p, Mutationen der Gene p53 und rb). Eine genetische Disposition zum Bronchialkarzinom kann sich ergeben, wenn ein Allel des Tumorsuppressorgens bereits in der Keimbahn mutiert ist. Viele dominante Onkogene leiten sich von Genen ab, die an der Wachstumsregulation beteiligt sind. Tumorsuppressorgene regulieren den Zellzyklus.
Zellen von Bronchialkarzinomen können Peptidhormone produzieren und Rezeptoren für diese Hormone exprimieren, so dass ein autokrin stimuliertes Zellwachstum zustande kommt. Krebszellen aller histologischen Typen exprimieren Rezeptoren für Nikotin. Die Nikotinbindung hemmt die Apoptose.
2.5.7.5 Klassifikation
Bronchialkarzinome entstehen anscheinend in pluripotenten Stammzellen des Bronchialepithels. Das würde ihre histologische Vielfalt erklären. Die WHO-Klassifikation unterscheidet folgende 4 Haupttypen der pulmonalen Neoplasien:
-
a)
Plattenepithelkarzinom (29 %)
-
b)
kleinzelliges Karzinom (28 %)
-
c)
Adenokarzinom (32 %) inkl. bronchoalveoläres Karzinom (3 %)
-
d)
großzelliges Karzinom
Den Rest bilden undifferenzierte Karzinome, Karzinoide, Tumoren der Bronchialdrüsen und seltene Tumortypen.
Unter therapeutischen Gesichtspunkten gruppiert man die Bronchialkarzinome in nichtkleinzellige (a, c, d) und kleinzellige (b).
Plattenepithelkarzinome entwickeln sich gewöhnlich in proximalen Segmentbronchien, wachsen langsam und metastasieren spät. Adenokarzinome haben oft einen peripheren Ursprung, doch selbst bei kleinen resezierbaren Tumoren besteht das Risiko okkulter Metastasen. Das bronchoalveoläre oder Alveolarzellkarzinom nimmt eine Sonderstellung ein (s. unten).
Kleinzellige Karzinome sind in >90 % der Fälle schon bei der Erkennung metastasiert. Sie entstehen meistens in den großen Atemwegen. Selten werden sie als kleine periphere Knoten angetroffen, die anscheinend weniger bösartig sind.
2.5.7.6 Lokalisation und Ausbreitung
-
Hilärer Tumor: Endobronchiales Wachstum bis zur Stenose mit lokalem Emphysem und zum Verschluss mit Atelektase, Retentionspneumonie und Abszess.
-
Rundherd: Mit Zerfall zur Tumorkaverne (Hämoptyse). Drainage durch Bronchus oder Einbruch in den Pleuraraum mit Erguss bzw. Empyem.
-
Ausbrechertumor an der Lungenspitze: Mit Einwachsen in den Plexus brachialis und die Gefäße (Pancoast-Syndrom ).
-
Ausbrechertumor ins obere Mediastinum: Mit Verlegung der V. cava superior (obere Einflussstauung, Stokes-Kragen).
-
Lymphogene Ausbreitung: Lymphknotenmetastasen (Bifurkation, axillar, supraklavikulär).
-
Hämatogene Ausbreitung: Fernmetastasen in allen Organen via V. pulmonalis (Knochen, Gehirn, Leber etc.).
2.5.7.7 Klinik
2.5.7.8 Screening
Bei starken Rauchern wurden mehrere Studien zur Früherfassung des Bronchialkarzinoms durchgeführt, meistens mit Röntgen- oder CT-Untersuchung des Thorax plus Sputumuntersuchung auf Tumorzellen alle 4 Monate, bei den Kontrollgruppen im jährlichen Abstand. Ergebnisse: Bei den häufig kontrollierten Probanden wurden mehr asymptomatische Frühstadien mit resezierbaren Tumoren erfasst, aber immer noch viele fortgeschrittene Tumoren (◘ Abb. 2.23). In der ersten Gruppe war die 5-Jahresüberlebensrate höher, die Gesamtmortalität war jedoch in beiden Gruppen gleich.


54-jähriger Raucher mit Adenokarzinom im rechten Unterlappen. a Röntgenbefund, b Computertomogramm
2.5.7.9 Lokale Symptome
Husten, oft als Raucherkatarrh verharmlost, „grippale Infekte“, Hämoptysen (◘ Abb. 2.24), lokale Bronchospastik, Dyspnoe, umschriebene pneumonische Prozesse mit Fieber und produktivem Husten. Brustschmerz bei Ausbreitung auf Pleura und Rippen, Pancoast-Syndrom (neuritische Schmerzen, vasomotorische Krisen an Hand und Arm), Heiserkeit durch Rekurrensparese, Horner-Symptomenkomplex (Sympathikuslähmung), Dysphagie durch Ösophaguskompression, Zwerchfellhochstand durch Phrenikusparese, Perikarditis, Herztamponade, finale respiratorische Insuffizienz.


Kleines Plattenepithelkarzinom bei einer 51-jährigen Frau. Die Indikation zur Bronchoskopie waren rezidivierende Hämoptysen
2.5.7.10 Metastatische Fernsymptome
Hirnmetastasen mit neurologischen Ausfallserscheinungen, Knochenmetastasen mit Schmerzen und Frakturen, Rückenmarkkompression, Knochenmarkinvasion mit Leukopenie oder Leukoerythroblastose, Lebermetastasen, Verschlussikterus, Lymphknotenmetastasen.
2.5.7.11 Paraneoplastische Syndrome
Können als initiale Erscheinungen auftreten und klinisch im Vordergrund stehen. Bei Kleinzellkarzinomen Cushing-Syndrom (ektopische ACTH-Bildung mit extremer Hypokaliämie), inadäquate ADH-Bildung mit Hyponatriämie und Eaton-Lambert-Syndrom (Myasthenie des Beckengürtels). Bei Plattenepithelkarzinomen ektopische Parathormonbildung mit Hyperkalzämie und Hypophosphatämie. Bei Adenokarzinomen Trommelschlegelfinger und Periostitis. Bei allen Formen Polymyositis, Dermatomyositis, Thrombophlebitis migrans, Endokarditis, disseminierte intravaskuläre Gerinnung, nephrotisches Syndrom.
2.5.7.12 Diagnostik
2.5.7.13 Tumorerfassung
Erfolgt primär gewöhnlich durch konventionelle Röntgenaufnahmen der Lunge. Eine optimale Darstellung des Tumors und seiner Ausbreitung ermöglicht die Computertomographie (◘ Abb. 2.25 und ◘ Abb. 2.26).


Bronchialkarzinom im linken Oberlappen mit Vergrößerung der linksseitigen Hiluslymphknoten (Sammlung Prof. Jend)


Tumor im linken Oberlappen bei einem 62-jährigen Mann. Histologisch ein Plattenepithelkarzinom. Im Staging Nachweis von Fernmetastasen
2.5.7.14 Histologische Sicherung und Klassifizierung
Materialgewinnung zur bioptischen Untersuchung je nach Lokalisation des Tumors durch Bronchoskopie, transbronchiale Biopsie, Mediastinoskopie oder transkutane Lungen- bzw. Pleurabiopsie, bei Rundherden durch Thorakotomie (◘ Abb. 2.27). Aus therapeutischen und prognostischen Gründen ist die Hauptsache eine Unterscheidung von kleinzelligen und nicht-kleinzelligen Karzinomen. Zytologische Präparate reichen dazu meistens nicht aus.


CT-gesteuerte Punktion einer peripheren Raumforderung links. Im histologischen Befund ergibt sich ein Adenokarzinom
2.5.7.15 Untersuchungen zur Stadieneinteilung
Erfassung der anatomischen Ausbreitung durch Röntgenaufnahme des Thorax in 2 Ebenen, ergänzt durch Computertomogramm zur Beurteilung von Pleura- und Mediastinumbefall. Bei positivem oder fraglichem Mediastinumbefund histologische Lymphknotenuntersuchung durch Mediastinoskopie. Erfassung extrathorakaler Absiedlungen des Tumors durch Hirn,- Leber- und Knochenszintigraphie, auch Sonographie der Leber, evtl. Laparoskopie. Bei Kleinzellkarzinomen Knochenmarkbiopsie (positiv bei 20–30 %). Röntgenuntersuchung des Ösophagus bei Kandidaten für eine Operation.
2.5.7.16 Stadieneinteilung (TNM-Staging)
Die Ausbreitungsdiagnostik (◘ Tab. 2.4) von Bronchialkarzinomen ist im Rahmen des Staging (◘ Tab. 2.5) für die Festlegung der Therapiestrategie von entscheidender Bedeutung.
Bei den kleinzelligen Bronchialkarzinomen werden nur 2 Stadien unterschieden, da die große Mehrzahl der Patienten initial chemotherapeutisch behandelt wird (◘ Tab. 2.6).
2.5.7.17 Therapie des nichtkleinzelligen Bronchialkarzinom
Potenziell kurativ sind Resektion und Strahlentherapie. Letztere wird auch als palliative Maßnahme eingesetzt. Die Chemotherapie ist nur palliativ wirksam, kann aber zu länger dauernden Remissionen führen.
2.5.7.18 Operative Therapie
Indiziert in den Stadien I und II, bei gutem Zustand der Patienten auch im Stadium IIIa. Der Tumor wird durch Lobektomie , Bilobektomie oder Pneumektomie unter Mitnahme der regionalen Lymphknoten reseziert. Wenn kein signifikanter mediastinaler Lymphknotenbefall vorliegt, werden auch mitbetroffene Teile von Thoraxwand, Perikard und Zwerchfell entfernt.
Jedem Eingriff geht eine gründliche Allgemeinuntersuchung voraus unter Einschluss von EKG und umfassender Lungenfunktionsprüfung. Die Operationsmortalität sollte bei Lobektomie unter 3 %, bei Pneumektomie unter 5 % liegen. 5 Jahresüberlebensraten: Stadium I 60–80 %, Stadium II 25–40 %, Stadium IIIa 10–30 % und Stadium IIIb <5 %. Solitäre Rundherde ohne Verkalkung sind bei Rauchern über 35 Jahre dringend karzinomverdächtig und deshalb zu resezieren. Bei Nichtrauchern unter 35 Jahren ist die Karzinomwahrscheinlickeit unter 1 % und ein abwartendes Verhalten unter regelmäßigen Kontrollen gerechtfertigt.
2.5.7.19 Strahlentherapie
Mit kurativer Intention indiziert im Stadium I–II, wenn die Operation verweigert oder wegen des Allgemeinzustandes nicht zumutbar ist, und im Stadium III. Die Standarddosis beträgt 60 Gy in 30 Dosen über 6 Wochen. Einen 9%igen Überlebensvorteil nach 2 Jahren hat die CHART-Methode (continuous hyperfractionated accelerated radiation therapy) mit 36 Bestrahlungen zu 1,5 Gy 3 × tgl. an 12 aufeinander folgenden Tagen (Gesamtdosis 54 Gy). Die mittlere Überlebenszeit von bestrahlten nicht operablen Patienten ist <1 Jahr, aber 6 % leben noch nach 5 Jahren. Durch nachfolgende Chemotherapie wird die Lebenserwartung etwas verbessert. Palliativbestrahlungen können gegen Brustschmerz, Hämoptysen, Dyspnoe und Husten hilfreich sein.
2.5.7.20 Chemotherapie
Auf die moderne kombinierte Chemotherapie sprechen 30–40 % der inoperablen Patienten mit fortgeschrittenem Bronchialkarzinom an, <5 % mit kompletten Remissionen. Die mittlere Überlebenszeit beträgt 9–10 Monate, die 1-Jahresüberlebensrate 40 %. Ökononische Analysen haben gezeigt, dass die Chemotherapie kosteneffektiv ist. Zum Einsatz kommen Kombinationen von Cisplatin oder Carboplatin mit Vinorelbin, Gemcitabin, Paclitacel, Docetaxel oder Topotpecan.
2.5.7.21 Therapie des kleinzelliges Bronchialkarzinom
Kleinzellige Bronchialkarzinome sind zum Zeitpunkt der Erkennung fast immer disseminiert und inoperabel. Sie sind jedoch strahlenempfindlich und sprechen auf Zytostatika an.
2.5.7.22 Chemotherapie
Die Ansprechrate ist >80 % mit kompletter Remission von 30–40 % im limitierten und 10–20 % im extensiven Stadium. Die mittlere Lebenserwartung der Patienten mit kleinzelligem Karzinom steigt durch die Chemotherapie von 6–17 auf 40–70 Wochen. Häufigstes Therapieschema: 4–6 Zyklen im Abstand von 3 Wochen mit Etoposid plus Cisplatin oder Carboplatin. Danach Wiederholung des Staging. Bei ungenügendem Ansprechen Übergang auf neue Kombinationen wie beim nichtkleinzelligen Karzinom beschrieben (▶ oben).
2.5.7.23 Strahlentherapie
Bei nachgewiesenen Gehirnmetastasen hochdosierte Bestrahlung (40 Gy). Prophylaktische Gehirnbestrahlung (24 Gy) nach Chemotherapie bei vollständiger Remission, da sonst innerhalb von 2 Jahren oft Hirnmetastasen auftreten können.
2.5.7.24 Kombinationstherapie
Im limitierten Stadium verbessert die Kombination von Chemotherapie und Bestrahlung des Lungenherdes die Lebenserwartung. Sie kann auch im extensiven Stadium indiziert sein. Geheilt werden 20–30 % der Patienten mit limitiertem Stadium und 1–5 % mit extensivem Stadium des Bronchialkarzinoms.
2.5.8 Bronchoalveoläres Karzinom
Seltene Lungengeschwulst (3 % aller pulmonalen Malignome), dessen Ausgangspunkt umstritten ist. Wahrscheinlich sind es alveolare Pneumozyten vom Typ II. Der Tumor wächst relativ langsam, in mehreren kubischen oder zylindrischen Zellschichten entlang der Alveolar- und Bronchiolarwand und scheint sich auf luminalem Wege in der Lunge auszubreiten. Die Größeren Luftwege bleiben offen. Auch das Interstitium, die Hiluslymphknoten und die Thoraxwand werden selten bzw. spät befallen. Geringe hämatogene Metastasierungstendenz. Tumorzellnachweis im Sputum schon 2 Jahre vor Röntgenbildveränderungen möglich.
2.5.8.1 Klinik
Keine vorausgehende oder begleitende obstruktive Raucherbronchitis wie beim Bronchialkarzinom, da keine Assoziation mit dem Rauchen besteht. Männer und Frauen erkranken im Verhältnis 1:1. In 20 % der Fälle sehr große Mengen schleimigen Sputums, das sekundär infiziert sein kann. Lungenfunktion durch Diffusionsstörung (Hypoxie) und restriktive Ventilationsstörung beeinträchtigt.
2.5.8.2 Diagnostik
2.5.8.3 Röntgenbild
Solitärer Rundherd oder multiple Fleckschatten in einer oder beiden Lungen. Gelegentlich konfluierende Verdichtung eines Lappens wie bei Lobärpneumonie.
2.5.8.4 Bronchoskopie
Materialgewinnung für zytologische Untersuchung.
2.5.8.5 Mediastinoskopie
Bei Hilusmetastasen.
2.5.8.6 Transbronchiale oder offene Lungenbiopsie
Zur Sicherung der Diagnose.
2.5.8.7 Therapie
Resektion solitärer Herde. Bestrahlung und Zytostatika sind unwirksam.
2.5.9 Lungenmetastasen
Sekundäre Geschwülste der Lunge sind häufig und kommen bei den verschiedensten Primärgeschwülsten vor (◘ Abb. 2.28). Die Absiedlung kann hämatogen, lymphogen oder durch infiltratives Wachstum erfolgen. Die Lymphangitis carcinomatosa mit diffusen Verschattungen in beiden Lungenfeldern mit und ohne Hilusbeteiligung kann vor Erkennung des Primärtumors (Magen, Mammae, Pankreas, Prostata) mit zunehmender Dyspnoe in Erscheinung treten. Es dominieren solitäre und multiple Rundherde. Diagnostisches Vorgehen bei solitären Rundherden wie beim Bronchialkarzinom. Nach Ausschaltung des Primärtumors und weiterer Metastasen werden einzelne Lungenmetastasen mit deutlicher Verlängerung der Lebenserwartung reseziert. Sehr gute Ergebnisse bei osteogenen Sarkomen.


