
Abstract
The family Arenaviridae currently comprises over 20 viral species, each of them associated with a main rodent species as the natural reservoir and in one case possibly phyllostomid bats. Moreover, recent findings have documented a divergent group of arenaviruses in captive alethinophidian snakes. Human infections occur through mucosal exposure to aerosols or by direct contact of abraded skin with infectious materials. Arenaviruses merit interest both as highly tractable experimental model systems to study acute and persistent infections and as clinically important human pathogens including Lassa (LASV) and Junin (JUNV) viruses, the causative agents of Lassa and Argentine hemorrhagic fevers (AHFs), respectively, for which there are no FDA-licensed vaccines, and current therapy is limited to an off-label use of ribavirin (Rib) that has significant limitations. Arenaviruses are enveloped viruses with a bi-segmented negative strand (NS) RNA genome. Each genome segment, L (ca 7.3 kb) and S (ca 3.5 kb), uses an ambisense coding strategy to direct the synthesis of two polypeptides in opposite orientation, separated by a noncoding intergenic region (IGR). The S genomic RNA encodes the virus nucleoprotein (NP) and the precursor (GPC) of the virus surface glycoprotein that mediates virus receptor recognition and cell entry via endocytosis. The L genome RNA encodes the viral RNA-dependent RNA polymerase (RdRp, or L polymerase) and the small (ca 11 kDa) RING finger protein Z that has functions of a bona fide matrix protein including directing virus budding. Arenaviruses were thought to be relatively stable genetically with intra- and interspecies amino acid sequence identities of 90–95 % and 44–63 %, respectively. However, recent evidence has documented extensive arenavirus genetic variability in the field. Moreover, dramatic phenotypic differences have been documented among closely related LCMV isolates. These data provide strong evidence of viral quasispecies involvement in arenavirus adaptability and pathogenesis. Here, we will review several aspects of the molecular biology of arenaviruses, phylogeny and evolution, and quasispecies dynamics of arenavirus populations for a better understanding of arenavirus pathogenesis, as well as for the development of novel antiviral strategies to combat arenavirus infections.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Arenaviruses as Both Important Human Pathogens and Highly Tractable Experimental Systems for the Investigation of Virus–Host Interactions
1.1 Arenaviruses and Their Impact in Human Health
Arenaviruses cause chronic infections of rodents across the world, and human infections occur through mucosal exposure to aerosols or by direct contact of abraded skin with infectious materials (Buchmeier et al. 2007). Both viral and host factors contribute to a variable outcome of arenavirus infection, ranging from virus control and clearance by the host defenses to chronic infection in the absence of clinical symptoms to severe disease. Several arenaviruses cause hemorrhagic fever (HF) disease in humans and pose a serious public health problem in their endemic regions. Thus, the Old World (OW) arenavirus, Lassa virus (LASV) , infects several hundred thousand individuals yearly in West Africa resulting in a high number of Lassa fever (LF) cases associated with high morbidity and mortality (Bray 2005; Geisbert and Jahrling 2004). The current LF endemic areas cover approximately 80 % of Sierra Leone and Liberia; 50 % of Guinea; 40 % of Nigeria; 30 % of each of Côte d’Ivoire, Togo, and Benin; and 10 % of Ghana (Fichet-Calvet and Rogers 2009), and recent studies have found evidence for expansion of LASV endemic regions into Mali, Senegal, and the Democratic Republic of Congo (Sogoba et al. 2012). Thus, it is quite likely that population at risk of LF includes most of West Africa and can be as high as 200 million people (Richmond and Baglole 2003); hence, with dengue fever exception, the estimated global burden of LF is the highest among viral HF (Falzarano and Feldmann 2013). Notably, increased traveling to and from endemic regions has led to the importation of LF cases into non-endemic metropolitan areas around the globe (Freedman and Woodall 1999; Isaacson 2001). Likewise, the New World (NW) arenavirus, Junin virus (JUNV) , causes Argentine HF (AHF) , a disease endemic to the Argentinean Pampas with hemorrhagic and neurological manifestations and a case fatality of 15–30 % (Peters 2002). In addition, evidence indicates that the worldwide-distributed prototypic arenavirus, lymphocytic choriomeningitis virus (LCMV) , is a neglected human pathogen of clinical significance (Jahrling and Peters 1992; Mets et al. 2000; Palacios et al. 2008; Peters 2006).
Public health concerns about arenavirus infections are exacerbated because of the lack of FDA-approved vaccines against HF arenaviral diseases and current therapy to combat arenavirus infections being limited to an off-label use of the nucleoside analogue ribavirin (Rib) (Huggins 1989; Kilgore et al. 1995, 1997; McCormick et al. 1986; McKee et al. 1988). However, Rib is only partially effective, and its use has several limitations including the need of intravenous (iv) and early administration for optimal efficacy, and significant side effects (Damonte and Coto 2002). Although the live-attenuated Candid1 strain of JUNV has been shown to be an effective vaccine against AHF (Enria and Barrera Oro 2002), outside Argentina Candid1 has only investigational new drug (IND) status and it does not protect against LF. Despite efforts dedicated to the development and testing of LASV vaccines, not a single LASV vaccine candidate has entered a clinical trial although several vaccine platforms, including the MOPV/LASV reassortant ML29 and recombinant VSV and vaccinia virus expressing viral antigens, have been successfully tested in non-human primates (Falzarano and Feldmann 2013; Lukashevich 2012).