Multiple Lungenmetastasen eines Prostatakarzinoms (Sammlung Prof. Jend)
2.5.10 Semimaligne und benigne Bronchialtumoren
2.5.10.1 Bronchialadenom e
2.5.10.1.1 Definition
Sammelbezeichnung für histologisch differente, vom Epithel ausgehende intrabronchiale Tumoren, die sich invasiv ausbreiten und metastasieren können und daher als potenziell maligne einzuordnen sind.
2.5.10.1.2 Klassifikation
-
Karzinoide (80–90 %): Das sind von endokrinen Zellen (APUD-System) der Bronchialschleimhaut ausgehende Bronchialadenome, die am häufigsten vorkommen. Produzieren gelegentlich ACTH oder ADH mit paraneoplastischem Syndrom, nach Metastasierung (in die Leber) auch ein typisches Karzinoid-Syndrom (▶ Kap. 4.4, Abschn. Dünndarmtumoren).
-
Adenozystische Tumoren (15 %): Auch als Zylindrome bezeichnet. Endobronchial wachsend, meistens zentral lokalisiert, sehr gefäßreich, daher bei Biopsie stark blutend.
-
Mukoepidermoide Tumoren (2–3 %): Die seltenste Form der Bronchialadenome, von den mukösen Drüsen ausgehend. Lokal destruktives Wachstum, selten Metastasen.
2.5.10.1.3 Klinik
Chronischer Husten, rezidivierende Hämoptysen, bei Obstruktion Pneumonitis und Atelektasen. Selten Cushing-Syndrom oder inadäquate ADH-Sekretion.
2.5.10.1.4 Diagnostik
Bronchoskopie und Biopsie, bei Rundherden histologisch nach der Resektion. Im Frühstadium kann das Röntgenbild unauffällig sein.
2.5.10.1.5 Therapie
In der Regel Resektion des befallenen Segmentes oder Lappens. Gelegentlich genügt eine Bronchotomie mit lokaler Exzision.
2.5.10.2 Benigne Bronchialtumoren
2.5.10.2.1 Definition
Gutartig wachsende Tumoren, die vom mesenchymalen Gewebe der Bronchien ausgehen.
2.5.10.2.2 Klassifikation
Chondrome, Hamartome, Osteome, Fibrome, Histiozytome, Lipome, Hämangiome, Neurofibrome.
2.5.10.2.3 Klinik
Die Tumoren entwickeln sich überwiegend in den zentralen Bronchien. Sie führen zu Stridor und Dyspnoe, Hustenanfällen und Blutungen. Nachweis durch Röntgenuntersuchung und Bronchoskopie mit Biopsie.
2.5.10.2.4 Therapie
Resektion oder endobronchiale Abtragung (Lasertechnik).
2.6 Erkrankungen der Lunge
Erkrankungen der Lunge
-
Pneumonien
-
Interstitielle Lungenkrankheiten
-
Idiopathische Lungenfibrose (UIP: usual interstitial pneumonia)
-
Iatrogene Lungenfibrosen
-
Strahlenschäden
-
Strahlenpneumonitis
-
Strahlenfibrose
-
-
Medikamenteninduzierte Lungenfibrose
-
Toxische Inhalationsschäden
-
Exogen-allergische Alveolitis
-
Eosinophile Pneumonien
-
Allergische bronchopulmonale Aspergillose
-
Parasitär bedingte eosinophile Pneumonie (Löffler-Syndrom)
-
Medikamentös induzierte eosinophile Pneumonie
-
Chronische eosinophile Pneumonie
-
Churg-Strauss-Syndrom (allergische Angiitis und Granulomatose
-
-
Goodpasture-Syndrom
-
Pneumokoniosen
-
Silikose
-
Anthrakosilikose
-
Asbestose
-
Andere Silikatosen
-
Berylliose
-
-
Sarkoidose
-
Tuberkulose
-
Lungentuberkulose
-
Extrapulmonale Tuberkulose
-
Miliartuberkulose
-
2.6.1 Pneumonie n
2.6.1.1 Definition
Durch akute Infektion verursachte Entzündung im Lungenparenchym, dem Abschnitt distal der terminalen Bronchiolen.
2.6.1.2 Klassifikation
Frühere Einteilungen der Pneumonien basierten auf dem Befallsmuster der Lunge oder auf den mikrobiologischen Befunden. Neuerdings hat sich nachstehende Klassifikation durchgesetzt, die Unterschiede im zu erwartenden Erregerspektrum widerspiegelt:
-
CAP: Community-acquired pneumonia/ambulant erworbene Pneumonie
-
HCAP: Health care-associated pneumonia/mit Plegeeinrichtungen assoziierte Pneumonie
-
HAP: Hospital-acquired pneumonia/nosokomiale Pneumonie
-
VAP: Ventilator-associated pneumonia/beatmungsassoziierte Pneumonie
-
2.6.1.3 Pathogenese
Eindringen des Erregers in den Alveolarraum. In den meisten Fällen geschieht dies durch Aspiration kontaminierter Tröpfchen aus dem Oropharynx. Auf dem Weg in die unteren Lungenbezirke müssen Mikroorganismen diverse Defensivmechanismen überwinden. Zu ihnen gehören der für Partikel über 10 µm sehr effektive Nasenfilter (Haare, Zilien, Schleim, Niesreflex), der Glottisschließ- und Hustenreflex und der mukoziliare Apparat des Bronchialbaumes. Gegen Mikroorganismen, die in die normalerweise sterilen unteren Lungenbezirke eingedrungen sind, wird das angeborene Immunsystem aktiv (▶ Kap. 8). Seine Komponenten sind Makrophagen (90 %) und Lymphozyten (10 %, überwiegend T-Zellen) im Alveolarraum, die Barriere des gut abgedichteten, bakterizide Substanzen bildenden respiratorischen Epithels und das subepitheliale Bindegewebe mit Neutrophilen, Lymphozyten und Gewebsmakrophagen als Abwehrzellen. Es wird unterstützt vom adaptiven Immunsystem, das Plasmazellen bereitstellt, die erregerspezifische Immunglobuline (IgA, IgG) sezernieren.
Unter folgenden Bedingungen ist das Angehen einer Pneumonie begünstigt:
-
Bewusstseinsstörungen: Alkoholrausch, Hirntraumen, zerebrale Anfälle oder Insulte, Schlafmittelvergiftungen, Narkosen (Aspiration bei gestörtem Husten- und Glottisreflex).
-
Thorakale Affektionen: Schmerzzustände nach Brustkorboperationen, schwere Kyphoskoliose, obstruktive Lungenkrankheiten, neuromuskuläre Atemstörungen, Intubation (Störung des Abhustens bzw. Wegfall des Nasenfilters).
-
Funktionsstörungen des mukoziliaren Apparates: Rauchen, Luftverunreinigungen, toxische Gase, chronische Bronchitis, Pertussis, Kartagener-Syndrom, Intubation, Alkoholismus.
-
Respiratorische Virusinfekte: Durch Desquamation und Schädigung des respiratorischen Epithels.
-
Atelektasen: Bronchialverschluss durch Tumor oder Fremdkörper, Narkose, Trauma (Sekretretention).
-
Flüssigkeitsansammlung in den Alveolen: Linksinsuffizienz, Lungenödem, Verlegung des Lymphabflusses durch Tumoren.
-
Störungen der Phagozytose: Neutropenie, Influenza, Asplenie, Schädigung der Alveolarmakrophagen durch Rauchen, Hypoxie, Hunger, Anämie.
-
Affektionen des Immunsystems: Immunsuppressive Therapie, Glukokortikoide, kongenitale und erworbene Agammaglobulinämie, AIDS, Komplementmangel, Sichelzellanämie, Myelom, hohes Alter, Diabetes mellitus.
2.6.1.4 Entzündungsreaktion im Lungenparenchym
Sie ist das Ergebnis der Erregerabwehr des Körpers. Wenn es den alveolaren Makrophagen nicht gelingt, die eingedrungenen Erreger zu eliminieren, werden sie durch T-Zellen zur massiven Freisetzung proinflammatorischer Zytokine (IL-1, TNFα) stimuliert, die zur lokalen Entzündung und zu den Allgemeinerscheinungen der Pneumonie führen. Die geschädigte alveoläre Kapillarmembran lässt Exsudat und anfangs auch Erythrozyten in den Alveolarraum austreten. Das Kapillarendothel exprimiert Chemokine, gefolgt von starker Neutrophilenansammlung, die den Entzündungsprozess noch verstärkt. Die Flüssigkeitsansammlung in der Lunge kann zur Hyperventilation mit respiratorischer Alkalose, zur schweren Hypoxämie und zur respiratorischen Insuffizienz führen.
2.6.1.5 Pneumonieerreger
Viele gehören dauernd oder zeitweise zur normalen oropharyngealen Flora und bleiben unschädlich, solange keine Aspiration und Haftung in den unteren Lungenabschnitten erfolgt. Andere werden von außen durch Inhalation aufgenommen und werden bei hoher Virulenz oder ungenügender Abwehr pathogen.
-
Erreger ambulant erworbener Pneumonien: Streptococcus pneumoniae, Mycoplasma pneumoniae, Haemophilus influenzae, Chlamydia pneumoniae, Chlamydia psittaci, Legionella pneumophila, orale Anaerobier, Moraxella catarrhalis, Staphylococcus aureus, Nocardia, Viren (Influenza-, Zytomegalie-, Respiratory-Syncytial-, Masern-, Varizellen-Zoster- und Hanta-Viren), Pilze (Histoplasma, Coccidioides, Blastomyces), Mycobacterium tuberculosis.
-
Erreger im Hospital erworbener (nosokomialer) Pneumonien: Aerobe gramnegative Bakterien (Klebsiella pneumoniae, der Erreger der Friedländerpneumonie, E. coli, Citrobacter u. a.), Pseudomonas aeruginosa, Staphylokoccus aureus, orale Anaerobier.
-
Erreger HIV-assoziierter Pneumonien: Pneumocystis carinii, M. tuberculosis, H. influenzae.
2.6.1.6 Pathologie
Nach Lokalisation und Ausdehnung unterscheidet man Lobärpneumonie n, die alle Alveolen eines Lungenlappens befallen und Bronchopneumonien , die in der Nachbarschaft von Bronchien lokalisiert sind, aber konfluieren können. Durch entzündliches Exsudat fallen die entzündeten Alveolen für den Gasaustausch aus. Es kann zu Einschmelzungen und Abszedierungen kommen. Bei einigen Viruspneumonien ist vorwiegend das Interstitium betroffen. Die pathologisch-anatomische Klassifizierung ist klinisch von geringer Relevanz, da sie keinen Rückschluss auf die Ätiologie zulässt.
2.6.1.7 Ambulant erworbene Pneumonie
2.6.1.7.1 Epidemiologie
In den USA kommen jährlich rund 4 Millionen Fälle von CAP vor. Davon werden 20 % ins Krankenhaus aufgenommen. Man rechnet mit einer Inzidenz von 12 Fällen auf 1000 Personen und bei den über 60-Jährigen mit einer Rate von 20/1000. Gefährdet sind Personen mit den oben erwähnten Risiken. Pneumonien nehmen in der Todesursachenstatistik den 6. Rang ein und verursachen von allen Infektionskrankheiten die meisten Todesfälle. Da keine Meldepflicht besteht, ist die Prävalenz nicht genau bekannt. Risikofaktoren sind Rauchen, Alkoholismus, chronische obstruktive Lungenkrankheit, Hypertonie und Diabetes mellitus. Besonders hoch sind Morbidität und Mortalität der nosokomialen Pneumonie auf Intensivstationen.
2.6.1.7.2 Ätiologie
Das Erregerspektrum ist breit. In Deutschland wurden folgende Anteile ermittelt: Streptococcus pneumoniae 35–50 %, Haemophilus (vor allem bei Rauchern) 20 %, Mykoplasmen 12–13 %, Viren (meist Influenza) bis zu 15 %, Chlamydophila pneumoniae <1 %. Mit Anaerobiern ist zu rechnen, wenn der Pneumonie eine Aspiration vorausging. Bei der Influenza-Pneumonie kommt eine Sekundärinfektion mit S. aureus vor. Selten wird eine CAP durch MRSA (Methicillin-resistenten S. aureus) verursacht.
2.6.1.7.3 Klinik
In der Regel starkes Krankheitsgefühl und große Körperschwäche. Die unterschiedliche Ausprägung der folgenden Hauptsymptome erlaubt differenzialdiagnostische Schlüsse:
-
Prodromi: Vorausgehender Infekt der oberen Luftwege bei Pneumokokkenpneumonie und anderen bakteriellen Pneumonien. Bei Mykoplasmen- und Viruspneumonien treten Symptome des oberen Respirationstraktes und Pneumonie gleichzei-tig auf.
-
Fieber: Plötzlicher Temperaturanstieg mit Schüttelfrost bei Pneumokokkenpneumonie und anderen grampositiven Pneumonien, ohne Schüttelfrost bei den übrigen Erregern. Bei alten und hinfälligen Patienten kann Fieber fehlen.
-
Husten: Meistens von großer Heftigkeit, oft mit retrosternalem Schmerz einhergehend. Kurze Dauer des Hustens spricht für bakterielle, virale oder Mykoplasmenpneumonie, Husten von mehrwöchiger Dauer für chronische Bronchitis mit Superinfektion, Pilzpneumonie oder Tuberkulose.
-
Auswurf: Eitrig, bei starkem Husten (infolge Ruptur kleiner Gefäße des Tracheobronchialbaums) oft blutig tingiert. Bei Pneumokokkenpneumonie zuerst grün-gelblich, im Lysestadium rostbraun (Erythrozytenaustritt in die Alveolen). Hämorrhagisch bei Staphylokokken- und Streptokokkenpneumonie und Tuberkulose (wegen nekrotisierender Wirkung der Erreger auf das Lungengewebe). Unproduktiver Husten bei Legionellen-, Mykoplasmen- und Grippepneumonie. Hier eitriges Sputum erst durch bakterielle Sekundärinfektion. Fötides Sputum bei Anaerobierinfektionen und Bronchiektasen.
-
Pleuritischer Brustschmerz: Mit atemabhängiger Intensitätszunahme bei Begleitpleuritis bakterieller Pneumonien häufig, seltener bei Mykoplasmen- und Virusinfektionen. Abdominalschmerz bei Reizung der Pleura diaphragmatica.
-
Dyspnoe: Abhängig vom Ausmaß des Lungenbefalls, besonders ausgeprägt bei lobärer Pneumokokkenpneumonie.
-
Weitere Symptome: Übelkeit, Erbrechen, Kopf- und Gliederschmerzen, Verwirrtheit, Benommenheit.
2.6.1.7.4 Diagnostik
2.6.1.7.5 Körperlicher Untersuchungsbefund
Die meisten Patienten machen einen schwerkranken Gesamteindruck und sind bettlägerig. Dem Fieber entsprechende Tachykardie, bei Kreislaufschwäche Hypotonie und Blässe. Tachypnoe, in schweren Fällen Nasenflügelatmen und Zyanose. Über den betroffenen Lungenabschnitten fein- bis mittelblasige klingende Rasselgeräusche, bei konfluierenden Prozessen Bronchialatmen, Bronchophonie, verstärkter Stimmfremitus und gedämpfter Klopfschall. Die erkrankte Seite kann bei der Atmung nachschleppen. Bei Pleuritis in- und exspiratorische Reibegeräusche, bei Pleuraerguss bzw. Empyem Dämpfung des Klopfschalls, Abschwächung bis Aufhebung des Atemgeräusches und des Stimmfremitus. Bei manchen Patienten, insbesondere bei Mykoplasmen- und Viruspneumonie, trotz ausgedehnter Veränderungen im Röntgenbild keine abnormen Befunde der physikalischen Untersuchung. Bei hämatogen entstandenen Pneumonien kann der Sitz der primären Infektion erkennbar sein (Gastrointestinaltrakt, Urogenitaltrakt etc.). Von einer Pneumonie ausgehende Bakteriämien können zu septischen Meningitiden, Arthritiden und pustulösen Hautveränderungen führen.
2.6.1.7.6 Röntgendiagnostik
Thoraxaufnahmen in zwei Ebenen sind die wichtigste Maßnahme zur Sicherung der Diagnose. Auf den Röntgenbildern können 3 Formen der Pneumonie unterschieden werden:
-
Alveoläre Pneumonie (Lobärpneumonie): Kompakte Verdichtungen, die meistens einen ganzen Lappen einnehmen und die Bronchien freilassen (Luftbronchogramm). Beginn der entzündlichen Exsudation in ein oder zwei peripheren Foci; von dort Ausbreitung des Exsudats über die interalveolären Kohn-Poren auf alle Alveolen eines Lappens. Typisch für Pneumokokken und Klebsiellenpneumonie (◘ Abb. 2.29 und ◘ Abb. 2.30).
Abb. 2.29 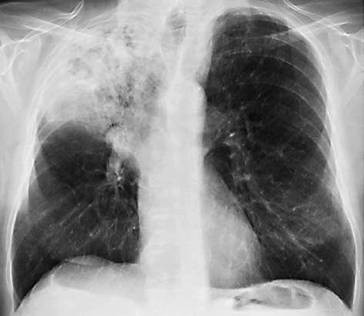
48-jähriger Patient mit Lobärpneumonie im rechten Oberlappen. In der Sputumanalyse Nachweis von Pneumokokken
Abb. 2.30 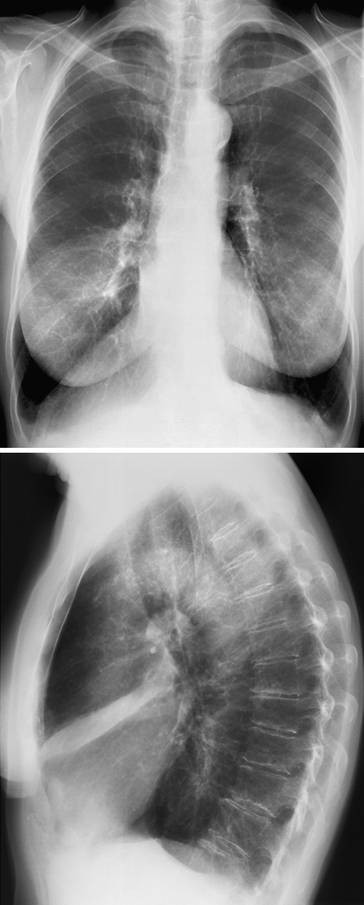
48-jährige Frau mit Mittellappenpneumonie
-
Bronchopneumonie: Multiple herdförmige Verdichtungen, die von peribronchialen Infiltraten ausgehen und wegen des primären Befalls der Bronchien segmental begrenzt bleiben und oftmals zur Atelektase führen. Häufigstes Bild bei Staphylokokkenpneumonie (◘ Abb. 2.31), Hämophilus-influenzae-Pneumonie, gramnegativen Pneumonien, und sekundärer Pneumonie bei chronischer obstruktiver Bronchitis.
Abb. 2.31 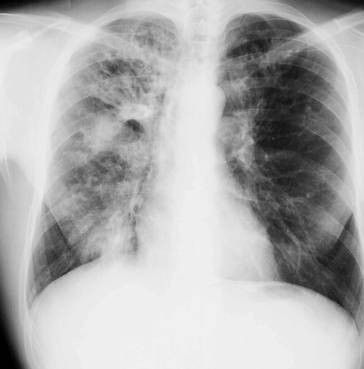
Abszedierende Staphylokokkenpneumonie bei einem 50-jährigen Mann
-
Interstitielle Pneumonie: Diffuse feinfleckige (noduläre), netzartige (retikuläre) oder peribronchiale („Doppelgleisschatten “) Verdichtungen. Sie entstehen durch ein Ödem und zelluläre Infiltration des Interstitium und können zu kleineren segmental begrenzten Herden konfluieren, an deren Peripherie die netzförmige Zeichnung noch sichtbar ist. Kommen bei Mykoplasma- und Viruspneumonien vor.






2.6.1.7.7 Labordiagnostik
-
Erregernachweis: Die ätiologische Klärung ist bei der ambulant erworbenen Pneumonie wegen geringer therapeutischer Konsequenzen nicht unbedingt erforderlich. Bei unerwarteten Erregern ermöglicht sie jedoch den gezielten Einsatz der Antibiotika.
Sputumuntersuchung und Kultur: Geeignetes Sputum ist oft nicht leicht zu gewinnen. Es sollte im Mikroskop bei geringer Vergrößerung >25 Neutrophile und <10 Plattenepithelien pro Blickfeld aufweisen. Mit der Gram-Färbung sind einige Bakterien zu identifizieren. Kulturen können eine Resistenzbestimmung ermöglichen. Bei Einschmelzungen ist nach Tuberkelbazillen zu fahnden.
Blutkulturen: Bei hospitalisierten Patienten nur selten positiv, daher in der Regel entbehrlich. Ausnahmen bei schwerer Neutropenie und Immunschwäche.
Antigentests: Antigene von Pneumokokken und Legionella pneumophilia lassen sich mit einem einfachen Test über längere Zeit im Urin nachweisen. Schnelltest gibt es auch zum Nachweis von Influenza- und RSV-Viren.
-
Sonstige Laboruntersuchungen: Die Blutgasanalyse ergibt gewöhnlich eine Hypoxie (Rechts-Links-Shunt des Blutes durch Perfusion der nicht ventilierten pneumonischen Bezirke) mit Hypokapnie und respiratorischer Alkalose. Hyperkapnie und Azidose nur bei respiratorischer Insuffizienz. Differenzialblutbild: Starke Leukozytose mit Linksverschiebung (15.000–25.000/mm3) bei den grampositiven Pneumonien, bei Mykoplasmapneumonie selten bis 56.000 mm3. Bei schwerster Pneumokokkenpneumonie auch Leukopenie. Weniger hohe Leukozytose bei gramnegativen und Pilzpneumonien. Normale Leukozytenzahl bei Psittakose, Q-Fieber und Grippepneumonie. Bei der Grippe weisen Leukozytosen über 15.000/mm3 auf bakterielle Sekundärinfektion hin. Entzündungsparameter: Blutsenkung, CRP, Serumelektrophorese (α2-Globuline), Immunglobulinbestimmung.
2.6.1.7.8 Differenzialdiagnosen
Abzugrenzen sind Pneumonien gegen folgende Lungenkrankheiten:
-
Tuberkulose: Subakuter oder chronischer Verlauf, Lokalisation und Struktur der Lungenherde im Röntgenbild, Erregernachweis im Sputum, Tuberculinreaktion.
-
Bronchialkarzinom: Bei endobronchialem Wachstum kommen Retentionspneumonien vor, die sich auch lösen können. Nachweis durch Bronchoskopie und Computertomogramm.
-
Fremdkörper: Können zu Retentionspneumonien und Atelektasen führen. Nachweis durch Bronchoskopie.
-
Lungenembolie: Abgrenzbar durch klinisches Bild (primäre Dyspnoe, kein oder spätes Fieber), Nachweis der Emboliequelle und durch Lungenszintigraphie oder thorakales CT.
-
Interstitielle Lungenkrankheiten: Können Husten, Fieber und Pleuraschmerz hervorrufen. Gekennzeichnet durch chronischen Verlauf, meistens ohne Fieber mit zunehmender Beeinträchtigung des Gasaustausches (Dyspnoe), disseminiertem Befall beider Lungen und Fehlen eines Erregers.
2.6.1.7.9 Allgemeine Maßnahmen
In mittelschweren und schweren Fällen Krankenhauseinweisung. Als Kriterium kann der CRB-65-Index mit folgenden Variablen dienen: Verwirrtheit, Atemfrequenz >30/min, Blutdruck systolisch <90 mmHg oder diastolisch <60 mmHg, Alter >65 Jahre. Patienten mit >2 Variablen sind stationär einzuweisen. Bettruhe bis zur Entfieberung, bei älteren immobilen Patienten unter Embolieprophylaxe (Low-Dose-Heparin mit 2×5000 IE s.c./Tag). Luftanfeuchtung, ausreichende Flüssigkeitszufuhr. Antitussiva bei unproduktivem Husten, Analgetika gegen Pleuraschmerz. Atemphysikalische Behandlung (Lagerung, Klopfmassagen). Kreislauf- bzw. Schockbehandlung bei septischem Verlauf. Sauerstoffinhalation bei deutlicher Hypoxie. Frühzeitige Beatmung bei fortschreitender respiratorischer Insuffizienz.
2.6.1.7.10 Antibiotikatherapie
2.6.1.7.11 Resistenzen
Sie betreffen hauptsächlich S. pneumoniae und MRSA (methicillinresistentem S. aureus). In den letzten 15 Jahren haben penicillin- und makrolidresistente Pneumokokken stark zugenommen. In Deutschland sind 5 % der Pneumokokkenstämme gegen Penicillin und 15 % gegen Makrolide resistent. Im internationalen Vergleich gibt es große Unterschiede. So beträgt die Resistenzrate gegen Pneumokokken in Frankreich 40,7 % und in den USA 22,2 %. Eine Pneumonie mit MRSA kann durch Kontakt mit Pflegeeinrichtungen erworben werden. Eine Infektion mit varianten MRSA-Stämmen kann aber auch ambulant erfolgen.
2.6.1.7.12 Empirische Antibiotikatherapie
In Abhängigkeit vom Erregerspektrum und der Resistenzlage ist das Vorgehen in den USA und europäischen Ländern unterschiedlich. Im Folgenden werden die Empfehlungen der S3-Leitlinien des deutschen CAP-Netzwerkes (CAPNETZ) wiedergegeben (Angaben in Tagesdosen).
-
Therapie ambulanter Patienten ohne Risikofaktoren:
-
1. Wahl: Amoxicillin 3×1 g p.o. (<70 kg 3×0,75 g) für 7–10 Tage
-
Alternativen: Azithromycin 1×500 mg für 3 Tage oder Clarithromycin 2×500 mg, dann 2×250 mg p.o. für 7–10 Tage oder Roxithromycin 1×300 mg p.o. für 7-10 Tage oder Doxycyclin 1×200 mg p.o. (<70 kg 1×200 mg ab 2. Tag 100 mg) für 7–10 Tage
-
-
Ambulante Patienten mit Risikofaktoren (Antibiotika-Vortherapie, Alter >65 Jahren + Komorbidität [Herz, Lunge, Niere, Leber], Alten + Pflegeeinrichtungen, Krankenhausvorbehandlung, COPD):
-
1. Wahl: Amoxicillin/Clavulan 3×2 g p.o. (<70 kg 3×1 g) für 7–10 Tage oder Sultamicillin 2×0,75 g für 7–10 Tage
-
Alternativen: Moxifloxacin 1×400 mg p.o. für 7–10 Tage oder Levofloxacin 1×750 mg, auch 2×500 mg für 7–10 Tage oder Cefpodoximproxetil 2×200 mg p.o. für 7–10 Tage oder Cefuroximaxetil 2×500 mg p.o. für 7–10 Tage
-
-
Hospitalisierte CAP-Patienten ohne Risikofaktoren für Pseudomonas:
-
1. Wahl Amoxicillin/Clavulin 3×2,2 g i.v. für 7–10 Tage oder Ampicillin/Sulbactam 3×3,0 g i.v. für 7–10 Tage oder Cefuroxim-Axetil 3×1,5 g i.v. für 7–10 Tage oder Ceftriaxon 1×2,0 g i.v. für 7–10 Tage oder Cefotaxim 3×2,0 g i.v. für 7–10 Tage evtl. plus Makrolid
-
Alternativ: Levofloxacin 2×0,5 g i.v. für 7–10 Tage oder Moxifloxacin 1×0,4 g i.v.
-
-
Hospitalisierte Patienten mit Risiko für P. aeruginosa: Durch Erkrankungen mit Veränderungen des Lungengerüstes, stationärer Aufenthalt in den letzten 30 Tagen, Steroidtherapie, Aspiration, Breitspektrumantibiotika >7 Tage in den letzen 30 Tagen, Malnutrition:
-
1. Wahl (7–10 Tage): Piperacillin/Tazobactam 3×4,5 g i.v. oder Cefepim 3×2,0 g i.v. oder Imipenem 3×1,0 g i.v. oder Merpenem 1×1 g i.v. evtl plus Makrolid
-
Alternativen: Levofloxacin 2×0,5 g i.v. oder Ciprofloxacin (nur in Kombination) 3×0,4 g i.v.
-
2.6.1.7.13 Komplikationen
Lungenabszess, Pleuraerguss bzw. Empyem, metastatische Infektionen (Hirnabszess, Endokarditis).
2.6.1.7.14 Prognose
Krankheitsdauer je nach Alter und Komorbidität 2–4 Wochen oder länger. Mortalität bei ambulant behandelten Patienten <1 %, bei stationär behandelten etwa 10 % (darunter 5 % Todesfälle an Pneumonie.
2.6.1.8 Beatmungs- bzw. Ventilator-assoziierte Pneumonie (VAP)
2.6.1.8.1 Definition
Sonderform der HAP und der mit Pflegeeinrichtungen assoziierten Pneumonie (HCAP: health care-associated pneumonia).
2.6.1.8.2 Epidemiologie
Auf Intensivstationen kommt es bei bis zu 15 % der intubierten Patienten zu einer VAP. Das Pneumonierisiko nimmt mit der Dauer der Beatmungstherapie zu. Bei demselben Patienten kann es auch zu Rezidiven kommen.
2.6.1.8.3 Erregerspektrum
Man unterscheidet Nicht-MDR- und MDR-Pathogene. MDR (multidrug-resistant) bedeutet Resistenz gegen drei oder mehr differente Antibiotikaklassen mit unterschiedlichen Wirkungsmechanismen.
-
Nicht-MDR-Erreger: S. pneumoniae, Haemophilus influenzae, MSSA, sensitive Enterobateriaceae, E. coli, KLepsiella pneumoniae, Proteus spp., Enterobacter spp., Serratia marcescens.
-
MDR-Erreger: Pseudomonas aeruginosa, MRSA, Acinebacter spp., resistente Enterobacteriacae, Enterobacter spp., ESBL (extendet-spectrum β-Lactamase)-positive Stämme, Klebsiella spp., Legionella pmeumoniae, Burkholderia cepacia, Aspergillus spp.
2.6.1.8.4 Pathogenese
Das Angehen einer Pneumonie durch Problemkeime bei Beatmungspatienten wird durch folgende Faktoren begünstigt: Kolonisation des Oropharynx mit pathogenen Mikroorganismen nach Antibotikaselektion, Mikroaspiration bei der Intubation und später entlang des Tubus, wenn sich Sekret über dem Ballon des Tubus gestaut hat, Schädigung der Trachealschleimhaut durch Absaugen, Kolonisation in der Trachea und im Tubus, die sich mit einem Glyoxalisbiofilm überziehen kann, Immunparalyse bei schwerkranken und septischen Patienten.
2.6.1.8.5 Klinik
Fieber, Tachypnoe, gesteigerte respiratorische Sekretion, Tachykardie, Schallverkürzung und Bronchialatmen über dem befallenen Lungenbezirk. Klärung durch Röntgenaufnahme, die technisch erschwert sein kann.
2.6.1.8.6 Diagnostik
2.6.1.8.7 Klinische Kriterien
Fieber >38,5° C oder <36° C, Leukozytose (>11.000 µl) oder Leukopenie (<4000 µl), Hypoxämie, Infiltrate im Röntgenbild, massiver Keimgehalt im Trachealaspirat (einheitliche Morphologie im Gram-Präparat). An alternative Fieberursachen denken!
2.6.1.8.8 Quantitative Erregerkultur
Zur Unterscheidung von Kolonisation und Infektion und Testung der Erreger auf Antibiotikaempfindlichkeit. Im mehr proximal durch blinde Absaugung gewonnenen Sekret sind Befunde ab 105 bis 106 CFU/ml (colony forming units) positiv, bei bronchoskopisch entnommenen distalen Sekret schon ab 103 CFU/ml. Durch die quantitative Kultur kann eine unnötige Antibotikatherapie vermieden werden. Eine vorausgegangene Antibiotikatherapie kann zu einem falsch negativen Ergebnis führen.
2.6.1.8.9 Antibiotikatherapie
Mit der Behandlung gleich nach Entnahme der Sekretproben beginnen. Da deren Auswertung einige Zeit in Anspruch nimmt, ist die Antibiotikastrategie zu nächst empirisch. Im Gegensatz zur CAP muss diese mögliche MDR-Pathogene berücksichtigen.
-
Antibiotika bei Patienten ohne Risikofaktoren für MDR-Erreger:
-
Ceftriaxone (2 g i.v./alle 24 h) oder Moxifloxacin (400 mg i.v. alle 24 h) oder Ciprofloxacin (400 mg alle 8 h) oder Levofloxacin (750 mg alle 24 h)
-
Alternativ: Ampicillin/Sulbactam (3 g i.v. alle 6 h) oder Ertapenem (1 g i.v. alle 24 h).
-
-
Dreifach-Antibiotikatherapie bei Patienten mit Risiko für MDR-Erreger und MRSA:
-
ein β-Laktam: Ceftzidime (2 g i.v. alle 8 h) oder Cefepime (2 g i.v. alle 8-12 h) oder Piperacillin/Tazobactam (4,5 g i.v. alle 6 h) oder Imipenem (500 mg i.v. alle 6 h) oder Meropenem (1 g i.v. alle 8 h) plus
-
ein zweiter Wirkstoff gegen gramnegative bakterielle Pathogene: Gentamicin oder Tobramicin (7 mg/kg i.v. alle 24 h) oder Amikacin (20 mg/kg alle 24 h) oder Ciprofloxacin (400 mg i.v. alle 8 h) oder Levofloxacin (750 mg i.v. alle 24 h) plus
-
ein Antibiotikum gegen grampositive bakterielle Pathogene: Linezolid (600 mg i.v. alle 6 h) oder Vancomycin (15 mg/kg bis 1 g i.v. alle 12 h).
-
Bei positiver Kultur Anpassung der Therapie an den Erreger. Bei negativer Kultur alle Antibiotika absetzen und nach anderer Fieberursache suchen. Wenn eine klinische Besserung eintritt, dann nach 48–72 h.
2.6.1.8.10 Prognose
In Abhängigkeit von der Grundkrankheit und dem Erreger ist mit einer Mortalität von bis 25 % und höher zu rechnen.
2.6.1.9 Hospital-akquirierte Pneumonie (nosokomiale HAP)
2.6.1.9.1 Definition
Es handelt sich um Pneumonien, die von nichtintubierten Patienten im Krankenhaus außer- oder innerhalb von Intensivstationen erworben werden.
2.6.1.9.2 Erregerspektrum
Im Unterschied zur VAP sind Nicht-MDR Pathogene bei der HAP häufiger. Außerdem ist die Abwehrlage der nicht intubierten Patienten besser. Im Erregerspektrum sind Anaerobier bei der HAP häufiger, weil es leichter zur Makroaspiration kommt.
2.6.1.9.3 Diagnostik
Die definitive Diagnose ist bei der HAP schwieriger, weil Sekret aus den unteren Lungenbezirken für eine aussagefähige quantitative Kultur schwer zu gewinnen ist.
2.6.1.9.4 Antibiotikatherapie
Erregerabhängig durchzuführen evtl. wie bei VAP. Oft kommt man mit einer Monotherapie (wie bei der CAP im Krankenhaus) aus. Nach einer deutlichen Aspiration ist wie bei vermuteten MDR-Pathogenen therapieren (▶ oben). Trotz bestehender Unsicherheiten versagen die Antibiotika selten. Auch die Mortalität ist deutlich niedriger als bei der VAP.
2.6.2 Interstitielle Lungenkrankheiten
Es handelt sich um eine große Gruppe von nichtneoplastischen und nichtinfektiösen Erkrankungen, die das Lungenparenchym befallen: Alveolen, Alveolarepithel, Kapillarendothel, den Raum zwischen diesen Strukturen sowie das perivaskuläre und lymphatische Gewebe. Mehr als 200 verschiedene Krankheitszustände mit diffuser parenchymatöser Lungenbeteiligung sind bekannt. Man hat eine Unterteilung in 2 große Gruppen vorgenommen:
-
Erkrankungen mit Alveolitis, interstitieller Entzündung und Fibrose und
-
Erkrankungen mit Granulombildung.
In jeder dieser beiden Gruppen wird zwischen den Krankheiten mit bekannter und denen mit unbekannter Ursache unterschieden (◘ Tab. 2.7). Die aufgeführten heterogenen Krankheiten haben ähnliche klinische, röntgenologische und pathophysiologische Manifestationen.
2.6.3 Idiopathische Lungenfibrose (UIP: usual interstitial pneumonia)
2.6.3.1 Ätiologie und Pathogenese
Häufigste Form der idiopathischen interstitiellen Pneumonien. Männer und Frauen erkranken gleich häufig, überwiegend im mittleren Lebensalter. Die Ursache ist ungeklärt, doch gibt es ein identisches Krankheitsbild auf genetischer Basis, die familiäre pulmonale Fibrose, die einen autosomal-dominanten Erbgang mit variabler Expressivität aufweist. Der initiale Entzündungsprozess im Bereich der Alveolen und des Interstitiums wird durch Immunkomplexe mit nicht identifizierter Antigenkomponente in Gang gesetzt. Er führt zur Stimulation der Fibroblasten und der glatten Muskelzellen im Interstitium, zur Bildung bindegewebiger Matrix und schließlich zur Fibrose mit Untergang der Alveolen, Kapillaren und kleinsten Blutgefäße.
Eine akute Verlaufsform mit Exitus innerhalb 1–6 Monaten wird als Hamman-Rich-Syndrom bezeichnet. Sie geht mit einer organisierenden diffusen Alveolarschädigung einher, die histologisch dem ARDS (adult respiratory distress syndrome) entspricht.
2.6.3.2 Klinik
Gewöhnlich schleichender Beginn der Symptome mit trockenem Husten und zunehmender Belastungsdyspnoe. Inspiratorische feinblasige Rasselgeräusche über den Unterfeldern dorsal. Herabsetzung der Vitalkapazität und der Compliance ohne Erhöhung des bronchialen Strömungswiderstandes. Hypoxie in Ruhe, bei Ergometerbelastung weiteres Absinken von pO2. Im Finalstadium dekompensiertes Cor pulmonale und respiratorische Insuffizienz.
2.6.3.3 Diagnostik
Ausschluss bekannter Grundkrankheiten, die zur Lungenfibrose führen können.
Im Röntgenbild manchmal ausgedehnte bilaterale Verschattungen (◘ Abb. 2.32), häufiger feines oder grobes retikuläres Netzmuster, im CT verstärkte diffuse retikuläre Zeichnung, oft Areale mit Milchglastrübung, Wabenmuster und Traktionsbronchiektasen (◘ Abb. 2.33).


Röntgenbild eines Patienten mit gesicherter idiopathischer Lungenfibrose (UIP)


72-jähriger Mann mit einer sich langsam entwickelten Dyspnoe. Typisches CT-Bild mit „honey combing“, Traktionsbronchiektasen und „ground glass pattern“ einer UIP
Weiteres diagnostisches Programm:
-
Videoassistierte Thorakoskopie: Zur Sicherung der Diagnose unentbehrlich.
-
Histologischer Befund: Proliferation der Fibroblasten und glatten Muskelzellen im Interstitium mit fibrotischer Verdickung der Alveolenwände, auch intraalveoläre Fibrose. Daneben entzündliche Ansammlungen von Makrophagen und Neutrophilen mit eingestreuten Lymphozyten und Eosinophilen. Immunhistologischer Immunkomplexnachweis. Abgrenzung eines Goodpasture-Syndroms und einer überwiegenden Vaskulitis sind möglich. Neutrophile und Makrophagen sind ein Indikator für die entzündliche Aktivität. Mit der transbronchialen Lungenbiopsie ist die diagnostische Ausbeute geringer.
-
Bronchoalveoläre Lavage: Ergibt eine erhöhte Zellzahl mit Dominanz der Makrophagen und Vermehrung der Neutrophilen bei normaler Lymphozytenzahl. Abgrenzung gegen allergische Alveolitis und Sarkoidose, die eine Vermehrung der Lymphozyten aufweisen.
2.6.3.4 Therapie und Verlauf
Langzeitbehandlung mit Glukokortikoiden. Die neutrophile Entzündungskomponente bessert sich durch Kombination mit Azathioprin oder Cyclophosphamid (Kontrolle durch Lavage). Als antifibrotische Mittel werden Colchicin und Interferon-γ-1b versucht. Bei progredienter Verschlechterung der Lungenfunktion ist eine Lungentransplantation indiziert. Ohne Transplantation sterben die meisten Patienten 4–5 Jahre nach Beginn der klinischen Symptome.
2.6.4 Iatrogene Lungenfibrosen
2.6.4.1 Strahlenschäden
Die Lunge gehört zu den langsam proliferierenden Geweben und reagiert auf ionisierende Strahlen mit Verzögerung. Toleranzdosen: 10 Gy als Einmalbestrahlung, 25 Gy in 2 Gy-Einzelfraktionen. Läsionen bei Überdosierung bzw. erhöhter Strahlenempfindlichkeit.
2.6.4.1.1 Strahlenpneumonitis
Sie kann nach 2–6 Monaten auftreten. Symptome sind trockener Husten, Dyspnoe, Fieber. Im Röntgenbild Lungenverdichtungen, die über das Bestrahlungsfeld hinausgehen können. Es besteht eine akute Alveolitis mit Ödem der Alveolarwand und Desquamation des Alveolarepithels. In schweren Fällen tödlicher Ausgang. Therapie: Glukokortikoide.
2.6.4.1.2 Strahlenfibrose
Tritt nach 6 Monaten bis zu einigen Jahren unabhängig von der Schwere der initialen Pneumonitis auf. Der Beginn ist schleichend mit Husten und Dyspnoe. Im Röntgenbild beginnen die Verdichtungen am Hilus und breiten sich von dort zur Peripherie aus. Pleuraverdickung und Schrumpfungstendenz mit Verziehung des Mediastinums. Auf das Bestrahlungsfeld begrenzt. Restriktive Ventilationsstörung. Steroide verhindern nicht das Fortschreiten der Fibrose. Im Verlauf partielle Remissionen möglich.
Vermeidbar durch richtig dosierte fraktionierte Bestrahlung.
2.6.4.2 Medikamenteninduzierte Lungenfibrose
Kommt bei Langzeitbehandlung mit Zytostatika (Busulfan und Bleomycin), Phenytoin, Amiodaron (◘ Abb. 2.34), Nitrofurantoin u. a. vor.


Fibrose durch Amiodaron nach langdauernder Einnahme des Medikaments (Sammlung Prof. Jend)
2.6.5 Toxische Inhalationsschäden
2.6.5.1 Luftverschmutzung (Smog)
Die allgemeine Luftverschmutzung durch Industrie-, Heizungs- und Autoabgase erreicht in Verbindung mit Nebel bei Störung des vertikalen Luftaustausches infolge bodennaher Kaltluft, also vor allem im Winter, einen gefährlichen Grad. Man spricht dann von Smog (aus smoke = Rauch und fog = Nebel).
Smog ist ein luftkolloidales System mit den toxischen Substanzen SO2, H2SO4, O3, CO und CO2. Toxizitätsparameter ist der SO2-Gehalt.
Gefährdet sind vor allem alte Menschen, Kleinkinder und Personen mit chronischen Herz- und Lungenkrankheiten. Es scheint durch das saure Inhalat vor allem zu obstruktiven Ventilationsstörungen zu kommen. 1952 bewirkte in London eine 14-tägige Smog-Phase einen Anstieg der Mortalität um 4000 über den Erwartungswert. Öffentliche Maßnahmen bei Smoggefahr: Beschränktes Heizen, Fahrverbot für PKW. Persönlicher Schutz für Gefährdete: In geschlossenen Räumen bleiben, Einsatz von Luftreinigern bzw. Klimaanlagen. In Notfällen Klinikeinweisung.
2.6.5.2 Rauchgasvergiftung
Fordert bei Bränden unter Feuerwehrleuten und vom Brand betroffenen Personen mehr Todesopfer als die Hitzeeinwirkung. Das Rauchgas enthält durch Kunststoffverbrennung Zyanidverbindungen und Salzsäure, daneben als schleimhautreizende Substanzen Schwefeldioxyd, Säuren, Akrolein und Formaldehyd. Durch Oxidation von Luftstickstoff entstehen giftige Nitrosegase, als Endprodukte der Verbrennung CO und CO2.
2.6.5.2.1 Klinik
In schweren Fällen tödliches Lungenödem nach wenigen Stunden. Ansonsten Luftnot, Husten, Reizung der oberen Luftwege, Hypoxie durch Kohlenmonoxyd, evtl. Zyanidvergiftung.
2.6.5.2.2 Diagnostik
Im Röntgenbild und CT bei 30 % der massiv Rauchgasvergifteten diffuse weiche Verdichtungen durch Flüssigkeitsansammlung, die sich nach Überleben der akuten Intoxikationsphase vollständig zurückbilden (◘ Abb. 2.35).