1.2 LCMV Infection of the Mouse as the Rosetta Stone of Virus–Host Interaction
Studies using LCMV infection of its natural host, the mouse, have led to major advances in virology and immunology that apply universally to other microbial infections and viral infections of humans including virus-induced immunopathological disease, MHC restriction, T cell-mediated killing, and effectiveness of adoptive immune T cell therapy in clearing viral infection (Zinkernagel 2002; Oldstone 2002). Likewise, the initial description of the contribution to viral persistence of negative immune regulators such as PDL-1 was made first with LCMV in mice (Barber et al. 2006). The outcome of LCMV infection of its natural host, the mouse, varies dramatically depending on the age, immunocompetence and genetic background of the host, as well as the route of infection, and the strain and dose of infecting virus (Oldstone 2002; Zinkernagel 2002; Buchmeier et al. 2007). Thus, iv inoculation of adult immunocompetent mice with LCMV Armstrong (ARM) strain results in an acute infection that induces a protective immune response that mediates virus clearance in 10–14 days, a process predominantly mediated by virus-specific CD8+ cytotoxic T lymphocytes (CTL). In contrast, iv inoculation with a high (≥2 × 106 PFU) dose of the immunosuppressive LCMV clone 13 (Cl-13) strain, which differs from ARM only at three amino acid positions, causes persistent infection associated with generalized immune suppression (Ahmed and Oldstone 1988; Ahmed et al. 1984, 1988). These dramatic phenotypic differences between genetically very closely related viruses provide investigators with a superb experimental system to examine both host immune mechanisms of virus control and viral counteracting strategies.
2 Molecular and Cell Biology of Arenaviruses
2.1 Arenavirus Genome Organization
Arenaviruses are enveloped viruses with a bi-segmented negative-stranded (NS) RNA genome and a life cycle restricted to the cell cytoplasm (Buchmeier et al. 2007). Virions appear pleomorphic when examined by cryoelectron microscopy, ranging in size from 40 to more than 200 nm in diameter. Each genomic viral RNA segment, L (ca 7.3 kb) and S (ca 3.5 kb), uses an ambisense coding strategy to direct the synthesis of two polypeptides in opposite orientation, separated by a noncoding IGR that has been shown to act as a bona fide transcription termination signal (Fig. 1). There are significant differences in sequence and predicted folded structure between the S and L IGR, but among strains of the same arenavirus species, the S and L IGR sequences are highly conserved. The S RNA encodes the viral NP and the glycoprotein precursor (GPC) that is posttranslationally cleaved by the cellular site 1 protease (S1P) to yield the two mature virion glycoproteins GP1 and GP2 that form the spikes that decorate the virus surface (Beyer et al. 2003; Lenz et al. 2001; Pinschewer et al. 2003). GP1 mediates virus receptor recognition and cell entry via endocytosis , whereas GP2 mediates the pH-dependent fusion event required to release the virus ribonucleoprotein (RNP) core into the cytoplasm of infected cells (Buchmeier et al. 2007). The L RNA encodes the viral RNA-dependent RNA polymerase (RdRp, or L polymerase) and the small RING finger protein Z that has functions of a bona fide matrix protein including directing virus budding (Perez et al. 2003; Strecker et al. 2003).

Arenavirus genome organization and virion structure. Arenaviruses are enveloped viruses with a bi-segmented NS RNA genome. Each genome segment uses an ambisense coding strategy to direct the synthesis of two viral polypeptides. The small (S, ca 3.5 kb) segment encodes for the glycoprotein precursor (GPC) and nucleoprotein (NP). Posttranslational processing of GPC by the cellular protease S1P results in the production of GP1 and GP2. The large (L, ca 7.3 kb) segment encodes for the virus RNA-dependent RNA polymerase (L) and a small RING finger protein (Z) that has the properties of the bona fide matrix proteins (M) found in many enveloped NS RNA viruses
Arenaviruses exhibit high degree of sequence conservation at the genome 3′-termini (17 out of 19 nt are identical), and, as with other NS RNA viruses, arenavirus genome termini exhibit terminal complementarity with the 5′- and 3′-ends of both L and S genome segments predicted to form panhandle structures (Buchmeier et al. 2007). This prediction is supported by electron microscopy (EM) data showing the presence within the virion particles of circular RNP complexes (Young and Howard 1983). It has been proposed, based on EM studies, that host ribosomes can be also incorporated into virions, but the biological significance of this remains to be determined (Muller et al. 1983).
2.2 Arenavirus Life Cycle
Cell entry
Arenavirus cell entry occurs via receptor-mediated endocytosis, which involves a pH-dependent fusion event between viral and cellular membranes within the acidic environment of the endosome (Rojek and Kunz 2008; Rojek et al. 2008a, b). The widely expressed cell surface receptor for extracellular matrix proteins (ECM), alpha-dystroglycan (αDG) , has been identified as a main receptor for LCMV, LASV, and several other arenaviruses (Kunz et al. 2002, 2004). To function as an arenavirus receptor, αDG needs to undergo several posttranslational modifications including a critical like-acetylglucosaminyltransferase (LARGE) -dependent glycosylation (Kunz et al. 2005). Intriguingly, recent genome-wide studies have uncovered evidence of positive selection of specific LARGE alleles within the Yoruba people living in regions of West Africa where LASV is endemic (Sabeti et al. 2007). Endocytic vesicles of the OW arenavirus LCMV were found to be non-coated, and LCMV cell entry process was reported to be cholesterol-dependent but clathrin-, dynamin-, caveolin-, ARF6-, flotillin-, and actin-independent (Rojek et al. 2008b). OW arenavirus cell entry follows an endocytic pathway that bypasses classical Rab5/EEA-1 positive early endosomes, but merges into the classical endocytotic pathway at the level of the multivesicular body (MVB) and is then delivered to the late endosome in an endosomal sorting complex required for transport (ESCRT)-dependent process (Pasqual et al. 2011). The unusual endocytotic pathway followed by LCMV and LASV may reflect the natural cellular trafficking of their receptor αDG. Nevertheless, it cannot be ruled out that binding of GP to αDG could trigger a novel route of entry for αDG that is tailored specifically to benefit virus cell entry as previously documented for other pathogens. Differently to OW arenaviruses, many NW arenaviruses, including the HFA JUNV, use the human transferrin receptor to initiate cell entry via clathrin-mediated endocytosis (Rojek et al. 2008b).