42-jähriger Mann mit akuter Dyspnoe nach massiver Rauchgasinhalation. Unter inhalativem Kortikoid gute Rückbildung in 10 Tagen
2.6.5.3 Weitere chemische Gasvergiftungen
2.6.5.3.1 Ammoniakvergiftung
Durch Ausströmen von Ammoniak aus Kühlanlagen und Transportbehältern. Auf den Schleimhäuten entsteht aus NH3 mit Wasser das stark alkalische NH4OH. Reizerscheinungen an Bindehäuten und oberen Luftwegen. Bei starker Inhalation Alveolenschädigung mit Lungenödem.
2.6.5.3.2 Vergiftung mit Halogenen (Chlor, Brom, Fluor)
Vergiftungen mit Halogenen kommen in chemischen Fabriken bei der Herstellung von Gummi, Kunststoffen, Desinfizienzien und Treibstoffen vor sowie beim Bleichen von Papier und Textilien. Symptome sind starke Irritation der oberen Luftwege mit Husten, Auswurf, Atemnot und Erbrechen. Bei massiver Inhalation entwickelt sich ein Lungenödem. Die Vitalkapazität bleibt 1–2 Jahre danach herabgesetzt.
2.6.5.3.3 Phosgenvergiftung
Carbonylchlorid (COCl2) war ein Kampfgas im ersten Weltkrieg. Substanz wird noch in der Farbstoff- und Pharmaindustrie verwendet. Toxische Wirkung durch Freisetzung der Karbonylgruppe. Leichter Geruch nach faulem Heu.
Keine Frühsymptome! Erst nach 3 h treten Atemnot und Zyanose auf sowie ein Lungenödem mit starkem blutigem Auswurf, das zur Exsikkose führt. In schweren Fällen Tod innerhalb 24 h.
Folgen einer Phosgenvergiftung sind häufig sekundäre bakterielle Pneumonien.
2.6.5.3.4 Vergiftung mit nitrosen Gasen (NO)
Kommen in der Düngemittelindustrie (Nitrierungsprozesse), in Bergwerksstollen (Entstehung bei Sprengungen) und in frisch gefüllten Getreidesilos (Silofüllerkrankheit), aber auch bei Großbränden (Oxydation von Luftstickstoff) vor. Eine besondere Gefahr geht von niedrigen Gaskonzentrationen aus, weil sie wegen schwachen Geruchs kaum wahrgenommen werden. Konzentrierte nitrose Gase riechen stechend und sehen braun-rot aus. Im Wasser entstehen HNO2 und HNO3, die zu Ätznekrosen der Bronchialschleimhaut und der Alveolen führen. Husten und Dyspnoe nach 4- bis 12-stündiger Latenzzeit, in schweren Fällen Lungenödem. Als Folge der akuten Intoxikation kann sich nach 2–6 Wochen eine obliterierende Bronchiolitis entwickeln.
2.6.6 Exogen-allergische Alveolitis
2.6.6.1 Definition
Durch inhalierte Allergene induzierte entzündliche Immunreaktion des Lungenparenchyms (Alveolenwand mit Kapillaren, Interstitium, terminale Atemwege), die 3 Stadien durchlaufen kann: Alveolitis, granulomatöse Entzündung, Fibrose. Im Respirationstrakt lassen sich somit 3 Zonen mit ihren eigenen allergischen Manifestationen unterscheiden:
-
Nase (allergische Rhinitis)
-
Bronchien (allergisches Asthma)
-
Alveolarraum (allergische Alveolitis)
2.6.6.2 Ätiologie
Für die meisten der zur Alveolitis führenden Allergene ist eine berufliche Exposition gegeben. Es handelt sich um Stäube und Dämpfe mit antigenhaltigem Material wie Tierproteinen, Schimmelpilzen, thermophilen Aktinomyzeten, Amöben und Chemikalien. Viele dieser Stoffe können bei atopischen Individuen auch zum Bronchialasthma führen. Um in die Alveolen zu gelangen, darf die Partikelgröße 5 µm nicht überschreiten. Auch für die Alveolitis scheint eine Disposition gegeben zu sein, da nicht alle Exponierten erkranken.
Eine Auswahl von exogen-allergischen Alveolitiden ist in ◘ Tab. 2.8 zusammengestellt.
2.6.6.3 Pathogenese
Die im angelsächsischen Schrifttum auch als „hypersensivity pneumonitis“ bezeichneten Läsionen entstehen durch Immunreaktionen vom Typ III (Bildung von Immunkomplexen aus zirkulierenden Antikörpern der Klasse IgG und dem Antigen unter Fixierung und Aktivierung von Komplement) und Typ IV (zellvermittelte Reaktionen vom verzögerten Typ, induziert durch antigenspezifische T-Lymphozyten). Darüber hinaus scheint eine direkte Komplementaktivierung auf dem alternativen Weg durch das verursachende Antigen vorzukommen. Im Gegensatz zum allergischen Asthma bronchiale mit Immunreaktionen vom Typ I sind IgE und Mastzellen nicht beteiligt. Auch eine Bluteosinophilie tritt nicht auf. Nur bei der bronchopulmonalen Aspergillose bestehen Bronchialasthma und Alveolitis nebeneinander.
Die immunpathologischen Abläufe bei der allergischen Alveolitis werden wie folgt interpretiert:
-
Alveolitis: Bildung von Immunkomplexen im Antigenüberschuss → Aktivierung von Komplement → Gewebsschädigung → Eindringen von T-Lymphozyten in die Alveolen, Reaktion mit dem Antigen, Freisetzung von Zytokinen und Mediatoren der Entzündung aus den T-Lymphozyten.
-
Granulombildung: Aktivierung der Alveolarmakrophagen durch T2-Helferzellen. Proliferation der Makrophagen unter Bildung von Epitheloid- und Riesenzellen.
-
Fibrose: Nach Stimulation der Fibroblasten im Interstitium durch 3 – von aktivierten Makrophagen gebildete – Proteine, die dabei zusammenwirken:
-
PDGF (platelet-derived-growth factor, auch in Thrombozyten enthalten)
-
Fibronektin (zugleich chemotaktisch für Mesenchymzellen, heftet sich an die bindegewebige Matrix)
-
Wachstumsfaktor der Alveolarmakrophagen (alveolar macrophage-derived growth factor).
-
2.6.6.4 Klinik
Das klinische Bild variiert in Abhängigkeit von der Häufigkeit und Intensität der Exposition mit dem verursachenden Agens, wahrscheinlich auch mit Unterschieden in der individuellen Reaktionsweise. Drei Verlaufsformen sind zu beobachten: Akute Form: Beginn 4–8 h nach gewöhnlich massiver Exposition (Staubwolke) mit trockenem Husten, Brustenge, Atemnot, Mattigkeit, Frösteln und Temperaturanstieg auf 38–40 °C. Rückbildung dieser Symptome nach einigen Stunden oder 1–2 Tagen. Während der Attacke Leukozytose bis 25×103/mm3, restriktive Ventilationsstörung. Röntgenbild normal oder mit peripheren Infiltraten oder feinnodulären Verdichtungen. Manchmal unmittelbar nach der Exposition vorübergehend Bronchospastik, gelegentlich Eosinophilie.
2.6.6.5 Subakute Form
Schleichender Beginn, oft über einige Wochen hindurch mit Husten und Dyspnoe, manchmal bis zur Zyanose und stationären Behandlungsbedürftigkeit. Die Rückbildung erfolgt Tage, Wochen oder einige Monate nach Beendigung der Exposition (◘ Abb. 2.36).


Farmerlunge vor und nach Exposition (Sammlung Prof. Jend)
2.6.6.6 Chronische Form
Nach langdauernder Antigenexposition mit oder ohne vorausgehende akute Schübe auftretend. Symptome ähnlich wie bei chronischer Bronchitis: Permanenter Husten mit wenig Auswurf, progrediente Dyspnoe, Zyanose, Polyglobulie, gelegentlich Cor pulmonale mit Rechtsinsuffizienz. Lungenfunktionsprüfungen: Restriktive Ventilationsstörung, Abnahme der Compliance und der Diffusionskapazität, finale respiratorische Insuffizienz. Im Röntgenbild Zeichen der diffusen Lungenfibrose. Die primär chronische Form scheint überwiegend bei chronischer Exposition mit geringen Mengen des Antigens zu entstehen. Der Zustand ist irreversibel.
2.6.6.7 Diagnostik
2.6.6.8 Anamnese
Ermittlung der meist beruflichen Exposition mit einem der bekannten Antigene. Beachtung des Zeitintervalls zwischen Exposition und Symptombeginn und zwischen Beendigung der Exposition und Abklingen der Symptome.
2.6.6.9 Lungenfunktionsprüfung
Kontrollen vor, während und nach der Exposition zum Nachweis des Kausalzusammenhanges. In Zweifelsfällen evtl. Provokationstest.
2.6.6.10 Immunologische Untersuchungen
In den meisten Fällen enthält das Serum präzipitierende Antikörper gegen das auslösende Antigen. Der Antikörpernachweis kann aber auch bei Gesunden positiv sein. Falsch negative Resultate kommen bei schlechter Qualität des Testantigens vor. Zum Nachweis der Reaktivität vom zellulären Typ ist der Leukozytenimmobilisationstest (LMIT: leucocyte migration inhibition test) geeignet. Hautteste ergeben Reaktionen vom Arthustyp (Ödem nach 6–7 h für 24–36 h).
2.6.6.11 Röntgenuntersuchung
Kann bei symptomatischen Patienten normal sein. In der akuten und subakuten Phase fleckige oder diffuse, oft nur feinkörnige diskrete Verdichtungen. In der chronischen Phase retikulonoduläre oder wabige Veränderungen. Die Röntgenzeichen sind jedoch unspezifisch.
2.6.6.12 Lungenbiopsie
Indiziert in unklaren Fällen zur Sicherung der Diagnose und ermöglicht außerdem das Stadium der Veränderungen festzustellen. Falls die transbronchiale Biopsie nicht genügt, wird eine offene Biopsie durchgeführt.
2.6.6.13 Therapie
Entscheidend ist die Expositionsprophylaxe. Bei der Farmerlunge z. B. Heueinbringung vor dem Trocknen, Belüftung des Arbeitsplatzes und Masken. Nötigenfalls Berufswechsel. In der akuten und subakuten Phase Kortikoide per os und als Inhalat (60–80 mg/Tag) bis zum Verschwinden der Symptome. Auch im chronischen Stadium Behandlungsversuch mit Kortikoiden, die aber bei Fortdauer der Exposition die Progredienz der Alveolitis nicht verhindern. Cromoglicat und Theophillin sind unwirksam.
2.6.7 Eosinophile Pneumonie n
2.6.7.1 Allergische bronchopulmonale Aspergillose
Kombination aus Asthma bronchiale und Alveolitis durch Immunreaktionen vom Typ I, III und IV gegen Aspergillus fumigatus, einem ubiquitär vorkommenden Schimmelpilz, der bei den Patienten die Bronchien besiedelt. Betroffen sind etwa 1 % der atopischen Asthmatiker.
2.6.7.1.1 Klinik
Es bestehen intermittierende subfebrile Temperaturen, Giemen, Husten mit bräunlichem geballtem Sputum, das Aspergillusmyzelien enthält, und eine Bluteosinophilie. Die Ventilationsstörung ist obstruktiv. Im Röntgenbild vorübergehende Infiltrate und infolge obturierender Schleimpfröpfe in den Bronchien auch Atelektasen. Das Serum enthält IgE- und IgG-Antikörper gegen Aspergillus fumigatus. Hauttests ergeben eine Frühreaktion vom Soforttyp und eine Spätreaktion vom Arthustyp.
2.6.7.1.2 Therapie
Pathogenetisch entscheidend sind die IgE-Antikörper. Dementsprechend sind Glukokortikoide indiziert und sehr effektiv. Sie beseitigen den Schleim, der den Pilzen als Nährboden dient und müssen lebenslang appliziert werden, um den Übergang in die prognostisch infauste Fibrose zu verhindern. Die Dosis richtet sich nach dem IgE-Titer, der erhöht bleibt, auf diesem Niveau aber konstant gehalten werden muss. Antimykotika sind wirkungslos. Nur bei immungeschwächten Patienten wird der Saprophyt invasiv mit der Gefahr einer Pneumonie und einer lebensbedrohlichen generalisierten Aspergillose.
2.6.7.2 Parasitär bedingte eosinophile Pneumonie (Löffler-Syndrom )
Flüchtige eosinophile Infiltrate bei Larvenwanderung parasitärer Würmer (Ascaris lumbricoides, Strongyloides stercoralis, Schistosomen und in den Tropen Wucheria bancrofti) durch die Lunge.
2.6.7.2.1 Klinik
Leichte bronchitische Symptomatik und Bluteosinophilie.
2.6.7.2.2 Diagnostik
Wiederholte Stuhluntersuchung auf Wurmeier.
2.6.7.2.3 Therapie
Entsprechende Wurmmittel.
2.6.7.3 Medikamentös induzierte eosinophile Pneumonien
Das typische Beispiel ist die akute Reaktion auf Nitrofurantoin, 2–10 Tage nach der Einnahme beginnend. Die gleichen Erscheinungen können auch andere Medikamente verursachen: Sulfonamide, Penicillin, Chlorpropamid, Thiazide, trizyklische Antidepressiva, Hydralazin, Goldsalze, Isoniazid u. a.
2.6.7.3.1 Klinik
Trockener Husten, Fieber und Dyspnoe.
2.6.7.3.2 Diagnostik
Das Röntgenbild zeigt fleckige oder diffuse Verdichtungen, auch eosinophile Pleuraexsudate.
2.6.7.3.3 Therapie
Die Behandlung besteht im Absetzen des Mittels. Im akuten Stadium helfen Glukokortikoide.
2.6.7.4 Chronische eosinophile Pneumonie
Ätiologisch unklares Krankheitsbild mit Fieberschüben, Nachtschweiß, Husten, Anorexie und Gewichtsabnahme, das einige Wochen oder Monate dauern kann. Das Röntgenbild zeigt periphere Infiltrate. Promptes Ansprechen mit Normalisierung des Lungenbefundes auf Kortikosteroide.
2.6.7.5 Churg-Strauss-Syndrom (allergische Angiitis und Granulomatose )
Auf dem Boden eines chronischen Asthmas entstehende granulomatöse Angiitis der mittleren und kleinen Arterien mit Einschluss der Venen und Venolen, die zu intra- und extravaskulären Granulomen und eosinophiler Gewebeinfiltration führt. Manifestiert sich bevorzugt in der Lunge, aber auch in den übrigen Organen, nicht selten in der Nase. Es handelt sich um eine Variante der Polyarteriitis nodosa, die im Gegensatz zum Churg-Strauss-Syndrom nur die mittleren Arterien befällt, die Lunge unbeteiligt lässt und nicht mit Asthma und Eosinophilie assoziiert ist. Das auslösende Agens ist unbekannt.
2.6.7.5.1 Klinik
Schweres Asthma, Fieberschübe, Myalgien, Hinfälligkeit, Gewichtsabnahme. An der Haut Purpuraeffloreszenzen ohne Thrombopenie, kutane und subkutane Knoten (70 %).
2.6.7.5.2 Diagnostik
Im Röntgenbild Lungeninfiltrate, nicht selten Nebenhöhlenverschattung. Gefäßveränderungen werden am Herzen, in den Nieren, im peripheren Nervensystem und im Gastrointestinaltrakt gefunden.
Hochgradige Bluteosinophilie (>1000/mm3). Sicherung der Diagnose durch Biopsie (Haut, Lunge).
2.6.7.5.3 Therapie und Prognose
Gute Langzeiterfolge mit Kortikoiden, besonders in Kombination mit Cyclophosphamid. Sehr schlechte Prognose unbehandelter Fälle.
2.6.8 Goodpasture-Syndrom
2.6.8.1 Ätiologie und Pathogenese
Es handelt sich um eine kombinierte immunologische Reaktion vom Typ II (zytotoxische Antikörperwirkung mit Komplementaktivierung) gegen die Basalmembran der Alveolen und der Glomeruli durch kreuzreagierende Antikörper, die zu pulmonalen Hämorrhagien und einer progredienten Glomerulonephritis führt. Als vorausgehende Ereignisse wurden Influenzavirusinfektionen und Inhalation von Kohlenwasserstoffen beobachtet. Primär könnte es demnach zu einer Veränderung der alveolären Basalmembran kommen, die danach als Antigen wirkt und eine Antikörperbildung auslöst.
2.6.8.2 Klinik
Initial Husten, Kurzatmigkeit und Hämoptoe, die zu-nächst gering, aber auch gleich massiv und bedrohlich sein kann. Längere Remissionen der pulmonalen Blutung können folgen. Schließlich kommt es zur Blutungsanämie. Die anfangs noch normale Nierenfunktion nimmt innerhalb weniger Monate rapide ab. Die Hälfte der Patienten stirbt an den Blutungen, alle kommen in die terminale Niereninsuffizienz.
2.6.8.3 Diagnostik
Nachweis zirkulierender Basalmembranantikörper im Serum. In der Nierenbiopsie immunhistologischer Nachweis linear angeordneter Ablagerungen von IgG an den Basalmembranen, z. T. auch von der Komplementkomponente C3. Im Röntgenbild der Lunge kommen perihiläre Verdichtungen vor. Meistens besteht eine Hypoxie.
2.6.8.4 Therapie
Plasmapherese und Immunsuppression mit hohen Dosen Methylprednisolon und Cyclophosphamid. Hämodialyse oder Nierentransplantation.
2.6.9 Pneumokoniosen
2.6.9.1 Definition
Durch Inhalation von anorganischem Staub verursachte entzündliche interstitielle Lungenkrankheiten, die zur Lungenfibrose führen. Im Gegensatz zu den Lungenkrankheiten durch organischen Staub sind die Pneumokoniosen nicht allergischer Natur.
2.6.9.2 Silikose
2.6.9.2.1 Ätiologie und Exposition
Die Silikose entsteht durch SiO2, das in den Alveolarraum inhaliert wird. Partikelgrößen von 5 µm abwärts. Die Gefährdung wächst mit der Reinheit des Quarzstaubs. Exponierte Berufsgruppen: Bergarbeiter vor Stein, Sandstrahlbläser, Steinbrecher, Ofenmaurer und Gießer, Arbeiter in Porzellan- und Steingutfabriken. Latenzzeit bis zum Krankheitsausbruch 10–20 Jahre, Sandstrahlbläser erkranken oft früher.
2.6.9.2.2 Pathogenese und Pathologie
Phagozytose der inhalierten Partikel durch alveolare Makrophagen. Abtransport mit den Zellen, durch den mukoziliaren Apparat der Bronchien und auf dem Lymphweg zu den hilären Lymphknoten. Ein Teil der Makrophagen wird durch das toxisch wirkende SiO2 zerstört und setzt dabei das Silicium sowie Enzyme und Mediatoren frei, die Fibroblasten zur Faserbildung stimulieren. Bei massiver Staubinhalation werden auch die Alveolarepithelien (Pneumozyten Typ 2) zerstört, wodurch surfactanthaltige Flüssigkeit in die Alveolen gelangt. Die Fibrose beginnt um die respiratorischen Bronchiolen herum oder in den Lymphknoten. Sie dehnt sich unter Zerstörung der Alveolen und Kapillaren allmählich aus. Es entstehen graue Knoten in beiden Lungen, vergrößerte, fibrotische, z. T. verkalkte Lymphknoten und fibröse Pleuraadhäsionen. Bei langdauernder starker Exposition kommt es häufig zur progressiven massiven Fibrose, die ganze Lappen erfasst und zu ausgedehnter narbiger Schrumpfung führt. Höhere Grade der Silikose sind manchmal mit Tuberkulose assoziiert (Silikotuberkulose).
2.6.9.2.3 Klinik
Eine akute Form bei Sandbläsern ohne Atemschutz kann innerhalb von 10 Monaten unter zunehmender Dyspnoe zum Tode führen. Die chronische Form ist lange asymptomatisch, später zunehmende Dyspnoe, trockener Husten. Durch progredienten Alveolenverlust und Narbenbildung entwickelt sich eine restriktive Ventilationsstörung mit Herabsetzung der Compliance und der Diffusionskapazität, keine Obstruktion. Überlagerung mit chronischer Raucherbronchitis häufig. Im Endstadium dekompensiertes Cor pulmonale und respiratorische Insuffizienz.
2.6.9.2.4 Diagnostik
Exposition und Röntgenbefund der Lunge, der die Grundlage für die Stadieneinteilung der Silikose als Berufskrankheit bildet:
-
Stadium I (leicht): Hilusvergrößerung, verstärkte maschenförmige Lungenzeichnung mit einzelnen kleinfleckigen Knötchen.
-
Stadium II (mittelgradig): Mittel- bis grobfleckige Knötchen in beiden Mittelfeldern und oberen Teilen der Unterfelder (Schneeflockenbild, ◘ Abb. 2.37).
Abb. 2.37 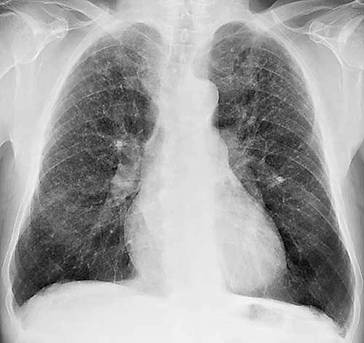
Silikose mit diffus verteilten kleinen, scharf begrenzten Herden über beiden Lungen (Sammlung Prof. Jend)
-
Stadium III (schwer): Flächenhafte Verschattungen und Schrumpfungsprozesse in den oberen Lungenpartien mit Pleurabeteiligung und Narbenemphysem, Schrumpfung des Mediastinums. Die Hilusschatten können verschwinden (◘ Abb. 2.38).
Abb. 2.38 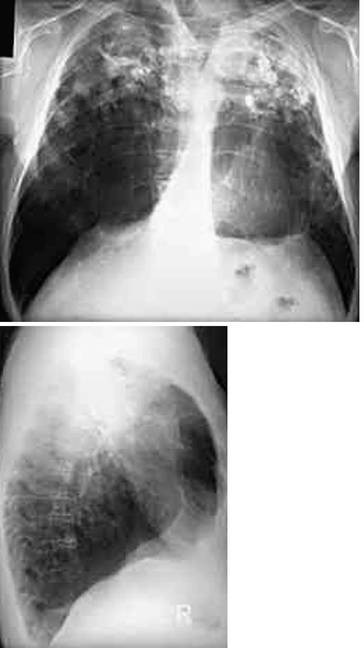
Schwere Silikose mit grobknotigen Konglomeraten in beiden Oberfeldern, Schrumpfung der Oberfelder und gerafften Hili. Kompensatorische Überblähung der rechten Lunge (Sammlung Prof. Jend)




2.6.9.2.5 Prophylaxe und Therapie
Arbeitsplatzhygiene. Bei ersten Silikosezeichen Entfernung aus dem staubhaltigen Milieu. Die Läsionen können trotzdem langsam fortschreiten. Tbc-Überwachung (Sputum), unspezifische Bronchitisbehandlung, keine Glukokortikoide. Die Silikose ist meldepflichtig und als Berufskrankheit entschädigungspflichtig. Das Rauchen als zusätzliche Noxe sollte unbedingt aufgegeben werden.
2.6.9.3 Anthrakosilikose
2.6.9.3.1 Ätiologie und Exposition
Die Anthrakosilikose ist eine Mischstaubsilikose. Entstehung durch Inhalation eines Gemisches von Kohlenstaub, SiO2, Silikaten und quarzfreien Stäuben. Das wirksame Agens ist SiO2. Exponiert sind Bergleute im Kohlebergbau und hier durch die Arbeit vor Stein. Die reine Kohlenstaubinhalation (Anthrakose), bei der Rußpartikel aus amorphem Kohlenstoff aufgenommen werden, bewirkt eine Pigmentierung, aber keine Erkrankung der Lunge.
2.6.9.3.2 Pathogenese und Pathologie
Phagozytose der in die Alveolen gelangten Partikel mit Makrophagenzerfall und Stimulation der Fibroblasten des Interstitiums zur gesteigerten Kollagenbildung wie bei der reinen Silikose. Der Quarzstaub liegt in den Knötchen, der Kohlenstaub in deren Randzone. Es resultiert eine schiefrig grauschwarze Färbung. Die Schwielenbildung ist stärker als bei alleiniger Silikose, auch die Tendenz zur progressiven massiven Fibrose.
2.6.9.3.3 Klinik
Jahrzehntelanger chronisch progredienter Verlauf, mit allmählich zunehmender Dyspnoe. Finalstadium wie bei der reinen Silikose. Häufige Überlagerung durch chronische Raucherbronchitis. Selten kommt eine Kombination mit rheumatoider Arthritis vor (Caplan-Syndrom). Man beobachtet sie auch bei Silikose und Asbestose.
2.6.9.3.4 Diagnostik
Röntgenbefunde ▶ Silikose.
2.6.9.3.5 Prophylaxe und Therapie
Wie bei Silikose. Es besteht kein erhöhtes Risiko für Karzinome der Lungen.
2.6.9.4 Asbestose
2.6.9.4.1 Ätiologie und Exposition
Krankheitsursache ist eingeatmeter „Asbeststaub“ mit Asbestfasern aus verschiedenen Silikaten gleichartiger Wirkung (Chrysotil, Amosit, Anthophyllit, Crocidolit). Schädigend wirken Fasern mit weniger als 3 µm Durchmesser und einer Länge über 10 µm. Diese Abmessungen ermöglichen einerseits das Vordringen in die Alveolen, andererseits erschweren sie die Beseitigung durch Makrophagen. Krankheitszeichen etwa 10 Jahre nach Beginn einer mittelschweren bis schweren Exposition.
2.6.9.4.2 Exponierte Berufsgruppen
Bergleute im Asbestabbau (Kanada, Russland, Südafrika), Hersteller und Benutzer von Asbestprodukten (Baumaterialien, Eternit, Isolierungen für Wärme- und Stromleitungen, Bremsbeläge, feuerfeste Kleidung und Decken etc.), Maler und Elektriker, die an asbestbeschichteten Leitungen arbeiten.
2.6.9.4.3 Pathogenese und Pathologie
Zerstörung der Makrophagen bei der Phagozytose des Asbestmaterials mit Freisetzung parenchymschädigender Enzyme und Fibroblasten stimulierender Mediatoren. Frühläsion ist eine peribronchiale Fibrose, davon ausgehend entsteht eine diffuse interstitielle Lungenfibrose. Typisch ist eine ausgeprägte Fibrose der Pleura, die auch isoliert auftreten kann. Etwa 12–17 % der Asbestosepatienten erkranken nach einer Expositionszeit von 16–18 Jahren am Bronchialkarzinom. Höheres Risiko bei gleichzeitigem Rauchen. Eine besondere Disposition besteht bei der Asbestose zum Mesotheliom der Pleura und des Peritoneums. Häufigkeitsmaximum 30–35 Jahre nach Beginn der Exposition, die nur wenige Jahre gedauert haben muss, da die Gewebeschädigung nach Beendigung der Exposition weiterwirkt. Etwa 80 % aller Mesotheliome sind mit Asbestose assoziiert. Beide werden als Berufskrankheit anerkannt.
2.6.9.4.4 Klinik
Husten, Dyspnoe, endinspiratorische feinblasige Rasselgeräusche, Zeichen von Pleuraergüssen, zunächst benignes und steriles, manchmal leicht blutiges Exsudat. Restriktive Ventilationsstörung mit Herabsetzung der Compliance und der Diffusionskapazität.
2.6.9.4.5 Diagnostik
Im CT zuerst pleurale Plaques (◘ Abb. 2.39), auch im Bereich der Herzkontur, später netzförmige bis grobstreifige Veränderungen in den Unterfeldern bis zur Wabenform. Kompensatorisches Emphysem der Oberfelder (◘ Abb. 2.40). Diagnose durch Nachweis von Asbestkörperchen im Sputum.


Asbestose. CT-Bild mit Pleuraplaques (Sammlung Prof. Jend)


CT-Bild einer Asbestose im Endstadium (Sammlung Prof. Jend)
2.6.9.4.6 Prophylaxe und Therapie
Kontrolluntersuchungen bei Exponierten. Bei ersten Krankheitszeichen. Arbeitsplatzwechsel. Manifest Erkrankte zur Früherkennung von Malignomen laufend kontrollieren.
Der Ersatz von Asbest durch andere Werkstoffe ist in der Industrie jetzt weitgehend abgeschlossen. Asbestverseuchte Gebäude wurden saniert.
Es gibt keine spezifische Behandlungsmöglichkeit, Glukokortikoide sind nicht indiziert.
2.6.9.5 Andere Silikatosen
2.6.9.5.1 Talkumlunge
Verursacht durch Talkstaub, der außer Talk (hydriertes Magnesiumsilikat) auch kleine Mengen Asbest und SiO2 enthält. Tritt bei Arbeitern in Talkumbergwerken, Talkummühlen, Gummi- und Schmiermittelindustriebetrieben auf.
Manifestation als asbestoseähnliche teils diffuse, teils noduläre Lungenfibrose mit Pleurabeteiligung. Auslösung wahrscheinlich durch SiO2- und Asbestanteile im Staub.
2.6.9.5.2 Kaolinlunge
Kaolin, ein Aluminiumsilikat, wird vor allem in der Keramikindustrie verwendet. Lungenfibrosen nach jahrelanger Staubexposition beruhen wahrscheinlich auf Quarzbeimengungen.
2.6.9.6 Berylliose
Verursacht durch Beryllium, ein hartes Leichtmetall, das im Flugzeug- und Atomreaktorenbau, für Präzisionsinstrumente und in der Röntgentechnik verwendet wird. Toxische Wirkungen vor allem bei Inhalation, aber auch beim Eindringen in die Haut (granulomatöse Ulzera).
2.6.9.6.1 Akute Berylliose
Bei massiver Exposition akutes Krankheitsbild mit Lungenödem, das in wenigen Tagen zum Tode führt.
2.6.9.6.1.1 Klinik
In den meisten Fällen Beginn der Symptome nach Wochen oder einigen Monaten mit Husten, Retrosternalschmerz, Dyspnoe, Schwäche und Gewichtsabnahme. Daneben auch Rhinitis Konjunktivitis und Dermatitis. Restriktive Ventilationsstörung, zunehmende Hypoxie durch Diffusionsstörung.
2.6.9.6.1.2 Diagnostik
Im Röntgenbild zarte, bilaterale Trübung der Unterfelder, später zu Konfluenz neigende Verdichtungen.
2.6.9.6.1.3 Therapie
Ansprechen auf hochdosierte Kortikoidtherapie, völlige Rückbildung möglich.
2.6.9.6.2 Chronische Berylliose
Entsteht bei kontinuierlicher, aber auch viele Jahre nach einer kurzdauernden Exposition. Systemerkrankung mit Läsionen an Lungen, Pleura, Myokard, Leber, Milz, Lymphknoten und Nieren.
2.6.9.6.2.1 Klinik
Das klinische Bild und der Röntgenbefund der Lunge entsprechen weitgehend der Sarkoidose (s. dort).
2.6.9.6.2.2 Diagnostik
Sicherung der Diagnose durch chemischen Berylliumnachweis in Probeexzisionen aus Leber, Lymphknoten oder Lunge. Berylliumnachweis im Urin auch bei gesunden Berylliumarbeitern oft positiv.
2.6.9.6.2.3 Therapie
Langzeitgabe von Glukokortikoiden. Prognose schlechter als bei Sarkoidose. Bei 25 % der Patienten entwickelt sich eine schwere Lungenfibrose mit dekompensiertem Cor pulmonale und respiratorischer Insuffizienz.
2.6.10 Sarkoidose
2.6.10.1 Definition
Systemerkrankung mit granulomatöser Immunreaktion in zahlreichen Organen durch ungeklärte Antigenstimulation.
Die Sarkoidose befällt in der Reihenfolge der Häufigkeit Lunge und Hiluslymphknoten, Leber, Milz, Haut, periphere Lymphknoten, Skelettmuskulatur, Augen, zentrales Nervensystem, Myokard, Knochen Tränen- und Speicheldrüsen.
2.6.10.2 Ätiologie
Trotz intensiver Suche konnte bisher kein belebtes oder unbelebtes Antigen als Ursache der Erkrankung identifiziert werden, so dass sich eine Aufzählung der verdächtigten Erreger oder Substanzen erübrigt. Es kommen auch unterschiedliche Antigene oder mehrere Epitope eines einzelnen Antigens als immunogene Stimulanzien in Betracht. Einige Gruppenerkrankungen in Gemeinschaftsunterkünften und Erkrankungen beider Ehepartner weisen auf eine exogene Ursache hin. Andererseits wurden familiäres Vorkommen und unter monozygoten Zwillingen eine häufigere Konkordanz beobachtet als bei dizygoten. Eine genetische Disposition ist daher zu bedenken.
2.6.10.3 Pathogenese und Pathologie
Der histologische und immunhistologische Befund der Granulome zeigt, dass es sich bei der Sarkoidose um eine Immunreaktion vom zellulären Typ handelt: Die Granulome bestehen aus aktivierten Makrophagen, die in Epitheloidzellen übergehen und mehrkernige Riesenzellen bilden. Sie sind von CD4-positiven TH1-Lymphozyten umrandet, von denen die Makrophagen zur Proliferation stimuliert werden. Die zelluläre Immunantwort verläuft in folgenden Phasen (▶ Kap. 8.1.2):
-
Aktivierung antigenspezifischer naiver CD4-positiver TH1-Lymphozyten durch antigenpräsentierende dendritische Zellen, die nach Phagozytose und Aufbereitung des Antigens in den regionalen Lymphknoten sesshaft geworden sind.
-
Danach klonale Proliferation der aktivierten TH1-Lymphozyten und Reaktion mit dem Antigenmaterial auf Makrophagen, die das Antigen ebenfalls phagozytieren, aufbereiten und zusammen mit MHC-Molekülen der Klasse II an ihrer Oberfläche präsentieren.
-
Durch diesen Kontakt werden die Makrophagen zu maximaler Aktivität gegen phagozytierte Erreger stimuliert und die TH1-Lymphozyten zur Sekretion von Interferon γ, das die Makrophagen zu Proliferation bringt. Außerdem kommt zu einer humoralen Immunantwort mit Vermehrung der Immunglobuline im Plasma.
Die Bildung von Granulomen weist im Allgemeinen auf einen erschwerten Abbau von phagozytiertem Material bzw. Krankheitserregern hin, wie z. B. bei der Tuberkulose. Im Gegensatz zu dieser kommt es bei der Sarkoidose jedoch nicht zur Verkäsung, wohl aber zur Fibrosierung der Läsionen. Indessen lässt die hohe Spontanheilungsrate der Sarkoidose erkennen, dass die immunologische Abwehrreaktion letztlich oft erfolgreich ist.
2.6.10.4 Epidemiologie
Die Sarkoidose kommt weltweit bei allen Rassen vor, in jedem Alter und bei beiden Geschlechtern. Am häufigsten erkranken Erwachsene zwischen dem 20. und 40. Lebensjahr. Die Prävalenz differiert von 2–40 Fällen unter 100.000 Einwohner. Wegen der Vielfalt der klinischen Manifestationen kann die Sarkoidose Ärzten verschiedener Fachrichtungen begegnen.
2.6.10.5 Klinik
Das Initialstadium ist bezüglich von Symptomen und Organmanifestationen sehr variabel:
-
Asymptomatischer Beginn: Zufällige Entdeckung der symmetrischen Hilusschwellung im Röntgenbild oder Feststellung einer schmerzlosen Halslymphknotenschwellung durch Arzt bzw. Patient (◘ Abb. 2.41).
Abb. 2.41a,b 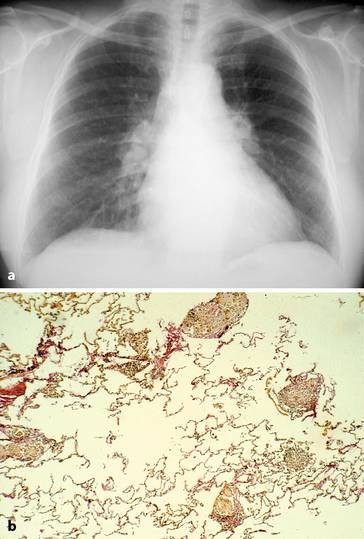
Sarkoidose Typ I. a Radiologischer Zufallsbefund bei einer 28-jährigen Frau: bihiläre Lymphadenopathie. b Histologische Sicherung der Diagnose (Granulome mit Riesenzellen) durch transbronchiale Biopsie
-
Akuter Beginn: Mit Fieber, Mattigkeit, Schwäche, Appetitlosigkeit, Gewichtsabnahme, manchmal Husten, Dyspnoe und Retrosternalschmerz. Zwei besondere Symptomenkomplexe sind hervorzuheben.
-
Löfgren-Syndrom: Erythema nodosum, Fieber, bilaterale hiläre Adenopathie, Arthritis (Entzündung des periartikulären Gewebes) an den Sprunggelenken (◘ Abb. 2.42).
Abb. 2.42 
Erythema nodosum bei einem 32-jährigen Mann mit Sarkoidose Typ I, der mit Fieber, Arthralgien und bihilärer Lymphknotenschwellung erkrankte. Remission unter Kortikoiden
-
Heerfordt-Waldenström-Syndrom: Fieber, Parotisschwellung, vordere Uveitis (Iridozyklitis) und Fazialisparese. Ohne Therapie dauern die akuten Symptome einige Wochen und bilden sich spontan restlos zurück.
-
-
Schleichender Beginn (chronische Sarkoidose): Über Monate hinweg Mattigkeit, leichter Husten, Belastungsdyspnoe, Gewichtsverlust. Es können sich mehrere Organmanifestationen entwickeln. Auch in diesen Fällen sind vollständige Remissionen möglich. Etwa 50 % behalten einen leichten Grad organischer Dysfunktionen, 10 % zeigen einen schweren Verlauf und enden tödlich.




2.6.10.6 Organmanifestationen
-
Unterer Respirationstrakt: Ist bei über 90 % der Patienten befallen. Charakteristisch sind eine interstitielle granulomatöse Lungenerkrankung mit Befall von Alveolen, Blutgefäßen und Bronchiolen und die symmetrische Schwellung der mit Granulomen durchsetzten Hiluslymphknoten. Im Röntgenbild sind folgende Typen abzugrenzen:
-
Typ I: Doppelseitige Hilusdrüsenvergrößerung ohne Parenchymveränderungen (◘ Abb. 2.41).
-
Typ II: Hilusvergrößerung mit diffusen Parenchymveränderungen, d. h. retikuläre Zeichnung und miliare Knötchen (◘ Abb. 2.43).
Abb. 2.43 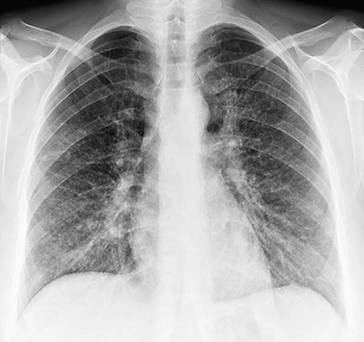
Sarkoidose Typ II. Mikronoduläre Zeichnungsvermehrung über beiden Lungen, Hili etwas verklumpt (Sammlung Prof. Jend)
-
Typ III: Diffuse Parenchymveränderungen ohne Hilusbeteiligung.
-
Typ IV: Chronische Parenchymveränderungen von mehr als zweijähriger Dauer mit Lungenfibrose (Strang- und Narbenbildung, Induration, Schrumpfung, Zysten und Emphysemblasen (◘ Abb. 2.44).
Abb. 2.44 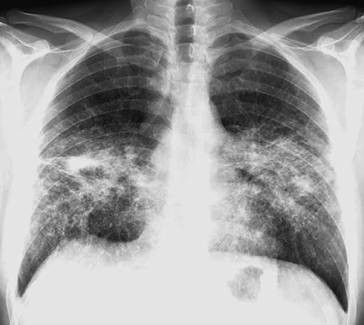
Sarkoidose Typ II/IV. Fibrosekonglomerate in den Mittel- und Unterfeldern beiderseits. Im Randbereich kleine Knötchen. Verdickte Hili (Sammlung Prof. Jend)
Fortgeschrittene Veränderungen im Röntgenbild können mit geringen Symptomen verbunden sein. Es überwiegt eine restriktive Ventilationsstörung mit herabgesetzter Vitalkapazität und Compliance, schließlich mit Hypoxie und Cor pulmonale. Bei stärkerem Bronchienbefall kommt es auch zur Obstruktion mit Verminderung der maximalen exspiratorischen Atemstromstärke und der FEV1.
-
-
Oberer Respirationstrakt: Behinderung der Nasenatmung in 20 % der Fälle, Tonsillen und Epiglottis sind seltener betroffen. Heiserkeit, Giemen und Stridor kommen vor.
-
Lymphknoten: Intrathorakal sind neben den Hilusdrüsen oft auch die paratrachealen und mediastinalen Lymphknoten vergrößert. Periphere Lymphadenopathie (zervikal, axillar, inguinal) wird oft angetroffen. Die Lymphknoten sind fest, von gummiartiger Konsistenz, gut verschieblich, nicht schmerzhaft und ulzerieren nie.
-
Haut: Erythema nodosum im akuten Initialstadium (10–30 % der FälIe), kombiniert mit Arthralgien. Relativ selten mikropapuläres Hautsarkoid mit gruppenweise angeordneten braun-roten Knoten in der Haut und Lupus pernio, das grobknotige Hautsarkoid, mit indurierten blauroten, frostbeulenähnlichen Läsionen an Nase, Wangen, Lippen, Ohren, Fingern und Knien.
-
Augen: Veränderungen an den Augen erleidet etwa jeder vierte Sarkoidosepatient. Es dominiert die vordere Uveitis (75 %) gegenüber der hinteren (25–35 %). Die akute, meist vordere Uveitis bildet sich gewöhnlich vollständig zurück. Die chronische granulomatöse Form befällt die gesamte Uvea und kann zu irreversiblen Schäden (hintere Synechien, Hornhauttrübung, Katarakt, Glaukom, Aderhautveränderungen) und auch zur Erblindung führen. Durch granulomatöse Läsionen der Tränendrüsen kann sich infolge verminderter Tränensekretion eine Keratokonjunctivitis sicca entwickeln. Retrobulbäre Granulome führen zur Protrusio bulbi.
-
Knochenmark und Milz: Beide Organe sind in 40–60 % der Fälle mit granulomatösen Veränderungen beteiligt, in der Regel ohne klinische Folgen. Eine tastbare Milzschwellung haben 10–25 % der Patienten.
-
Leber: In 20–30 % der Fälle besteht eine Hepatomegalie. Bioptisch findet man bei 60–90 % granulomatöse Veränderungen, gewöhnlich periportal gelegen. Von den Enzymen ist am ehesten die alkalische Phosphatase erhöht. Transaminasen und Bilirubin steigen nicht oder wenig. Eine cholestatische Zirrhose mit portaler Hypertension gehört zu den Seltenheiten.
-
Niere und Calciumstoffwechsel: Klinisch manifeste granulomatöse Nierenläsionen sind selten, obwohl an den Tubuli, Glomeruli und Nierenarterien Sarkoidbefall beschrieben wurde. Bemerkenswert ist eine Hyperkalzämie mit Hyperkalziurie, die zur Nephrokalzinose führen kann. Sie beruht auf gesteigerter Bildung von 1,25-Dihydroxyvitamin D. Vorkommen in etwa 2 % aller Fälle, verbunden mit Intoxikationsgefahr durch exogenes Vitamin D.
-
Knochen, Gelenke, Muskeln: Am Hand- und Fußskelett können durch Granulationsgewebe aus dem Knochenmark pseudozystische Herde (Ostitis multiplex cystoides: Morbus Perthes-Jüngling) oder gitterförmige Atrophien entstehen, begleitet von schmerzhafter Weichteilschwellung und nicht selten von Lupus pernio der Haut. Gesamthäufigkeit: 5 % aller Fälle von Sarkoidose. Arthralgien oder Arthritiden werden bei 25–50 % der Patienten beobachtet. Trotz häufiger Granulome in den Muskeln bleiben die Ausfallserscheinungen gering (Knoten, Polymyositis, Myopathie).
-
Herz: Myokardsarkoidose mit Rhythmusstörungen und Funktionsstörung des linken Ventrikels. Plötzlicher Herztod durch Kammerflimmern bei latenter Myokardsarkoidose. Cor pulmonale bei Lungenfibrose.
-
Endokrines System: Durch Befall der Hypophyse gelegentlich partielle Vorderlappeninsuffizienz oder Diabetes insipidus. Selten Nebennierenunterfunktion. Schwangerschaften sind möglich und bringen vorübergehende Besserung durch gesteigerte Glukokortikoidsekretion.
-
Exokrine Drüsen: Klinisch manifester Parotisbefall mit doppelseitiger Drüsenschwellung und Xerostomie bei weniger als 10 % aller Fälle.
-
Nervensystem: In etwa 5 % der Fälle ist das Nervensystem betroffen. Am häufigsten ist die Fazialisparese, gefolgt von Seh-, Hör- und Schluckstörungen. Gelegentlich granulomatöse basale Meningitis und zerebrale Anfälle. Multiple Läsionen können an multiple Sklerose erinnern.