Expression and replication of the viral genome
The fusion between viral and cellular membranes within the late endosome compartment releases the viral RNP into the cytoplasm where both RNA replication and gene transcription of the genome take place (Buchmeier et al. 2007). The NP and L coding regions are transcribed into a genomic complementary mRNA, whereas the GPC and Z coding regions are not translated directly from genomic RNA, but rather from genomic sense mRNAs that are transcribed using as templates the corresponding antigenome RNA species, which also function as replicative intermediates. Arenavirus mRNAs are non-polyadenylate and contain at their 5′-ends a short (4- to 5-nucleotide) non-templated sequence and a cap structure that are likely obtained from cellular mRNAs via a cap-snatching mechanism whose details remain to be determined, but the endonuclease activity associated with the arenavirus L polymerase likely contributes to this process (Morin et al. 2010). Transcription termination of viral mRNAs was mapped to multiple sites within the distal side of the IGR (Meyer et al. 2002; Meyer and Southern 1994; Tortorici et al. 2001), suggesting that the IGR acts as a bona fide transcription termination signal for the virus polymerase, which has been confirmed using cell-based minireplicon systems (Pinschewer et al. 2005).
NP and L are the only viral trans-acting factors required for efficient arenavirus RNA synthesis, whereas Z has been shown to exert a dose-dependent inhibitory effect on the RNA biosynthetic activities of the arenavirus polymerase complex (Cornu and de la Torre 2001, 2002). The arenavirus L protein has the characteristic sequence motifs conserved among the RdRp (L proteins) of NS RNA viruses (Poch et al. 1989). Bioinformatic analysis and functional assays have provided evidence that LASV L protein is organized into three distinct structural domains and that the N- and C-parts of L are able to functionally trans-complement each other (Brunotte et al. 2011). Notably, the recent EM characterization of a functional MACV L protein has revealed a core ring domain decorated by appendages, which may reflect a modular organization of the arenavirus polymerase (Kranzusch and Whelan 2011). The NP is the main structural element of the viral RNP that directs viral RNA synthesis. NP exhibits also a type I interferon (IFN-I) counteracting activity that was mapped to its C-terminus (Martinez-Sobrido et al. 2006, 2007, 2009). Two recently determined crystal structures of LASV NP at 1.80 Å (Qi et al. 2010) and 1.5 Å (Hastie et al. 2011) resolution identified distinct N- and C-terminal domains connected by a flexible linker. The N-terminal domain was proposed to contain a potential cap-binding site that could provide the host-derived primers to initiate transcription by the virus polymerase (Qi et al. 2010). However, analysis of the structure of the N-terminal domain of LASV NP in complex with ssRNA suggests that this originally proposed cap-binding site likely corresponds to a binding site for the viral genome (Hastie et al. 2011). The C-terminal of NP has a folding that mimics that of the DEDDH family of 3′–5′ exoribonucleases (Hastie et al. 2011; Qi et al. 2010). Functional studies confirmed the 3′–5′ exoribonuclease activity of LASV NP, which was proposed to be critical for the anti-IFN activity of NP but dispensable for the role of NP on replication and transcription of the viral genome (Hastie et al. 2011; Qi et al. 2010). However, LCMV with a mutant NP lacking the 3′–5′ exoribonuclease had a large decrease in fitness during its replication in IFN-deficient Vero cells (Martinez-Sobrido et al. 2009).
Assembly and budding
Assembly and cell release of infectious arenavirus progeny require both Z and correct processing of GPC into GP1 and GP2 by the cellular site 1 protease (S1P) (Kunz et al. 2003b; Perez et al. 2003; Pinschewer et al. 2003; Strecker et al. 2003; Urata et al. 2006). GP1 and GP2 together with a stable 58 amino acid long signal peptide (SSP) form the functional GP complex. SSP has been implicated in different aspects of the trafficking and function of the viral envelope glycoproteins (Saunders et al. 2007; York and Nunberg 2006, 2007; York et al. 2004). GP1 located at the top of the spike and held in place by ionic interactions with the N-terminus of the transmembrane GP-2 mediates virus interaction with host cell surface receptors, whereas GP2 exhibits similarities with fusion-active domains of other viral envelope proteins. The arenavirus RING finger protein Z is a structural component of the virion that has no homologue among other known NS RNA viruses. In LCMV-infected cells, Z has been shown to interact with several cellular proteins including the promyelocytic leukemia (PML) protein and the eukaryote translation initiation factor 4E (eIF4E), which have been proposed to contribute to the non-cytolytic nature of LCMV infection and repression of cap-dependent translation, respectively (Borden et al. 1998; Campbell Dwyer et al. 2000; Djavani et al. 2001; Volpon et al. 2010). For most enveloped viruses, a matrix (M) protein is involved in organizing the virion components prior to assembly. Interestingly, arenaviruses do not have an obvious counterpart of M. However, structural and functional studies indicated that Z was the arenavirus counterpart of the M protein found in many other enveloped NS RNA viruses that play a critical role in assembly and cell release of mature infectious virions (Perez et al. 2003; Strecker et al. 2003; Urata et al. 2006). Accordingly, Z has been shown to be the main driving force of arenavirus budding, a process mediated by the Z late (L) domain motifs : PTAP and PPPY similar to those known to control budding of several other viruses including HIV and Ebola virus, via interaction with specific host cell proteins (Perez et al. 2003; Strecker et al. 2003; Urata et al. 2006). Consistent with this observation, Z exhibited features characteristic of bona fide budding proteins: (1) ability to bud from cells by itself and (2) substituted efficiently for other L domain. Targeting of Z to the plasma membrane, the location of arenavirus budding, strictly required its myristoylation (Perez et al. 2004; Strecker et al. 2006). Results derived from cryoelectron microscopy of arenavirus particles (Neuman et al. 2005) were also consistent with the role of Z as a functional M protein.