2.6.10.7 Diagnostik
Dazu gehören:
-
Sicherung des Vorliegens einer Sarkoidose
-
Bestimmung ihres Aktivitätsgrades
-
Feststellung der Schwere der entstandenen Funktionsstörungen
2.6.10.8 Röntgenuntersuchung der Lunge
Standardaufnahmen lassen die bereits beschriebenen Typen der Sarkoidose erkennen, die aber keine konsekutiven Stadien der Erkrankung repräsentieren. Infiltrate sind vieldeutig, aber eine doppelseitige Hilusvergrößerung ist diagnostisch wegweisend. Computertomographie: Perilobuläre scharf begrenzte Knötchen. Mattscheibenartige Trübungen bei akuter Alveolitis. Früher Fibrosenachweis. Genaue Erfassung des Lymphknotenbefalls (Hili, Mediastinum).
2.6.10.9 Biopsie
Materialgewinnung von dem am leichtesten zugänglichen Organ. Haut- und transbronchiale Lungenbiopsie haben die beste Ausbeute. Biopsien sind auch mittels Mediastinoskopie und Leberpunktion zu gewinnen. Der Nachweis von nicht verkäsenden Granulomen mit Riesenzellen sollte die Diagnostik nicht beenden. Auszuschließen sind noch weitere granulombildende Erkrankungen (Tuberkulose, Histoplasmose, Toxoplasmose, Lues), Hypersensitivitätspneumonitis durch organische und anorganische Substanzen (Medikamente, Beryllium), Kollagenkrankheiten und Neoplasien (Karzinome, Lymphome).
2.6.10.10 Lungenfunktionsprüfung
Bestimmung der Diffusionskapazität für CO (Spirometrie) und Blutgasanalyse in Ruhe und unter Belastung.
2.6.10.11 EKG
In Ruhe und 24-h-Aufzeichung zur Erfassung von Rhythmusstörungen.
2.6.10.12 Ophthalmologische Untersuchung
Mit der Spaltlampe.
2.6.10.13 Laboruntersuchungen
Entzündungsparameter (BKS, Elektrophorese), Blutbild, Leberwerte, Vitamin D, Serumcalcium.
2.6.10.14 Selektive Methoden von unsicherer Spezifität
-
Kveim-Siltzbach-Hauttest: Intradermale Injektion eines Extraktes aus sarkoidosebefallenen Milzzellen. Nach 4 Wochen ergibt die Biopsie an der Injektionsstelle bei 70–80 % der Patienten sarkoidoseähnliche Läsionen. Nicht standardisiert und unspezifisch, in den USA nicht zugelassen. Gilt als obsolet.
-
Bestimmung der ACE-Aktivität im Serum: Erhöhte Werte in 65 % der Fälle; 5 % aller positiven Fälle haben keine Sarkoidose.
-
67 Gallium-Szintigraphie: Mangels Spezifität nicht länger indiziert.
2.6.10.15 Therapie
Glukokortikoide sind als einzige Pharmaka überzeugend wirksam, solange noch keine Fibrosierung eingetreten ist. Sie supprimieren die abnorme Aktivität der TH1-Lymphozyten und Makrophagen und sind auch gegen die Komplikation der Hyperkalzämie das geeignete Mittel. Dosierung: 1 mg/kg Prednison/Tag per os für 3–6 Wochen, danach allmähliche Dosisreduktion auf 7,5–20 mg/Tag für 2–3 Monate. Solche Zyklen können nach Bedarf wiederholt werden.
Zu berücksichtigen ist aber, dass man eine überschießende Abwehrreaktion dämpft, ohne die Krankheitsursache zu beseitigen, dass die Granulome eine relativ geringe destruktive Tendenz haben und sich im Durchschnitt 40 % der Fälle spontan zurückbilden. Beim Löfgren-Syndrom beträgt die spontane Heilungsrate über 80 %.
Unter Abwägung von Nutzen und Gefahren der Glukokortikoide propagieren viele Therapeuten zunächst für 2–3 Monate den Spontanverlauf abzuwarten und erst nach Ausbleiben einer Spontanremission mit der Therapie zu beginnen. Fieber und Arthralgien können in dieser Zeit mit nichtsteroidalen Antiphlogistika beseitigt werden.
Eine Soforttherapie mit Glukokortikoiden ist bei Lungenveränderungen im Stadium II, bei kardialer, okulärer und neurologischer Sarkoidose angezeigt. Die Inhalation von Glukokortikoiden ist ungenügend wirksam. Eine beginnende Uveitis und Hautveränderungen lassen sich lokal mitbehandeln.
Einzelbeobachtungen über günstige Effekte von Methotrexat und Hydroxychloroquin (bei Hautaffektionen) sind bisher nicht durch placebokontrollierte Studien gesichert.
Im Finalstadium des pulmonalen Prozesses kommt eine Lungentransplantation in Betracht.
Wegen der Hyperkalzämiegefahr ist auf ausreichenden Sonnenschutz zu achten.
2.6.10.16 Verlauf und Prognose
Der Krankheitsverlauf ist engmaschig unter Einschluss der Lungenfunktionsprüfung mit Blutgasanalyse zu kontrollieren. Rezidive sind mit Glukokortikoiden zu behandeln. Die meisten akuten Fälle heilen spontan ab. Etwa 50 % aller Patienten behalten leichte stabile Dysfunktionen zurück. In 15–20 % der Fälle nimmt die Erkrankung einen chronischen progredienten Verlauf mit Rezidiven und bei 10 % endet sie an pulmonalen oder kardialen Komplikationen tödlich.
2.6.11 Tuberkulose
2.6.11.1 Definition
Infektionskrankheit durch Mycobacterium tuberculosis. Der verwandte Erreger der Rindertuberkulose (M. bovis) kann auch den Menschen befallen, spielt aber nach weitgehender Ausrottung der Rindertuberkulose heute nur noch in Entwicklungsländern eine begrenzte Rolle.
2.6.11.2 Epidemiologie
2.6.11.3 Verbreitung
Weltweit wird noch mit über 8 Millionen Tuberkulosefällen pro Jahr gerechnet, von denen mehr als 95 % auf die Entwicklungsländer entfallen. Dort ist die Tuberkulose stark unter HIV-Infizierten verbreitet. In den westlichen Ländern liegt die Inzidenzrate bei 22/100.000. Hier wurde sie durch infizierte Immigranten relativ hoch gehalten.
2.6.11.4 Disposition
Nur etwa 5 % der mit Tuberkelbazillen Infizierten erkranken manifest. Ein erhöhtes Erkrankungsrisiko besteht für Säuglinge und sehr alte Menschen, besonders Pflegeheimbewohner, ferner für Unterernährte, Diabetiker, Alkoholiker und Patienten mit schwächenden Allgemeinerkrankungen und allen Formen der Immunschwäche.
2.6.11.5 Übertragung
Sie erfolgt von Mensch zu Mensch auf dem Luftweg durch Tröpfcheninfektion. Kleine Tröpfchen mit Tuberkelbazillen als Kern können bis in die Alveolen vordringen. Die Ansteckung mit M. bovis erfolgt durch rohe Milch von erkrankten Kühen (Euterbefall).
2.6.11.6 Ätiologie und Pathogenese
2.6.11.7 Erreger
M. tuberculosis ist ein aerober, fakultativ intrazellulärer Parasit, der in Form säurefester Stäbchen intra- und extrazellulär existieren kann. Er besitzt eine lipidreiche Zellwand mit immunreaktiven Komponenten, die seine Phagozytose fördern und seine Persistenz in den Makrophagen erleichtern.
2.6.11.8 Abwehrmechanismen
-
Angeborene (unspezifische) Immunantwort: Nachdem die Tuberkelbazillen in die Alveolen gelangt sind, werden sie von Alveolarmakrophagen mittels unspezifischer Rezeptoren für Pathogene phagozytiert. Die damit verbundene Aktivierung der Makrophagen reicht oft zur Bewältigung der Infektion aus. Wenn das nicht gelingt und sich die Erreger sogar intrazellulär vermehren, werden Bakterienantigene zusammen mit MHC-Molekülen der Klasse II an der Zelloberfläche exprimiert, um eine adaptive Immunreaktion auszulösen.
-
Adaptive (spezifische) Immunantwort: Antigenspezifische T-Helferzellen vom Typ TH1 werden zunächst von antigenpräsentierenden dendritischen Zellen aktiviert. Sie proliferieren und treten dann mit den antigenpräsentierenden Alveolarmakrophagen in Reaktion. Es resultiert 2–8 Wochen nach der Infektion eine maximale Aktivierung der Makrophagen, die zu Granulomen proliferieren und eine fieberhafte entzündliche Allgemeinreaktion induzieren. Bei guter Abwehrlage heilt die Primärinfektion unter Verkapselung ab. Andernfalls dehnt sich der Prozess unter Verkäsung der Granulome und Kavernenbildung in der Lunge aus.
2.6.11.9 Stadieneinteilung
2.6.11.10 Stadium I
Lokale Primärinfektion. Entzündliche Reaktion am Ort der Erregerhaftung in der Lunge mit Beteiligung der Lymphbahn und der regionalen Hiluslymphknoten. Dauer: einige Tage bis 2 Wochen. Klinisch gewöhnlich stumm. Wenn es mit Beginn der adaptiven Immunantwort nach 2–8 Wochen zu manifesten Herden kommt, spricht man von primärer Lungentuberkulose.
2.6.11.11 Stadium II
Generalisationsstadium. Lymphohämatogene Aussaat der Tuberkelbazillen (Hiluslymphknoten Ductus thoracicus obere Hohlvene) wenige Wochen nach der Lokalinfektion mit Absiedlungen in vielen Organen und Geweben, vor allem der Lungen (Spitzen), Lymphknoten, Knochen und Nieren. Dieses sehr häufige Stadium, vor der Immunantwort beginnend, bleibt klinisch meistens stumm. An Manifestationen, die noch der primären Tuberkulose zugerechnet werden, kommen vor: Erythema nodosum, Pleuritis exsudativa, Simmon-Lungenspitzenherde. In anderen Organen entstehen schlafende Herde, die später zu Erkrankungen dieser Organe führen können.
2.6.11.12 Stadium III
Organtuberkulose: Postprimäre Tuberkulose der Lunge und anderer Organe nach Abheilung der lokalen Primärinfektion und des Generalisationsstadiums. Ausgangspunkt sind reaktivierte Tuberkuloseherde.
2.6.11.13 Lungentuberkulose
2.6.11.13.1 Primäre Lungentuberkulose
Folgende Manifestationen kommen vor:
-
Gewöhnlicher Primärinfekt: Entweder unbemerkt oder mit leichtem Fieber und trockenem Husten einhergehend. Im Röntgenbild umschriebene Verdichtung, meistens im Unterlappen, und Hilusschwellung (sog. Primärkomplex), nach 1–2 Jahren kleine Kalkherde in diesen Bereichen. Die pulmonalen Herde liegen in der Regel subpleural. Nach 2–4 Wochen klinische Abheilung.
-
Hiluslymphknotentuberkulose: Vor allem bei Kleinkindern, deren Lymphknoten stärker reagieren. Weiterschwelen der Hilusläsionen mit Beteiligung der Bronchial- und Bifurkationslymphknoten, auch doppelseitig. Klinisch symptomarm mit subfebrilen Temperaturen, bei Kindern nächtlicher Husten. Selten Lymphknoteneinbruch in das Bronchialsystem mit pneumonischer Lungentuberkulose oder Bronchusstenose mit Atelektase.
-
Progredienter Primärherd (Primärherdphthise): Käsige Pneumonie im Bereich des Primärherds mit Kavernenbildung und intrakanalikulärer Ausbreitung. Gefahr einer solchen primär progressiven Form bei Kindern unter 3 Jahren.
2.6.11.13.2 Pleuritis tuberculosa
2.6.11.13.2.1 Pathogenese
Pleurabefall entweder vom Primärherd aus über den Lymphweg oder hämatogen im Generalisationsstadium, ausnahmsweise durch hämatogene Streuung postprimärer Tuberkuloseherde. Am häufigsten entsteht die Pleuritis exsudativa bei der Erstinfektion Erwachsener (Späterstinfektion) und gibt dann dringend Anlass zur Suche nach der Infektionsquelle.
2.6.11.13.2.2 Klinik
Hohes Fieber mit relativ gutem Allgemeinbefinden. Schallverkürzung und fehlendes Atemgeräusch über der betroffenen Lungenseite. Im Röntgenbild Erguss bis in die Spitze reichend. Nach Rückbildung meistens keine erkennbaren Herdschatten.
2.6.11.13.2.3 Diagnostik
Proteingehalt im Exsudat (im Gegensatz zu Transsudaten >3 g/dl). Nachweis von spezifischem Granulomgewebe durch Pleurabiopsie. Tuberculinreaktion bei 30 % der Patienten noch negativ. Erregernachweis mikroskopisch und kulturell nicht immer möglich, aussichtsreicher mit der Polymerase-Kettenreaktion (PCR).
2.6.11.13.3 Postprimäre Lungentuberkulose
2.6.11.13.3.1 Pathogenese
Meistens endogene Reaktivierung der hämatogen entstandenen Simmon-Lungenspitzenherde durch resistenzmindernde Faktoren (Hunger, Abmagerung, Diabetes mellitus, Alkoholismus etc.). Exogene Reinfektion sehr selten, wohl nur durch massives Inokulum und Abnahme der Tuberculinallergie möglich. Die hohe intraalveoläre O2-Konzentration in den Lungenspitzen scheint das Wachstum der Tuberkelbazillen zu begünstigen. Der pulmonale tuberkulöse Entzündungsprozess tendiert zu apiko-kaudalem Fortschreiten und schubweisem, chronischem Verlauf. Das Ausmaß der Läsionen reicht von kleinen, in Vernarbung übergehenden Spitzenherden bis zu ausgedehnten käsigen Pneumonien mit großkavernösem Zerfall. Das im Zentrum der Tuberkel nekrotisierende Granulationsgewebe nimmt käsige Beschaffenheit an und verflüssigt sich mangels proteolytischer Enzyme nur partiell. Wenn der Zerfallsprozess Anschluss an einen Bronchus gewinnt, entleert sich das nekrotische Material nach außen und trägt die Tuberkelbazillen in andere, noch nicht befallene Lungenabschnitte. Zurück bleiben Kavernen genannte Hohlräume, die zur Quelle massiver tödlicher Blutungen werden können. Bei Ruptur in die Pleura entsteht ein tuberkulöses Empyem. In alten Kavernen entwickeln sich manchmal Aspergillome. Wird ein verkästes Infiltrat ohne Gewebseinschmelzung durch Granulationsgewebe abgegrenzt, entsteht ein tumorähnlicher tuberkulöser Rundherd. Erhält er eine Bindegewebskapsel, spricht man von einem Tuberkulom. Beide können bei Reaktivierung einschmelzen und in die Lunge streuen. Parenchymschwund, schrumpfende Narben und sekundäres Emphysem führen zu einer progredienten restriktiven Ventilationsstörung, zur Verteilungsstörung und zum Cor pulmonale. Respiratorische Insuffizienz und Rechtsinsuffizienz kennzeichnen das Finalstadium, das durch rechtzeitige Behandlung abwendbar ist.
2.6.11.13.3.2 Klinik
Gewichtsabnahme, am Abend Temperaturerhöhung, Nachtschweiß, Bewegungstemperaturen, die nach 30 Minuten nicht abklingen. Chronischer Husten, Hämoptysen, Auswurf. Uncharakteristische Oberbauchbeschwerden. Beschleunigung der BKS, oft leichte Anämie. Unbehandelt erreicht der Lungenprozess innerhalb weniger Wochen seine volle Ausdehnung.
2.6.11.13.3.3 Diagnostik
2.6.11.13.3.4 Röntgendiagnostik
Standardaufnahmen in 2 Ebenen, ergänzt durch Computertomographie (◘ Abb. 2.45). Initial ist häufig ein sog. infraklavikuläres Frühinfiltrat nachzuweisen, ein unscharf begrenzter bronchopneumonischer Herd, der schnell verkäsen und einschmelzen kann. Abgeheilte inaktive Herde erscheinen narbig-fibrös und enthalten oft Kalkablagerungen. In progredienten Fällen kommt es zu einer knotig-infiltrativen Oberlappenspitzentuberkulose, die nach kaudal fortschreitet. Unter Kavernenbildung und Fibrosierung wird das Lungengewebe zunehmend zerstört.


Kavernöse Lungentuberkulose. a Exsudative kavernöse Lungentuberkulose mit wolkigen, unscharfen Verdichtungen in beiden Oberfeldern. Kaverne apikolateral rechts (Sammlung Prof. Jend). b CT-Bild einer massiven kavernösen Lungentuberkulose links mit Streuung nach rechts. c 68-jähriger Mann mit behandelter kavernöser Lungentuberkulose. Komplikation durch Besiedlung mit Aspergillen
2.6.11.13.3.5 Klassifikation
Unter Einbeziehung von Körpertemperatur, BKS, bakteriologischem Sputumbefund und Röntgenbild werden folgende Kriterien in der Diagnose zusammengefasst:
-
aktiv (Fieber, erhöhte BKS, Sputum positiv, weiche Schatten) oder inaktiv?
-
offen (Sputum positiv), fakultativ offen (Einschmelzungen bei negativem Sputumbefund) oder geschlossen (Sputum mikroskopisch und kulturell negativ)?
-
Lokalisation: rechtsseitig, linksseitig oder doppelseitig (Ober-, Mittel- oder Untergeschoss bzw. Lappen)?
-
Qualität der Läsionen: exsudativ, produktiv (knotig), indurativ, zirrhotisch oder kavernös?
2.6.11.14 Extrapulmonale Tuberkulose
2.6.11.14.1 Tuberkulöse Perikarditis
Entstehung hämatogen, manchmal gleichzeitig mit einer Pleuritis tuberculosa, oder durch Einbruch eines Lymphknotens ins Perikard. Akutes Initialstadium mit Fieber, Präkordialschmerz und Perikarderguss, mit Lokomotivgeräusch bei der Auskultation. Relativ häufig Übergang in chronische konstriktive Perikarditis. Die Diagnose kann ohne Perikardbiopsie schwierig sein.
2.6.11.14.2 Kehlkopftuberkulose
Kommt bei schwerer offener Lungentuberkulose durch massive Bazillenabsiedlung auf der Schleimhaut vor, selten bei geringen Lungenbefunden. Beginn mit Laryngitis, die Husten und Heiserkeit verursacht, im Verlauf Granulome und Ulzera.
2.6.11.14.3 Nierentuberkulose
Manifestation 5–20 Jahre nach der Infektion. Bazillenabsiedlung während des Generalisationsstadiums in der gut durchbluteten (O2-reichen) Nierenrinde. Dort entstehen primär kleine, narbig abheilende Tuberkel. Nach Reaktivierung kanalikuläre und lymphogene Keimverschleppung. Entstehung von Tuberkeln in Kelchnischen und Papillen, Kavernenbildung, Befall von Ureteren und Harnblase. Nicht selten Hydronephrosen durch Ureterobstruktion.
2.6.11.14.3.1 Klinik
Dysurische Beschwerden, Rückenschmerzen, auch Koliken und Makrohämaturie. Verdächtig sind Pyurie und Mikrohämaturie bei sterilem Harn. Retrograd kann die kontralaterale Niere mitbefallen werden.
2.6.11.14.3.2 Diagnostik
Erregernachweis durch Kultur. Sonogramm und i.v. Pyelogramm zur Erfassung der Läsionen. Zystoskopischer Nachweis der tuberkulösen Zystitis.
2.6.11.14.4 Genitaltuberkulose
2.6.11.14.4.1 Beim Mann
In der Regel sekundär bei Nierentuberkulose, durch absteigende kanalikuläre Infektion entstehend. Betroffen werden Prostata, Samenblasen und Nebenhoden, die langsam anschwellen, ohne auf Druck zu schmerzen.
2.6.11.14.4.2 Bei der Frau
Primärsitz in den Tuben oder im Endometrium. Keine enge Assoziation mit Nierentuberkulose. Entsteht am häufigsten, wenn die Erstinfektion im geschlechtsreifen Alter erfolgt. Kanalikuläre Ausbreitung, lokale Peritonitis, Leibschmerzen und Menstruationsstörungen. Langsame Progredienz.
2.6.11.14.5 Skeletttuberkulose
Im Kindes- und Erwachsenenalter vorkommend. Entsteht durch hämatogene Streuherde, an der Brustwirbelsäule nicht selten durch Übergreifen pulmonaler Prozesse (auf dem Lymphweg). Diagnostisch wegweisend sind die gleichzeitigen Lungenherde.
2.6.11.14.5.1 Knochentuberkulose
Spondylitis tuberculosa der mittleren Brustwirbel mit Gibbus (Pott-Gibbus), Paresen und kalten Abszessen (◘ Abb. 2.46). Bei Kindern Absiedlungen in Epiphysennähe mit subperiostaler Abszedierung, die in die Weichteile durchbricht und zu kalten Abszessen führt. Diagnose durch Biopsie und Kultur (nur in 40 % der Fälle positiv wegen geringen Keimgehaltes im Eiter).