2.3 Arenavirus Reverse Genetics
In contrast to positive-stranded RNA viruses, deproteinized genomic and antigenomic RNAs of NS RNA viruses, such as LCMV, cannot function as mRNAs and are not infectious. This reflects the fact that the template of the polymerases of NS RNA viruses is exclusively a nucleocapsid consisting of the genomic RNA tightly encapsidated by the NP, which associated with the virus polymerase proteins forms a RNP complex. Thus, generation of biologically active synthetic NS viruses from cDNA requires trans-complementation by all viral proteins involved in virus replication and transcription. These considerations hindered the application of recombinant DNA technology to the genetic analysis of these viruses. However, significant progress has been made in this area, and for members of all families of NS RNA viruses, it has become possible to develop cell-based minireplicon systems and to rescue infectious virus from cloned cDNAs (Neumann and Kawaoka 2004). The use of reverse genetics approaches has revolutionized the analysis of cis-acting sequences and trans-acting proteins required for virus replication, transcription, maturation, and budding. In addition, the possibility to generate predetermined specific mutations within the virus genome and analyze their phenotypic expression in vivo in the context of the virus natural infection is contributing very significantly to the elucidation of the molecular mechanisms underlying virus–host interactions at the cellular and molecular levels, which has provided investigators with novel and powerful approaches for the investigation of viral pathogenesis. In addition, these developments have also paved the way for engineering these viruses for vaccine and gene therapy purposes (Subbarao and Katz 2004; von Messling and Cattaneo 2004).
Arenavirus minireplicon (MR) systems
Cell-based minireplicon (MR) systems have been developed for a variety of arenaviruses (Emonet et al. 2011a), and their use provided conclusive evidence that NP and L are the minimal viral trans-acting factors required for efficient RNA synthesis mediated by the arenavirus polymerase complex (Hass et al. 2004; Lee et al. 2000; Lopez et al. 2001). Moreover, LCMV MR assays uncovered that oligomerization of L was required for the activity of the LCMV polymerase (Sanchez and de la Torre 2005). Consistent with this finding, LASV L protein was shown to contain both N- and C-termini domains that mediate L–L interaction (Brunotte et al. 2011). MR-based assays facilitated mutation-function studies that identified residues within the N-terminus of L that played a critical role in synthesis of viral mRNA but not in RNA replication (Lelke et al. 2010). Moreover, MR-based assays together with structural data uncovered within the N-terminus of LASV L protein, an endonuclease functional domain that was critically required for arenavirus transcription (Morin et al. 2010). Mutation-function analysis of the genome 5′- and 3′-termini using LCMV and LASV MR-based assays demonstrated that the activity of the arenavirus genomic promoter requires both sequence specificity within the highly conserved 3′-terminal 19 nt of arenavirus genomes and the integrity of the predicted panhandle structure formed between the 5′- and 3′-termini of viral genome RNAs (Hass et al. 2006; Perez and de la Torre 2002). Likewise, MR-based assays provided direct experimental confirmation that the IGR is a bona fide transcription termination signal (Pinschewer et al. 2005). These studies also uncovered that Z exhibited a dose-dependent inhibitory effect on both RNA replication and gene transcription by the arenavirus polymerase complex (Cornu and de la Torre 2001, 2002; Cornu et al. 2004; Hass et al. 2004; Lopez et al. 2001).
Generation of infectious recombinant arenaviruses
Generation of infectious virus from cloned cDNAs has been reported for LCMV (Flatz et al. 2006; Sanchez and de la Torre 2006), PICV (Lan et al. 2009), JUNV (Albarino et al. 2009; Emonet et al. 2011b), and LASV (Albarino et al. 2011). Both RNA polymerase I and T7 RNA polymerase (T7RP)-based systems haven been successfully used, and with similar efficiencies, to direct intracellular synthesis of L and S genome RNA species required for the rescue of infectious recombinant arenaviruses (Emonet et al. 2011a). Intriguingly, the rescue of PICV and JUNV using the T7RP-based system did not require plasmid-supplied viral NP and L proteins, indicating that T7RP-mediated RNA synthesis produced both viral antigenome RNA species that were substrate for encapsidation and replication, and mRNAs that serve as template to produce levels of NP and L sufficiently high to facilitate virus rescue. This phenomenon has been reported for several other negative-sense RNA viruses (Emonet et al. 2011a). Reverse genetics approaches similar to those used to rescue infectious LCMV, PICV, JUNV, and LASV from cloned cDNAs should be now applicable to other arenaviruses. The ability to generate recombinant arenaviruses with predetermined specific mutations and analyze their phenotypic expression in the context of the natural course of infection has opened new opportunities to investigate arenavirus–host interactions that influence a variable infection outcome, ranging from virus control and clearance by the host defenses to long-term chronic infection associated with subclinical disease, and severe acute disease including HF. Thus, the use of rLCMV/VSVG uncovered the arenavirus GP as a viral Achilles’ heal and provided the foundations for a novel strategy to develop safe and effective live-attenuated arenavirus vaccines (Pinschewer et al. 2003). Likewise, rLCMV/VSVG was very instrumental in facilitating studies aimed at understanding the regulation of CD8+ T cell function within the infected brain (Pinschewer et al. 2010), as well as how viruses can induce organ-specific immune disease in the absence of molecular mimicry and without disruption of self-tolerance (Merkler et al. 2006). Other engineered rLCM viruses have been used to study LASV cell entry pathway (Rojek et al. 2008b) and the role of NP in the inhibition of induction of IFN-I by LCMV (Martinez-Sobrido et al. 2009). Similarly, studies aimed at examining the critical role played by the S1P-mediated processing of arenavirus GPC in the virus life cycle were aided by the use of recombinant viruses where the S1P cleavage site within GPC was replaced by a canonical furin cleavage site (Albarino et al. 2009; Rojek et al. 2010; Urata et al. 2011). The rescue of attenuated and virulent forms of PICV (Lan et al. 2009; Liang et al. 2009) or the Docile and Aggressive strains of LCMV (Chen et al. 2008) has allowed for the identification of specific genetic determinants of virus virulence. Moreover, it has been possible to generate single-cycle infectious, reporter-expressing, recombinant LCMV in which the GPC open reading frame (ORF) was replaced by GFP (rLCMVDGP/GFP) (Rodrigo et al. 2011). Genetic complementation with plasmids or stable cell lines expressing arenavirus GPCs of interest produces the corresponding GPC-pseudotyped rLCMV∆GP/GFP that can be used to evaluate antibody responses to HF arenaviruses using a Biosafety Level 2 (BSL2) platform.
Despite recombinant arenaviruses could be generated very efficiently, the ability to rescue arenaviruses expressing additional genes of interest posed unexpected difficulties. This problem was overcome by the generation of tri-segmented rLCMV (r3LCMV) containing 1L and 2S segments. Each of the two S segments was altered to replace one of the two viral ORF, GPC, or NP, by a gene of interest (GOI) (Emonet et al. 2009b). The rationale behind this approach was that the physical separation of GP and NP into two different S segments would represent a strong selective pressure to select and maintain a virus capable of packaging 1L and 2S segments. A variety of r3LCMV has been rescued that express one or two additional GOI. Depending on the GOI expressed (protein function, size of the gene), these r3LCMV showed little or no attenuation in cultured cells and they exhibited long-term genetic stability as reflected by unaltered expression levels during serial virus passages of the GOI incorporated into the S segment. The use of r3LCMV expressing appropriate reporter genes should facilitate the development of chemical screens to identify antiviral drugs, as well as siRNA-based genetic screens to identify host cell genes contributing to the different steps of the arenavirus life cycle. However, the use of r3LCMV to study virus–host interactions in mice has encountered some limitations due to the observation that r3LCMV with WT growth properties in cultured cells usually exhibits attenuation in mice due to reasons that remain to be determined.
3 Genetic Variability of Arenaviruses
3.1 Organization and Phylogenetic Relationships of the Arenaviridae Family
Until recently, the family Arenaviridae has consisted of a single genus, Arenavirus, that includes 25 recognized species. Based on their antigenic properties, arenaviruses are divided into two distinct groups: The OW arenaviruses (Lassa–lymphocytic choriomeningitis serocomplex) include viruses indigenous to Africa and the ubiquitous LCMV, and the NW arenaviruses (Tacaribe serocomplex) include viruses indigenous to the Americas. Subsequent genetic analyses based on sequencing data supported very well the previously defined antigenic grouping (Bowen et al. 1997). The monogeneric phylogenetic view of arenaviruses has recently shifted dramatically with the discovery of a divergent group of arenaviruses in captive alethinophidian snakes (Bodewes et al. 2013; Hetzel et al. 2013; Stenglein et al. 2012). Preliminary phylogenetic studies indicate that these reptilian arenaviruses constitute a sister clade to mammalian arenaviruses, and have been proposed to represent a new genus within the family Arenaviridae.
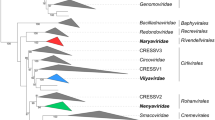
OW arenaviruses form one monophyletic group that is deeply rooted to the three identified NW arenavirus phylogenetic groups, clades A, B, and C. Clade B contains the human pathogenic viruses Chapare (CHPV), Guanarito (GTOV), JUNV, Machupo (MACV), and Sabiá (SABV), as well as several nonpathogenic including Tacaribe virus (TCRV) (Fig. 2). Among OW arenaviruses, LASV, Mobala virus (MOBV), and Mopeia virus (MOPV) are monophyletic, while Ippy virus (IPPYV) and LCMV are more distantly related. The recently discovered Lujo virus (LUJV), endemic in Zambia, is most closely related to OW arenaviruses, but its GPC contains elements of NW arenavirus sequences. Both OW and NW arenaviruses have been evolved from a common ancestor, developing a wide range of antigenic specificity, host species range, pathogenicity, and virulence (Zapata and Salvato 2013).

Rooted tree of arenavirus S segment. Phylogeny reconstruction by neighbor joining with 10,000 bootstrap replications under maximum composite likelihood substitution model was done with Mega 6 (Tamura et al. 2013). Bootstrap values below 80 % are hidden. A similar tree was obtained for the L segment. GenBank accession numbers of S segment (all complete sequences) used to generate the tree, as well as species names of rodent reservoirs were obtained from previously published data (King et al. 2012), with the following exceptions: Skinner tank virus (SKTV, EU123328.1) from Neotoma mexicana (USA); Tonto creek virus (TTCV, EF619034.1) from Neotoma albigula; Big brushy tank virus (BBTV, EF619035.1) from Neotoma albigula, Arizona (USA); Catarina virus (CTNV, DQ865244) from Neotoma micropus (Cajimat et al. 2013); and Luna virus (LUNV-LSK-1, AB693148.1) from Mastomys natalensis, Zambia (Ishii et al. 2012). Colored dots next virus isolates indicate reservoir species: green, neotominae subfamily; blue, sigmodontinae subfamily; red, murinae subfamily; cian, the bat Artibeus spp, proposed reservoir of Tacaribe virus (TCRV-p2b2); and magenta, the Boa constrictor snake, reservoir of the recently reported putative arenaviruses University of Helsinki virus (UHV-1) and inclusion body disease virus (IBDV-NL3) (Bodewes et al. 2013; Hetzel et al. 2013). Absence of colored dot indicates unknown reservoir
As with other RNA viruses, arenavirus replication is mediated by an error-prone polymerase complex, which together with reassortment events of its segmented genome generates the genetic variability that provides arenavirus with the potential for rapid evolution and high capability of adaptation to new environments. The contribution of recombination to arenavirus genetic variability has not been explored at length, but similarly to other NS RNA viruses, recombination events among arenavirus genomes appear to be extremely rare. Nevertheless, both reassortment and recombination events need to be taken into account to understand the evolution and population dynamics of arenaviruses (Albarino et al. 1998; Archer and Rico-Hesse 2002; Charrel and de Lamballerie 2003; Charrel et al. 2001, 2003). It is worth noting that arenaviruses exhibit a superinfection inhibition feature that diminishes the possibilities for recombination and reassortment events since both are dependent on two different arenaviruses infecting the same cell (Ellenberg et al. 2004, 2007; Moreno et al. 2012b). The mechanisms underlying virus superinfection inhibition vary among different viruses (Geleziunas et al. 1994; Horga et al. 2000; Huang et al. 2008; Morrison and McGinnes 1989). In the case of arenaviruses, high levels of Z protein inhibit de novo formation of a functional polymerase complex thus leading to inhibition of viral RNA synthesis (Cornu and de la Torre 2001, 2002; Cornu et al. 2004; Kranzusch and Whelan 2011; Loureiro et al. 2011). Hence, infected cells may express levels of Z that do not permit synthesis of RNA by the polymerase complex of a second infecting arenavirus (Ellenberg et al. 2004, 2007; Moreno et al. 2012b).