Spondylitis tuberculosa. Verschmälerung des Zwischenwirbelraumes. Destruktion der Abschlussplatten der benachbarten Wirbelkörper
2.6.11.14.5.2 Gelenktuberkulose
Betroffen sind vorwiegend die großen gewichttragenden Gelenke (Hüften und Knie). Früher oft durch M. bovis bedingt, jetzt selten. Chronische, nichttraumatische Bursitiden und Tendovaginitiden lassen an Tuberkulose denken.
2.6.11.14.6 Nebennierentuberkulose
Hämatogener Befall, vor allem der Nebennierenrinde. Kommt meistens nur bei schwerer Lungentuberkulose vor. Klinisch Bild der Nebenniereninsuffizienz.
2.6.11.14.7 Darmtuberkulose
Früher häufige Primärinfektion durch M. bovis mit Verkalkung regionaler Lymphknoten (noch in Entwicklungsländern anzutreffen). Infektion durch M. tuberculosis bei schwerer offener Lungentuberkulose durch Verschlucken bazillenreichen Sputums. Es entsteht eine dem Morbus Crohn ähnliche Ileitis terminalis mit Fistelbildung und Periproktitis. Eine Peritonitis mit Aszites kann hämatogen oder durch Übergreifen einer Darm- oder einer Adnextuberkulose entstehen.
2.6.11.14.8 Hauttuberkulose
2.6.11.14.8.1 Erythema nodosum
Typischerweise in den ersten Wochen der Primärinfektion nach dem Generalisationsstadium auftretend. Stets mit stark positiver Tuberculinreaktion assoziiert. Kommt gelegentlich auch bei postprimärer Tuberkulose vor. Kennzeichen: Blaurote, schmerzhafte, knotige Infiltrate an der Streckseite beider Unterschenkel, zu Schüben neigend, aber ohne Narben abheilend. Sehr unspezifisch.
2.6.11.14.8.2 Lupus vulgaris
Hämatogen entstehende granulomatöse Hauttuberkulose mit kleinen rötlich-gelben Knötchen als Primäreffloreszenz und ausgedehnter Vernarbung. Ausbreitung flächenhaft mit Defektbildung, vor allem in den mittleren Gesichtsanteilen. Gewöhnlich nur bei langdauernder unbehandelter Tuberkulose zu beobachten.
2.6.11.14.9 Augentuberkulose
Uveitis und Chorioretinitis sind die häufigsten Manifestationen. Schwierig gegen andere Ursachen abzugrenzen. Wegweisend kann eine Keratoconjunctivitis phlyctaenularis sein.
2.6.11.14.10 Meningitis tuberculosa
Manifestiert sich überwiegend wenige Wochen nach der Infektion im frühen Generalisationsstadium (30 % noch tuberculinnegativ), weshalb Kinder und junge Erwachsene am häufigsten betroffen sind. Primärtuberkel in der Leptomeninx verkäsen und brechen in den Subarachnoidalraum ein. Die tuberkulöse Meningitis kann im Rahmen einer Miliartuberkulose, aber auch durch hämatogene Streuung bei einer postprimären Tuberkulose auftreten. In 70 % der Fälle findet man spezifische Lungenveränderungen. Betroffen ist die Hirnbasis mit den durchziehenden Hirnnerven und deren Arterien, die entzündlich verschlossen werden können.
2.6.11.14.10.1 Klinik
Schleichender Beginn mit Kopfschmerzen, subfebrilen Temperaturen und Reizbarkeit. Danach folgen Übelkeit, anhaltendes Erbrechen, Meningismus und Trübung des Sensoriums. Doppelbilder, Fazialisschwäche und Schwerhörigkeit zeigen Hirnnervenbeteiligung an. Adhäsionen mit Zisternenverschluss können zum Hydrocephalus externus führen. Bei der selteneren spinalen Meningitis beobachtet man Paraplegien und Wurzelsyndrome.
2.6.11.14.10.2 Diagnostik
Liquoruntersuchung: Klar, farblos, mit Spinnwebgerinnsel (Fibrin). Lymphozytäre Pleozytose mit Zellzahlen bis 1200/3. Liquorzucker unter 40 mg/dl (stark erniedrigte Werte sonst nur bei eitriger Meningitis). Durch inadäquate ADH-Sekretion kann eine Hyponatriämie entstehen. Erregernachweis im Liquor durch Kultur.
Die Diagnose muss umgehend gestellt werden (Klinik, Lungenbefund, Liquorzucker), weil die Therapie nicht verzögert werden darf. Ohne spezifische Therapie besteht eine Letalität von 100 %.
2.6.11.15 Miliartuberkulose
2.6.11.15.1 Pathogenese
Massive hämatogene Bazillenaussaat in alle Organe mit Ausbildung unzähliger hirsekorngroßer Tuberkel, die im Zentrum verkäsen können. Bei Kindern und jungen Erwachsenen erfolgt die Streuung meistens im frühen Generalisationsstadium nach der Primärinfektion, bei älteren Menschen in der Regel aus reaktivierten Herden einer alten Organtuberkulose, gewöhnlich der Lungen. Ausschlaggebend ist eine primäre Schwäche oder ein später Zusammenbruch der Abwehrmechanismen. Immunsupprimierte Patienten können eine besonders schwere, nichtreaktive Generalisation der Tuberkulose bekommen, bei der sich kaum Granulationsgewebe entwickelt, sondern nur überall Bazillen ablagern.
2.6.11.15.2 Klinik
Bei jüngeren Patienten mit Primärtuberkulose wird das Vollbild der Erkrankung früher erreicht als bei älteren. Aber auch die mehr akute Form beginnt schleichend mit allgemeiner Schwäche, Appetitmangel, subfebrilen Temperaturen und Gewichtsverlust. Pulmonale Zeichen sind Atemnot, trockener Husten, schließlich Zyanose.
Der Auskultationsbefund bleibt wegen der interstitiellen Lokalisation des Prozesses unauffällig!
Früher oder später kommen die Zeichen der Meningitis hinzu, die Milz ist meistens mäßig geschwollen. Im Alter besteht oft ein schwer deutbares Krankheitsbild mit rezidivierendem Fieber, Anämie, Lymphknotenschwellungen, Pleura- und Bauchhöhlenexsudat, auch mit Knochenherden und Augenbeteiligung.
2.6.11.15.3 Diagnostik
Schwierigkeiten ergeben sich aus der häufigen Tuberculinallergie, der 2–3-wöchigen Latenzzeit bis zum Auftreten der miliaren Herde im Röntgenbild und dem oft negativen Sputumbefund. Die BKS steigt allmählich an, eine Leukozytose ist nicht obligatorisch. Spezifisches Granulationsgewebe kann durch Leber-, Knochenmarks- und transbronchiale Lungenbiopsie gewonnen werden. Entscheidend ist in der Praxis meistens das Röntgenbild der Lunge. Unbehandelt endet die Miliartuberkulose fast immer tödlich.
2.6.11.15.4 Diagnostik
2.6.11.15.5 Bakteriologie
Die Untersuchung auf Tuberkelbazillen ist notwendig zur Sicherung der Diagnose, zur Beurteilung der Ansteckungsfähigkeit und zur Kontrolle der Therapie. Untersuchungsmaterial: Sputum, Kehlkopfabstrich und Magennüchternsaft bei Lungentuberkulose, Urin bei Nieren- und männlicher Genitaltuberkulose, Menstrualblut bei weiblicher Genitaltuberkulose, Pleuraergüsse, Aszites und Liquor bei Tuberkuloseverdacht und bioptisch gewonnenes Gewebe aus verdächtigen Herden. Folgende Methoden kommen zum Einsatz:
-
Mikroskopie: Untersuchung auf säurefeste Stäbchen mit der Färbung nach Ziehl-Neelsen (Carbolfuchsin, Salzsäure-Alkohol) oder zeitsparend mit einer Auramin-Färbung im Fluoreszenzmikroskop. Geeignet für die Untersuchung von Sputum und Biopsiematerial, nicht absolut spezifisch.
-
Kultur: Wird 8 Wochen beobachtet. Züchtung auf Festmedium (Lövenstein-Jensen) oder Flüssigmedium (MB-Redox-Indikatorsystem).
-
Identifizierung: Mittels DNA-Gensonde. PCR, wenn mikroskopisch positiv bei Aids oder Verdacht auf Tb-Meningitis.
-
Resistenzbestimmung: Dauert 4–6 Wochen.
2.6.11.15.6 Röntgenuntersuchung
In den Röntgenaufnahmen (a.p. und evtl. seitlich) Darstellung von miliaren Herden. In der Regel erster und wichtigster Hinweis auf das Vorliegen einer Tuberkulose. Die jeweiligen Befunde zeigen die Ausdehnung und das Ausmaß der Läsionen und ihr Ansprechen auf die Behandlung.
2.6.11.15.7 Tuberculin-Test
Testung der Reaktion auf intrakutan applizierte Proteinderivate aus Tuberkelbazillen, zur Erfassung der Tuberculinallergie, einer lokalen zellulären Immunreaktion vom verzögerten Typ.
-
Testmaterial: Als gereinigte Präparate stehen Tuberculin GT und Tuberculin PPD Mérieux zur Verfügung.
-
Mendel-Mantoux-Test: Mit Tuberculin GT, das in Ampullen mit Trockensubstanz für 1 IE, 10 IE, 100 IE und 1000 IE geliefert wird. Wenn der Ampulleninhalt in je 1 ml Lösungsflüssigkeit aufgelöst wird, enthalten 0,1 ml die auf der Charge angegebenen Einheiten. Die Standardtestdosis beträgt 10 IE. Man injiziert 0,1 ml mit dieser Dosis streng intrakutan an der desinfizierten Volarseite des Unterarmes. Positiv ist die Reaktion, wenn sich nach 72 h eine Induration von mindestens 2 mm Durchmesser mit umgebender Rötung gebildet hat. Eine Rötung allein bedeutet nichts. Bei negativem Ergebnis kann der Test mit 100 IE wiederholt werden. Liegt ein akutes Krankheitsbild vor, beginnt man mit 1 IE. Es kann sonst eine sehr heftige Kutanreaktion geben.
-
Stempeltest: Der Teststempel enthält 5 IE PPD-Tuberculin Mérieux. Er wird an der desinfizierten Volarseite des Unterarmes unter Straffung der Haut 2 Minuten lang fest aufgedrückt, was einer intrakutanen Injektion gleichkommt.
-
Diagnostische Bedeutung: Die Tuberculinreaktion wird 2–10 Wochen nach der Erstinfektion positiv und bleibt es meistens lebenslang, wenn auch im Alter eine Abschwächung vorkommt. Ein positiver Test zeigt also eine Tuberkuloseinfektion an. Er sagt aber nichts über die Aktivität des Prozesses aus. Eine negative Reaktion kommt im Frühstadium, bei Pleuritis tuberculosa, Meningitis tuberculosa, Miliartuberkulose und Aids häufig vor. Ferner bei anergischen Patienten (Sarkoidose). Bis 1945 waren über 90 % der 20-Jährigen tuberculinpositiv, meistens infolge einer stummen Infektion im Kindesalter. Heute sind die jungen und mittleren Jahrgänge dank der erfolgreichen Tuberkulosebekämpfung weitgehend tuberculinnegativ (Ausnahme: positive Reaktion nach BCG-Impfung). In einem Ansteckungsfall kann daher mit der Tuberculinreaktion mit guten Erfolgsaussichten nach der Infektionsquelle gefahndet werden.
2.6.11.15.8 Therapie
2.6.11.15.9 Allgemeine Maßnahmen
Isolierung auf einer Tuberkulosestation bis zur Beseitigung der Kontagiosität (3 negative Sputumproben). Meldung an das staatliche Gesundheitsamt, das Umgebungsuntersuchungen veranlasst. Bettruhe bei Fieber, später Liegekuren.
2.6.11.15.10 Chemotherapie
Sie ermöglicht eine wirksame Behandlung der Tuberkulose mit allen ihren Manifestationen. Die langsame Vermehrung der Tuberkelbazillen erfordert prinzipiell eine Langzeitbehandlung und zur Erzielung einer optimalen Wirkung die Verabreichung der Tagesdosis aller Medikamente in einer Einzelgabe auf nüchternen Magen, am besten vor dem Frühstück. Gegen jedes Tuberkulosemittel werden in der Erregerpopulation einige Keime durch Mutation resistent. Sie würden bei einer Monotherapie herausgezüchtet und zum Rezidiv führen. Obligatorisch ist deshalb die Kombination von mindestens zwei, am besten drei der wirksamen Mittel (◘ Tab. 2.9).
-
Medikamente der 1. Reihe: Isoniazid, Rifampin, Pyrazinamid Ethambutol, Streptomycin. Alle sind bakterizid und reduzieren schnell die Zahl der lebenden Tuberkelbazillen (Sputum wird negativ) und töten im Verlauf einiger Wochen auch ruhenden Bakterien ab (Sterilisation der Organe). Bis auf Streptomycin werden sie oral appliziert. Die Verträglichkeit ist im Bereich empfohlenen Tagesdosen im Allgemeinen gut. Streptomycin allerdings führt nach längerer Anwendung zu irreversiblen Gehörschädigungen und renalen Funktionsstörungen. Obligatorische Indikationen für Streptomycin: Miliartuberkulose, Meningitis, Perikarditis.
-
Medikamente der 2. Reihe: Die Aminoglykoside Amikacin und Kanamycin, Capreomycin, Paraaminosalicylsäure (PAS), Thiacetazon, Viomycin, Cycloserin. Neuere Mittel: Rifabutin, Rifapentin, Ofloxacin, Ciprofloxacin. Diese Mittel kommen bei Resistenzen gegen jene der 1. Reihe zum Einsatz. Ihre Verträglichkeit ist deutlich geringer.
-
Glukokortikoide: Unter bakterizidem antibiotischem Schutz werden sie zur Dämpfung der entzündlichen Aktivität bei tuberkulöser Meningitis und Perikarditis gegeben.
-
Kontrollen: Bei unzuverlässigen Patienten Überwachung der Medikamenteneinnahme. Sputumkontrollen nach 2 und 4 Monaten zur Sicherung des Behandlungserfolges. Röntgenkontrolle am Behandlungsende und nach einem Jahr. Während der Behandlung Kontrollen von Blutbild, Blutsenkung, Leberfermenten.
-
Therapieversagen: Wenn Sputumkulturen nach 3 Monaten noch positiv sind. Nach erneuter Testung der Erregerempfindlichkeit Umstellung auf eine geänderte Kombinationstherapie über längere Zeit. Hoffnung auf ein neues Mittel gegen resistente oder wenig empfindliche Tuberkelbazillen weckt die bisher nur an Mäusen erfolgreich getestete Substanz R207910, ein Diarylchinolin, das die ATP-Synthase der Tuberkelbazillen hemmt und gegen mehrere Stämme von M. tuberculosis aktiv war (Andries, K. et al. Science 2005;307:223-7).
2.6.11.15.11 Operative Therapie
Drainage beim tuberkulösen Pleuraempyem, Dekortikation bei ausgedehnten Pleuraschwarten im jüngeren Alter. Teilresektionen bei persistierenden Kavernen, Bronchiektasen und Rundherden, auch wenn der Tuberkulomverdacht nahe liegt, da Narbenkarzinome in solchen Herden vorliegen können. Alle Eingriffe unter Abdeckung mit INH und RMP; medikamentöse Nachbehandlung für 2–4 Monate zur Verhütung einer postoperativen Reaktivierung.
2.6.11.15.12 Prophylaxe
2.6.11.15.13 BCG-Impfung
Immunisierung Nichtinfizierter mit lebenden Keimen von Bacillus Calmette-Guérin, einem stark abgeschwächten Mycobacterium-bovis-Stamm. Streng intrakutan werden 0,1 ml der Vakzine (100.000–300.000 Keime je Dosis) injiziert. Die Impfung sollte am besten kurz nach der Geburt erfolgen, weil noch keine Ansteckung erfolgt sein kann. Die Tuberculin-Probe wird nach wenigen Wochen positiv. Erreicht wird ein Ansteckungsschutz von über 80 %. Auf jeden Fall werden die schweren disseminierten Formen eliminiert. Anwendung in gefährdeten Regionen und beim Pflegepersonal. Massenimpfungen würden die Früherfassung der Tuberkulose durch den Tuberculin-Test erschweren.
2.6.11.15.14 Chemoprophylaxe
Sie kann die Infektion mit Tuberkelbazillen nicht verhindern, aber bei richtigem Einsatz den Übergang von der Infektion zur Krankheit. Die Durchführung erfolgt mit täglicher Gabe von 300 mg Isoniazid und ist in folgenden Situationen indiziert:
-
Bei tuberculinnegativen Personen, die engen Kontakt mit Patienten mit offener Tuberkulose hatten (Haushalt, Arbeitsplatz etc.). Wird die Tuberculinreaktion nach 2 Monaten positiv, Fortsetzung der INH-Einnahme, andernfalls Beendigung.
-
Bei älteren tuberculinpositiven schwerkranken Patienten mit länger zurückliegender Tuberkulose, die niemals Tuberkulosemittel erhalten haben. Dadurch soll der im Alter relativ häufigen Reaktivierung begegnet werden.
-
Eventuell bei tuberculinpositiven Patienten mit echtem Risiko der Reaktivierung (Kortikoidbehandlung, Immunsuppression).
2.7 Erkrankungen von Pleura , Mediastinum und Zwerchfell
Erkrankungen von Pleura, Mediastinum und Zwerchfell
-
Pneumothorax
-
Pleuritis
-
Pleuraempyem
-
Pleuraerguss
-
Pleuratumoren
-
Primäre Pleuratumoren
-
Metastatische Pleuratumoren
-
-
Mediastinitis
-
Mediastinaltumoren
-
Lage- und Funktionsanomalien des Zwerchfells
2.7.1 Pneumothorax
2.7.1.1 Definition
Ansammlung von Luft im Pleuraspalt mit partiellem oder totalem Lungenkollaps.
-
Geschlossener Pneumothorax ohne Verbindung zur Außenluft.
-
Offener Pneumothorax mit Verbindung zum Bronchialsystem oder durch die Brustwand nach außen.
2.7.1.2 Ätiologie
2.7.1.3 Spontanpneumothorax
Umfasst alle nicht durch äußere Gewalteinwirkung entstehenden Fälle von Pneumothorax:
-
Idiopathische Form: Ursache unklar. Überwiegend bei jungen Erwachsenen zwischen dem 20. und 40. Lebensjahr. Es platzen kleinste Bläschen, meistens in der Lungenspitze. Keine organische Pleura- oder Lungenkrankheit. Neigung zu Rezidiven.
-
Sekundäre Formen: Ruptur der Pleurablätter bei Asthma, Emphysem, Bronchialkarzinom, Lungenmetastasen, Ösophaguskarzinom, Staphylokokkenabszessen, Tuberkulose, Alveolitis, Sarkoidose, Pneumokoniosen, Marfan-Syndrom, Ehlers-Danlos-Syndrom u. a. Lungenaffektionen.
2.7.1.4 Traumatischer Pneumothorax
Entsteht durch äußere Gewalteinwirkung bei verschiedenen Anlässen:
-
Unfall: durch perforierende Brustkorb- und Lungenverletzungen und stumpfe Traumen mit Rippenbrüchen
-
iatrogen bedingt: Brustkorberöffnung, Thorakoskopie, perkutane und transbronchiale Lungenbiopsie, Subklaviapunktion, Pleurapunktion, Pleuradrainage, kardiale Reanimation, transdiaphragmale Leberbiopsien
-
Barotrauma: Überdruckbeatmung
2.7.1.5 Pathogenese und Pathophysiologie
Jedes Öffnen des Pleuraspalts nach innen oder außen bewirkt, dass die Lunge mit ihrer elastischen Retraktionskraft Luft in den Pleuraraum ansaugt und dabei an Volumen verliert, bis sie völlig kollabiert ist. Je nach dem Grad des Pneumothorax wird der physiologische Unterdruck im Pleuraraum teilweise oder ganz aufgehoben. Durch die elastischen Zugkräfte der nicht betroffenen Lunge verlagert sich das Mediastinum nach der gesunden Seite. Beim Ventilpneumothorax wird inspiratorisch Luft in den Pleuraraum angesaugt und nicht wieder abgegeben. Steigt der Luftdruck im Pneumothorax über den athmosphärischen Druck, werden das Mediastinum mit seinen Gefäßen und die gesunde Lunge komprimiert. Ein solcher Spannungspneumothorax führt zu einer akut bedrohlichen Abnahme des Herzschlagvolumens und der Lungenfunktion. Bedrohlich ist auch die selten vorkommende Einblutung in den geöffneten Pleuraraum. Sie kann zum hämorrhagischen Schock führen.
2.7.1.6 Klinik
Beim Spontanpneumothorax oft plötzlicher stechender Schmerz. Schweres Heben oder ein Hustenanfall können vorausgehen. Ruhedyspnoe und Tachykardie nur in schweren Fällen und bei vorbestehender Lungenkrankheit. Beim Spannungspneumothorax progrediente Dyspnoe, Tachykardie, Schweißausbruch, Hautblässe, große Unruhe und Angst, schließlich Kollaps. Auf der betroffenen Seite Klopfschall hypersonor, Atemgeräusch abgeschwächt oder aufgehoben. Keine physikalischen Zeichen bei kleinem Pneumothorax.
2.7.1.7 Diagnostik
Durch Röntgenuntersuchung der Lunge (◘ Abb. 2.47). Abgrenzung der kollabierten Lunge vom luftgefüllten Pleuraraum, dem die Lungenzeichnung fehlt. Bei der Durchleuchtung kann Mediastinalflattern sichtbar werden.