3.2 Mechanisms of Arenavirus Genetic Diversity
3.2.1 Arenavirus Mutation Frequencies
RNA viruses have high mutation rates, in the range of 10−3 to 10−5 (Drake and Holland 1999) due to the error-prone nature of their viral RdRp (Domingo 2007). Accordingly, early studies already suggested high genetic variability of arenavirus populations based on the observation that CTL escape mutants could be readily selected both in vivo (Pircher et al. 1990) and in cell culture (Aebischer et al. 1991; Lewicki et al. 1995). Likewise, emergence of neutralization-resistant LCMV mutants in persistently infected mice was found to be a rather frequent event associated with single point mutations (Ciurea et al. 2000, 2001). More recent studies have examined the mutation frequencies (the number of nucleotide substitutions and indels per nucleotide sequenced) in LCMV populations recovered after one or several passages in BHK-21 cells. These studies involved sequencing of multiple clones of several regions of the GP, NP, and L genes and found mutation frequencies in the range of 1.0 × 10−4 to 2.7 × 10−4 and 1.0 × 10−4 to 5.7 × 10−4 for populations after one and nine, respectively, cell culture passages (Grande-Perez et al. 2002). The highest mutation frequency detected corresponded to the NP region and the lowest to the L region. These values correlated with the corresponding values of Shannon entropy, parameter that measures the genetic heterogeneity of the viral population (Domingo 2006). Other studies have documented mutation frequencies for arenaviruses in the range of 2.6 × 10−4 to 5.5 × 10−4 mutations per nucleotide during serial passages in cell culture (Ruiz-Jarabo et al. 2003). In addition, mutation frequencies in the range of 1.2 × 10−4 to 3.5 × 10−4 substitutions per nucleotide were estimated for both the intracellular and released viral populations during LCMV persistence in BHK-21 cells (Grande-Perez et al. 2005b). These figures are consistent with a large body of literature documenting the genetic variability of both negative- and positive-stranded RNA viruses (Domingo 2007) and support the view that arenavirus populations are ensembles of genetically closely related mutant genomes, called quasispecies, subjected to natural selection and genetic drift (Sevilla and de la Torre 2006).
3.2.2 RNA Recombination in Arenaviruses
Discrepancies between NP- and GPC-based phylogenetic trees observed between clades A and B arenaviruses and North American (lineage A/Rec) arenaviruses including White Water Arroyo virus (WWAV) and the two newly discovered North America arenaviruses, Catarina and Skinner Tank viruses, led to hypothesize that a recombination event between members of clades A and B led to the emergence of lineage A/Rec (Archer and Rico-Hesse 2002; Charrel et al. 2001, 2002, 2003; Fulhorst et al. 2001). Additional recombination events have been suggested to explain the evolutionary relationships between Oliveros (OLIV) and Pichinde (PICV) viruses with OW arenaviruses (Albarino et al. 1998). However, the generation of recombinant arenaviruses requires cells to be simultaneously infected by two different arenaviruses, and although coinfection with two different arenaviruses has been reported in cultured cells (Ellenberg et al. 2004, 2007; Lukashevich et al. 2005; Moreno et al. 2012b; Riviere et al. 1986), in nature it may be less likely and further restricted by the superinfection exclusion feature associated with arenavirus infection (Ellenberg et al. 2004, 2007; Lukashevich et al. 2005; Moreno et al. 2012b; Riviere et al. 1986). Some arenaviruses share a common geographic localization, such as Guanarito and Pirital (PIRV) viruses in Venezuela (Fulhorst et al. 1999; Weaver et al. 2000); Junin and Oliveros viruses in Argentina (Mills et al. 1996); and Machupo, Latino, and Chapare viruses in Bolivia (Delgado et al. 2008). Likewise, different arenaviruses can some times infect the same rodent species, such as Guanarito and Pirital viruses, which were isolated both in cotton rats Sigmodon alstoni and in cane mice Zygodontomys brevicauda (Weaver et al. 2000). However, despite an extensive genetic characterization, the existence of a GTOV-PIRV recombinant virus has never been demonstrated (Cajimat and Fulhorst 2004; Fulhorst et al. 2008), which might be explained by superinfection exclusion among arenaviruses (Damonte et al. 1983; Ellenberg et al. 2004, 2007). Since no A/Rec arenavirus has been isolated in South America and neither clade A nor clade B arenavirus has been isolated in North America, it is difficult to explain how the recombination occurred. It is plausible that the two parental arenaviruses were originally present in North America but disappeared, either by natural extinction, migration or by being outcompeted by the recombinant arenavirus. The migration to North America of a recombinant arenavirus generated in South America seems improbable because South America arenaviruses most likely descend from an ancestral arenavirus located in North America (Cajimat et al. 2007). Moreover, North America arenaviruses have a long-term coevolutionary relationship with rodents from Neotominae subfamily, which are almost exclusively found in North America (Cajimat et al. 2007). It should be noted that clade C arenaviruses also exhibit a shift between GPC- and NP-based trees (Charrel et al. 2008), and it seems unlikely that such a rare event would be the origin of two different lineages. It seems more plausible that the phylogenetic information derived from arenavirus GPC is influenced by specific selection pressures (Cajimat et al. 2007). The reasons why GPC would be under a selection pressure different from the other arenavirus genes need to be determined. Likewise, the statistical significance of the recombination event found in North America and clade C arenaviruses using recombination detection softwares should be validated using an up-to-date arenavirus genetic database.
The recently discovered arenaviruses associated with cases of inclusion body disease (IBD) in snakes (Bodewes et al. 2013; Hetzel et al. 2013; Stenglein et al. 2012) might represent a very special case of heterologous recombination between an arenavirus and a filovirus within a coinfected host (snake). Phylogenetic analysis showed that snake-related arenaviruses constitute a monophyletic lineage separated from NW and OW arenaviruses (Bodewes et al. 2013; Stenglein et al. 2012). Intriguingly, the GPC of snake-related arenaviruses was more closely related to the GPC of filoviruses than to arenaviruses, but the natural history of events favoring this potential recombination event between filo- and arenaviruses remains to be elucidated.
3.2.3 Arenavirus Genomic Reassortments
Reassortment events require, like recombination, a coinfection event that can be readily recreated in cultured cells (Ellenberg et al. 2004, 2007; Lukashevich 1992; Lukashevich et al. 2005; Moreno et al. 2012b; Moshkoff et al. 2007; Riviere et al. 1986; Riviere and Oldstone 1986). However, in nature, there is only one documented case of an arenavirus reassortant event (Palacios et al. 2008). Extensive phylogenetic analysis of NW and OW arenaviruses revealed no proof of detectable genomic reassortment events during their evolution (Charrel et al. 2003), with the exception of an arenavirus isolated from three fatal cases of LCMV infection in transplantation patients who received organs from a single donor (Palacios et al. 2008). Phylogenetic analysis revealed a new arenavirus, in which L and S segments were related to LE and M1-M2 LCMV strains, respectively, suggesting that likely a reassortment event was involved in the generation of this arenavirus. The lower reassortant frequency in arenaviruses may be due to the superinfection exclusion exhibited by the members of this family (Ellenberg et al. 2004, 2007; Moreno et al. 2012b).
Since reassortment of arenaviruses in nature seems to be restricted to some specific combinations of L and S segments (Lukashevich 1992; Riviere et al. 1986), and recombination in arenaviruses is extremely infrequent (Archer and Rico-Hesse 2002; Charrel et al. 2002), mutations during genome replication by an error-prone polymerase arenaviruses are thought to be the main contributing factor to arenavirus genetic variability.
3.3 Arenavirus Hypermutation
Interferon-inducible adenosine deaminase that acts on double-stranded RNA (ADAR1) has been suggested to play a role in the innate immune response against LCMV (Zahn et al. 2007). ADARs are a family of dsRNA-binding enzymes present in animals that deaminate adenosine to inosine, and are involved in the regulation of a variety of posttranscriptional processes as well as in an antiviral response (Nishikura 2010). Since inosine and adenosine have different base-pairing properties, editing can alter protein-coding transcripts and noncoding targets including microRNA, small interfering RNA viral transcripts, and repeat elements (Hundley and Bass 2010; Mallela and Nishikura 2012). Upon LCMV infection of murine L929 cells, both expression and activity of ADAR1-L were upregulated and its editing activity was not inhibited by viral replication (Zahn et al. 2007). An adenosine-to-guanosine (A-to-G) transition bias was found in cDNA clones of the GPC gene derived from LCMV RNA isolated from L929 cells at seven days post-infection. Likewise, an A-to-G/U-to-C transition bias was observed in clones of LCMV genomic RNA species derived from RNA isolated from spleen of LCMV-infected C57BL/6 mice at four days post-infection, whereas no G-to-A/C-to-U transition was observed. Moreover, two of 54 analyzed clones showed an A-to-G or U-to-C hypermutation pattern in a very short stretch of the GP where 38 % of all adenosines or 14 % of uracils were mutated. In LCMV clones isolated from mice, no hypermutated LCMV clones were found. Approximately 50 % of the clones with an amino acid mutation were non-functional, suggesting that ADAR1-L-induced mutations in LCMV RNA reduce viral infectivity and protein function, thus supporting a contribution of ADAR1-L to the innate antiviral immune response. Nevertheless, multiplication of LCMV was not increased in mouse embryonic fibroblasts derived from ADAR1−/− mice, which raises some questions about a direct role of ADAR1 in restricting LCMV multiplication (Ward et al. 2011).