Pneumothorax im rechten Unterfeld mit CT-Bild (Sammlung Prof. Jend)


64-jährige Frau mit therapiertem Mammakarzinom. Nach 5 Jahren Ausbildung eines Pleuraergusses. Thorakoskopisch Nachweis einer Lymphangiosis carcinomatosa
2.7.1.8 Therapie und Verlauf
2.7.1.9 Spannungspneumothorax
Der Spannungspneumothorax erfordert eine sofortige Punktion (auch ohne Röntgenuntersuchung) mit dicker Kanüle erfordert (im 3. ICR und Anschluss einer halbgefüllten 20-ml-Spritze ohne Stempel). Nach der Punktion Pleuradrainage, evtl. chirurgische Versorgung.
Der Spannungspneumothorax ist eine Notfallsituation!
2.7.1.10 Spontanpneumothorax
Besonders bei der idiopathischen Form ist mit rascher Schließung im kollabierten Zustand der Lunge zu rechnen. Wenn die Luftansammlung nicht breiter als ein Querfinger ist und der intrapleurale Druck nicht über 5 cm Wassersäule, kann die Spontanresorption abgewartet werden, die wenige Wochen benötigt. Bei größerem Pneumothorax oder intrapleuralem Druck über 5 cm Wassersäule ist eine Pleurasaugdrainage anzulegen. Bleibt die Lunge innerhalb von 8 Tagen nicht entfaltet, muss chirurgisch eingegriffen werden.
2.7.2 Pleuritis
2.7.2.1 Definition
Entzündung der Pleura, die ohne Exsudatbildung (Pleuritis sicca) oder mit Exsudatbildung (Pleuritis exsudativa) einhergehen kann.
2.7.2.2 Ätiologie und Pathogenese
Es kann sich um bakterielle und nichtbakterielle Entzündungen handeln. Der Entzündungsprozess kann von den benachbarten Organen (Lunge, Zwerchfell, Mediastinum, Ösophagus, Brustwand) auf die Pleura übergreifen oder von der Pleura ausgehen. Die wichtigsten Ursachen sind in ◘ Tab. 2.10 aufgeführt.
2.7.2.3 Klinik
2.7.2.4 Pleuraschmerz
Scharfe, oft heftige Bruststiche, bei der Atembewegung auftretend oder an Intensität eindeutig zunehmend. Sowohl bei trockener wie bei beginnender exsudativer Entzündung.
2.7.2.5 Pleurareiben
In- und exspiratorisches Reibegeräusch bei der Auskultation deutlich wahrzunehmen. Bei trockener und beginnender exsudativer Pleuritis.
2.7.2.6 Klopfschalldämpfung und Aufhebung des Atemgeräusches
Nur bei exsudativer Pleuritis mit stärkerer Ergussbildung.
2.7.2.7 Diagnostik
2.7.2.8 Röntgenuntersuchung
Nachweis der Exsudation und narbiger Restzustände bzw. nachfolgender Pleuraschwarten. Feststellung primärer Lungenerkrankungen. Zusätzliche Klärung durch Computertomographie.
2.7.2.9 Sonographie
Erlaubt den Nachweis kleiner, im Röntgenbild nicht eindeutig zu erfassender Ergussmengen.
2.7.2.10 Pleurapunktion
Gewinnung des Exsudats zur chemischen, mikroskopischen, zytologischen, bakteriellen und PCR-Untersuchung.
Abgrenzung gegen Transsudate (Stauungsergüsse):
-
Exsudat: Spez. Gewicht >1016, Proteingehalt >3 g/dl, Leukozyten >103/mm3
-
Transsudat: Die für Exsudat genannten Grenzwerte werden unterschritten.
2.7.2.11 Thorakoskopie und Pleurabiopsie
In vielen Fällen für die Klärung der Diagnose entscheidend (Tuberkulose, Tumor).
2.7.2.12 Therapie
Behandlung der Grundkrankheit. Bei stärkerer Beeinträchtigung der Atmung können mehrmalige Entlastungspunktionen oder eine Drainage erforderlich werden. Bei ausgedehnter Schwartenbildung chirurgische Dekortikation.
2.7.3 Pleuraempyem
2.7.3.1 Ätiologie
Eiteransammlungen im Pleuraraum kommen fast nur als Zweiterkrankung vor, am häufigsten als para- oder metapneumonisches Empyem. Weitere Ursachen sind: perforierter Lungenabszess, septische oder aseptische Lungenoperationen, Nahtdehiszenz nach Ösophagusoperationen, Infektion bei Drainagebehandlung eines nichteitrigen Pleuraergusses oder Pneumothorax, Lungentuberkulose, perforierende oder penetrierende Verletzungen, septische Metastasen (Staphylokokkensepsis). Als Erreger sind Aerobier und Anaerobier von Bedeutung: Staphylokokken, Klebsiella pneumoniae, E. coli, Bakteroides, Peptostreptokokkus, Proteus, Pseudomonas. Weniger häufig findet man Streptokokken und Pneumokokken oder die alleinige Besiedlung mit Candida albicans.
2.7.3.2 Pathogenese
Der Keimbesiedlung der Pleura folgt die Absonderung eines leukozyten- und fibrinreichen Exsudats, das durch Gerinnung gallertartig wird und in einen dicken Fibrinbelag übergeht, der die Pleurablätter verklebt. Einwandernde Fibroblasten verstärken die Wandhaftung. Zwischen verfestigten Arealen bleiben Eiteransammlungen bestehen (gekammertes Empyem). Unbehandelt erfolgt der Übergang in die chronische Phase mit Schrumpfung der Schwiele und Verkleinerung der befallenen Brustkorbhälfte, Mediastinalverziehung und Zwerchfellraffung. Der zentrale Anteil des Schwartensacks bleibt meistens mit Eiter gefüllt. Die wesentlichen Empyemfolgen für den Patienten sind: chronischer toxischer Infektherd (Amyloidoserisiko), Thoraxdeformität, restriktive Ventilationsstörung und sekundäres Cor pulmonale.
2.7.3.3 Klinik
Brustschmerz, Husten, später Fieber, Nachtschweiß, Leukozytose und Anstieg der Blutsenkung, auch Thrombozytose. Empyeme können sich als erkennbare Exazerbation einer Grundkrankheit entwickeln (meta- oder postpneumonisch), aber auch larviert, z. B. nach einer zirkumskripten Aspirationspneumonie, die nicht registriert wurde. Symptomarm und leicht zu übersehen sind Empyeme, die sich unter einer Antibiotikabehandlung entwickeln.
2.7.3.4 Diagnostik
Den entscheidenden Hinweis gibt das Röntgenbild, manchmal durch ein Computertomogramm zu ergänzen (◘ Abb. 2.49). Man sieht homogene Pleuraverschattungen, die verschiebbar sind, auch gekammerte und hängende Ergüsse, im Spätstadium Pleuraschwarten. Sicherung der Diagnose durch gezielte Punktion. Weitere Abklärung durch bakteriologische Untersuchung einschließlich PCR. Steriler Eiter ist tuberkuloseverdächtig.


42-jähriger Alkoholiker mit verschleppter Fiebersymptomatik und Ausbildung eines Empyems. a CT-Bild, b Bild der Thorakoskopie
2.7.3.5 Therapie
Punktion bis zur vollständigen Entleerung. Bei Bakterienwachstum im Punktat Spül-Saug-Drainage am tiefsten Punkt. Zusätzlich systemische Antibiotikabehandlung nach Empfindlichkeit der Erreger. Beim gekammerten, nicht zu entleerendem Empyem Thorakotomie und Ausräumung.
2.7.4 Pleuraerguss
2.7.4.1 Physiologie
Die geringe physiologische Flüssigkeitsmenge im Pleuraspalt wird von der parietalen Pleura fortlaufend sezerniert und von der viszeralen Pleura rückresorbiert. Der niedrige Kapillardruck im Lungenkreislauf (5–10 mmHg) bewirkt bei einem onkotischen Druck des Plasmas von 28 mmHg, dass an der viszeralen Pleura ein Resorptionssog von etwa 20 mmHg entsteht. Makromoleküle und etwas Flüssigkeit werden über Lymphgefäße der parietalen Pleura abgeleitet, die sich im basalen und diaphragmalen Bereich in den Pleuraraum öffnen.
2.7.4.2 Pathogenetische Klassifikation
Bei den Pleuraergüssen unterscheidet man 2 Formen, die sich in ihrer Entstehung und Zusammensetzung unterscheiden:
-
Exsudat: Entsteht in der erkrankten Pleura durch Steigerung der Kapillarpermeabilität der parietalen Pleura. Es hat einen erhöhten Eiweiß- und Zellgehalt und wird schlechter als die normale Pleuraflüssigkeit resorbiert.
-
Transsudat: Entsteht in der gesunden Pleura, vor allem bei kardialer Stauung. Durch die Erhöhung des Kapillardrucks im großen Kreislauf wird die Sekretion der parietalen Pleura gesteigert, durch die Erhöhung des Kapillardrucks im gestauten kleinen Kreislauf gleichzeitig die Rückresorption der viszeralen Pleura herabgesetzt. Auch der Lymphabfluss ist vermindert. Bei Pleuraexsudaten durch Hypalbuminämie (Leberzirrhose, nephrotisches Syndrom) bewirkt der Abfall des onkotischen Drucks, dass die Flüssigkeitsfiltration aus der parietalen Pleura zunimmt und die Rückresorption durch die viszerale Pleura nachlässt.
2.7.4.3 Ätiologie
Exsudate entstehen durch Entzündungen der Pleura (◘ Abb. 2.50) und bei Tumorbefall, Transsudate bei kardialer Stauung, Störungen des Lymphabflusses und bei Herabsetzung des onkotischen Drucks im Plasma (▶ pathogenetische Klassifikation). Sonderfälle einer Flüssigkeitsansammlung sind der Hämatothorax (stumpfe oder penetrierende Verletzungen, Spontanpneumothorax, hämorrhagische Diathesen, Antikoagulanzienbehandlung) und der Chylothorax (Lymphabfluss aus dem Ductus thoracicus in den Pleuraspalt bei Verletzung oder Obstruktion des Duktus durch Fibrose oder Tumoren, selten bei Lymphangiomyomatose).


Lungenabszess mit Empyem nach Pleuradurchbruch. Spiegelbildung durch Eindringen von Luft in den Pleuraraum (Pyopneumothorax). (Sammlung Prof. Jend)
2.7.4.4 Klinik
Schmerzen bei Pleuritis und Tumorbefall. Ganz allgemein Atemnot durch die Behinderung der Respiration. Dazu die Symptome der Grundkrankheit.
2.7.4.5 Diagnostik
Lokalisation und Ausdehnung werden durch die Röntgenuntersuchung des Thorax erfasst. Abgrenzung vom Exsudat durch Punktatuntersuchung (▶ Abschn. 2.7.2). Beim Vorliegen eines Exsudats führen folgende Untersuchungen diagnostisch weiter: Pleurabiopsie, zytologische Untersuchung (Tumorzellen, Eosinophile, auch Asbeststäbchen), Bakteriologie, Amylasebestimmung (erhöhte Werte bei Pankreatitis).
2.7.5 Pleuratumoren
2.7.5.1 Primäre Pleuratumoren
2.7.5.1.1 Pleurafibrom
Seltener lokalisierter gut abgekapselter Tumor, der meistens zufällig entdeckt wird. Gelegentlich besteht dumpfer Brustkorbschmerz. Oft sind Trommelschlegelfinger vorhanden. Chirurgische Entfernung ist in jedem Fall angezeigt, da manche der Tumoren sich wie langsam wachsende Malignome verhalten.
2.7.5.1.2 Malignes Mesotheliom
Sehr bösartiger, aggressiver Tumor, der selten in einer lokalisierten, dafür häufiger in einer diffusen Form vorkommt (◘ Abb. 2.51). Enge Assoziation mit Asbest-Exposition. Unbehandelt überleben die meisten Patienten weniger als ein Jahr. Therapie verbessert die Prognose um einiges: Extrapleurale Pneumonektomie, danach Chemotherapie (Cisplatin) und Radiotherapie.


Pleuraerguss rechts bei einem Patienten mit langjährigem Asbestkontakt. Nach Ablassen des Ergusses im Ultraschall solide Raumforderung. Mittels ultraschallgesteuerter Punktion Nachweis eines Mesothelioms
2.7.5.2 Metastatische Pleuratumoren
Kommen vor allem bei Mamma-, Pankreas-, Magen-, Uterus-, Ovarial-, Nieren- und Blasenkarzinomen vor. Am häufigsten ist das auf die Pleura übergreifende Bronchialkarzinom. Ein Pleurabefall bedeutet meistens Inoperabilität. Zytostatische Behandlung je nach Primärtumor, evtl. mit lokaler Instillation in den Pleuraraum.
2.7.6 Mediastinitis
2.7.6.1 Definition
Entzündungsprozesse im Mediastinum, dem zwischen beiden Pleurahöhlen gelegenen Gebiet. Man unterscheidet einen vorderen und hinteren Anteil. Als Grenze wird die Hinterwand der Trachea angenommen. Im vorderen Anteil werden oberes und unteres Mediastinum unterschieden. Die Grenze liegt in Höhe der Bifurkation der Trachea.
2.7.6.2 Ätiologie und Pathogenese
Häufigste Ursachen sind Perforationen des Ösophagus durch Ulzera, Tumoren, Divertikel und instrumentelle Verletzungen (Endoskopie, Bougierung, postoperative Nahtinsuffizienz). Außerdem können Entzündungsprozesse vom Tracheobronchialsystem, der Lunge und der mediastinalen Pleura (Tumoren, Abszesse etc.), von den Halsweichteilen (peritonsillitischer Abszess, Halsphlegmonen) sowie von Sternum und Wirbelsäule (Osteomyelitis) auf das Mediastinum übergreifen.
2.7.6.3 Klinik
2.7.6.4 Akute Mediastinitis
Äußerst bedrohliches Krankheitsbild mit Fieber, Tachykardie, Tachypnoe, Retrosternalschmerz, Schluckbeschwerden, Husten, Erbrechen und Singultus. Eine obere Einflussstauung (Thrombose der V. cava superior) und ein Mediastinalemphysem mit Aufsteigen in die Halsregion können hinzukommen.
2.7.6.5 Chronische Mediastinitis
Meistens nur mit geringen Beschwerden verbunden, vor allem Hustenreiz und retrosternale Sensationen. Mykose, Tuberkulose und Lues sind die Hauptursachen.
2.7.6.6 Diagnostik
Die Diagnose ergibt sich aus der Vorerkrankung und dem klinischen Bild, ergänzt durch die Röntgenuntersuchung des Thorax (Verbreiterung des Mediastinums) und des Ösophagus (mit wasserlöslichem Kontrastmittel). Die genaueste Darstellung erlaubt die Computertomographie.
2.7.6.7 Therapie
Bei akuter Mediastinitis Antibiotika und Drainage des Mediastinums nach kollateraler, parasternaler oder posteriorer Mediastinotomie. Hohe Letalität. Bei chronischer Mediastinitis Behandlung des Grundleidens.
2.7.7 Mediastinaltumoren
2.7.7.1 Klassifikation
Im medizinischen Sprachgebrauch werden mit der Bezeichnung „Mediastinaltumor“ alle raumfordernden Gebilde im Mediastinum zusammengefasst, obwohl nicht alle Geschwülste sind. Sie stellen sich im Röntgenbild als umschriebene Verschattungen im Mediastinum dar und sind wie folgt zu unterteilen:
-
Vorderes oberes Mediastinum
-
Retrosternale Struma: Überwiegend durch einen Gefäßstrang mit der Schilddrüse verbunden, selten als aberrierende Struma isoliert.
-
Thymom: Meistens gutartige, von epithelialen Zellen ausgehende Geschwülste, die noch T-Lymphozyten produzieren können (◘ Abb. 2.52). Mit dem Durchbrechen der Kapsel werden sie maligne.
Abb. 2.52 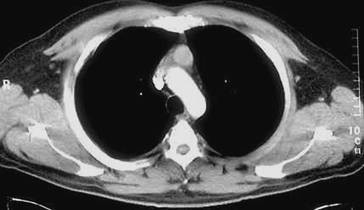
Thymom im vorderen oberen Mediastinum (Sammlung Prof. Jend)
-
Paraneoplastische Syndrome: Myasthenia gravis (10–15 %), aplastische Anämie (5 %), Thrombopenie und Leukopenie (30 %), Hypogammaglobulinämie (5–10 %). Nur in einem Drittel der Fälle bilden sich diese Manifestationen nach der Thymektomie zurück. In jedem Fall Totalresektion, zusätzlich evtl. kombinierte Chemotherapie.
-
Lipomatose: Bei Adipositas, Kortikoidtherapie oder idiopathisch.
-
Aneurysma der Aorta ascendens: ▶ Kap. 1.
-
-
Hilusbereich des Mediastinums: hiläre und mediastinale Lymphknotenvergrößerung bei malignen Lymphomen (◘ Abb. 2.16b), Leukosen, Lymphknotenmetastasen pulmonaler und extrapulmonaler Primärtumoren. Ferner bei Sarkoidose und Tuberkulose.
-
Vorderes unteres Mediastinum: teratoide Tumoren (Dermoidzysten, Teratome), Perikardzysten.
-
Diaphragmaler Bereich des Mediastinum: Hiatushernien.
-
Hinteres oberes Mediastinum: neurogene Tumoren (Neurinome, Neurofibrome, Sympathogoniome, Sympathikoblastome).
-
Hinteres unteres Mediastinum: Bronchialzysten, paravertebrale Abszesse, Aneurysmen der Aorta descendens.


2.7.7.2 Klinik
Durch Verdrängung, Kompression oder Läsion benachbarter Strukturen können mediastinale Prozesse folgende Störungen hervorrufen:
-
Obere Einflussstauung: Kompression oder Verschluss der V. cava superior mit Venenstauung und Zyanose der oberen Körperhälfte, sowie Anschwellung des Halses (Stoke-Kragen)
-
Husten, Stridor: Kompression des Tracheobronchialsystems
-
Schluckstörungen: Verdrängung und Kompression des Ösophagus
-
Heiserkeit: Rekurrenslähmung
-
Horner-Symptomenkomplex: Grenzstrangschädigung
-
Zwerchfellparese: Phrenikuslähmung
-
Singultus: Tumorinfiltration des Zwerchfells
-
Herzrhythmusstörungen: Tumorinfiltration des Herzens, Vaguslähmung
-
Magen-Darm-Atonie: Vaguslähmung
2.7.7.3 Diagnostik
2.7.7.4 Röntgenuntersuchung des Thorax
Die meisten raumfordernden Gebilde des Mediastinums werden durch die Standardaufnahmen in zwei Ebenen erfasst. Von den Thymomen entgehen nicht wenige dem Nachweis.
2.7.7.5 Computertomogramm
Indiziert bei abnormen Befunden im Standardröntgenbild, weil damit eine Unterscheidung zwischen Gefäßen, Zysten, Fettgewebe und Weichteilgewebe möglich ist. Sicherstes Zeichen der Gutartigkeit ist der Nachweis von Fettgewebe, das als Lipomatose häufig vorkommt. Außerdem ist durch das CT auch eine genaue Lokalisation möglich.
2.7.7.6 Weitere diagnostische Verfahren
Je nach der Lokalisation des Tumors und den klinischen Daten kommen in Betracht: Schilddrüsenszintigraphie, digitale Subtraktionsangiographie, Röntgenuntersuchung des Ösophagus, Bronchoskopie, Mediastinoskopie, Skalenus-Biopsie.
2.7.7.7 Therapie
Chirurgische Entfernung aller abgrenzbaren Tumoren, die Verdrängungserscheinungen bewirken. Bei malignen Prozessen Zytostatika und Röntgenbestrahlung, falls Operabilität nicht gegeben ist.
2.7.8 Lage- und Funktionsanomalien des Zwerchfells
2.7.8.1 Beidseitiger Zwerchfellhochstand
In den meisten Fällen durch Hochdrängung vom Abdomen her verursacht, z. B. bei Adipositas, Aszites, starker Lebervergrößerung, Schwangerschaft und sonstiger intraabdominaler Raumforderung. Ferner bei doppelseitiger Zwerchfelllähmung neurogener (Poliomyelitis, Polyradikulitis, amyotrophischer Lateralsklerose, Porphyrie) oder muskulärer (Myasthenia gravis, Muskeldystrophien, Curarevergiftung) Genese.
2.7.8.2 Einseitiger Zwerchfellhochstand
-
Verkleinerung einer Thoraxhälfte bzw. Volumenverminderung einer Lunge: Lungenembolie, Schrumpfungsprozesse.
-
Relaxatio diaphragmatica: Halbseitige Zwerchfellparese durch Phrenikusschädigung oder myogene Schwäche. Bei der Inspiration bewegt sich die gelähmte Seite (paradox) nach aufwärts (Röntgen-Durchleuchtung mit Schnupfversuch).
-
Reflektorische Relaxatio: Subphrenischer Abszess, Pankreatitis, Tumorinfiltration.
-
Hochdrängung vom Abdomen ausgehend: Magen- oder Kolonüberblähung (Ileus), Lebertumoren oder Zysten, große Milztumoren.
-
Zwerchfelltumoren: Lipome, Fibrome und Neurofibrome, selten maligne Fibrosarkome.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Piper, W. (2013). Krankheiten der Atmungsorgane. In: Innere Medizin. Springer-Lehrbuch. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-33108-4_2
Download citation
DOI: https://doi.org/10.1007/978-3-642-33108-4_2
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-33107-7
Online ISBN: 978-3-642-33108-4
eBook Packages: Medicine (German Language)