Hypermutation has been related to posttranscriptional editing by host adenosine deaminases of viral genomes from persistent infections of other RNA viruses such as measles virus (Cattaneo and Billeter 1992), human parainfluenza virus (Murphy et al. 1991; Rima et al. 2014), and respiratory syncytial viruses (Rueda et al. 1994). In these studies, the estimated mutation frequencies of U → C or A → G transitions were in the range of 3.3 × 10−2 to 1.0 × 10−1 mutations per nucleotide. The existence of hypermutated genomes had been hypothesized as proof of the genetic melting that occurs when the error catastrophe threshold is crossed as a result of chemical mutagenesis (Grande-Perez et al. 2002). However, evidence of hypermutated viral genomes has not been found in preextinction populations. In particular, hypermutated sequences of LCMV from 5-fluorouracil (FU) mutagenized populations were not amplified despite using a battery of oligonucleotide primers with different degrees of degeneracy targeting LCMV genomic RNA (Grande-Perez et al. 2005a). In this study, only one sequence was found that harbored nine mutations, six U → C transitions and the other three A → G, C → A, and G → A in an amplicon of 558 nucleotides (excluding primers), which renders a mutation frequency (relative to the consensus sequence of the sample from which it was obtained) of 1.6 × 10−2 mutations per nucleotide sequenced. No further analyses were conducted to find out whether this hypermutated molecule was the result of chemical mutagenesis or host adenosine deaminase. However, in a subsequent study (Martin et al. 2010), the participation of ADAR-like enzymes in the 5-FU-mediated increase of A → G transitions of LCMV genome was ruled out because the context in the immediate neighborhood of the observed A → G transitions was not preferred by the dsRNA adenosine deaminase (Polson and Bass 1994).
4 Contribution of Genetic Variability to Arenavirus–Host Interactions and Associated Disease
4.1 Coevolution of Arenaviruses and Their Natural Reservoirs: Intra- versus Inter-host Genetic Variation of Arenaviruses
Arenaviruses establish long-life persistent infections in their natural reservoirs in the absence of noticeable clinical symptoms (Childs and Peters 1993). Arenavirus natural reservoirs include rodent species from the family Muridae (Salazar-Bravo et al. 2002), but a notable exception is Tacaribe virus whose natural host is thought to be the fruit-eating bat Artibeus sp. (Downs et al. 1963). Coevolution of the arenaviruses with their natural hosts has likely been a major driving evolutionary force that has generated the currently existing arenavirus–host associations (Bowen et al. 1997), with groups of genetically closely related arenaviruses infecting closely related rodent species. Thus, OW arenaviruses are found in rodents from the subfamily Murinae, whereas NW arenavirus reservoirs belong to rodents from subfamilies Sigmodontinae and Neotominae (Fig. 2). Furthermore, inside the OW arenaviruses, the LASV complex (LASV, MOBV, MOPV, and IPPYV) is associated with Praomys rodents, while LCMV and Kodoko virus are found in rodents from the genus Mus (Lecompte et al. 2007). Likewise, within NW arenaviruses, the reservoirs of most North American arenaviruses are mainly rodents from the subfamily Neotominae, whereas the reservoirs of the South American arenaviruses belong to the subfamily Sigmodontinae (Emonet et al. 2009a). However, this coevolution hypothesis , which assumes also occasional host switching, could not be tested empirically due to the lack of detailed knowledge about rodent phylogeny. Studies trying to reconciliate the viral and host phylogenies for both OW (Hugot et al. 2001; Coulibaly-N’Golo et al. 2011) and NW (Jackson and Charleston 2004) arenaviruses did not find evidence of congruence between both phylogenies. Recent evidence has suggested that for OW arenaviruses, coevolutionary associations may have been concealed by the occurrence of multiple host switching and extinction events (Coulibaly-N’Golo et al. 2011). In addition, studies based on the recent availability of molecular data from both NW arenaviruses and their rodent reservoirs found evidence of arenavirus evolution via host switching , rather than codivergence between these arenaviruses and their hosts (Irwin et al. 2012).
Studies on the interaction between the surface glycoprotein GP1 of several NW arenaviruses and their cell entry receptor, the transferring receptor 1 (TfR1) (Abraham et al. 2009; Choe et al. 2011; Demogines et al. 2013; Flanagan et al. 2008; Radoshitzky et al. 2007, 2008), have suggested that arenaviruses might have exerted some degree of positive selection in their rodent reservoirs. These studies suggest that the dynamic of GP1–TfR1 interaction has had a direct impact in sequences of these viral and host genes. Interestingly, identified rapidly evolving positions within TfR1 were located within the TfR1 region that mediates interaction with the virus GP1. Mutations at these TfR1 residues altered the interaction with the viral GP1 without affecting TfR1 expression or function. As a consequence of the introduction of genetic divergence, this rodent receptor locus might have the potential to act as a species barrier to viral transmission limiting both the cross-species and zoonotic transmission of these viruses. Nevertheless, there is evidence that JUNV can enter cells very efficiently in a TfR1-independent manner, raising some questions about the true impact of the TfR1 gene as a key determinant of JUNV host range. NW arenaviruses, Junin virus, Machupo virus, and Guanarito virus, have acquired the ability to bind human TfR1 and are currently emerging in South America as zoonosis (Choe et al. 2011; Flanagan et al. 2008) associated with severe morbidity (Peters 2002). Intriguingly, a mutation within TfR1 predicted to affect infection by NW arenaviruses that use this receptor for cell entry was identified in Asian population (Demogines et al. 2013).
Genomic analysis of full-length genome sequences of 29 strains of LCMV collected from a variety of geographic regions at different times showed these viruses to be genetically highly diverse (Albarino et al. 2010). Several distinct lineages of LCMV could be identified, but there was little correlation with time or place of isolation. Bayesian analysis enabled estimation of the rate of evolution of the S and L genome segments of LCMV and Kodoko virus sequences and found them to be similar to other negative-stranded RNA viruses. The molecular evolutionary rate was estimated to be 3.3 × 10−4 and 3.7 × 10−4 substitutions per nucleotide and year for the S and L segments, respectively. The Bayesian analysis also indicated that the most recent common ancestor for LCMV S- and L genome RNA segments was estimated to be 1235 and 5142 years ago, respectively. These data indicate that LCMV is quite ancient, and the extensive diversity of the virus has accumulated over the past 1000–5000 years. Also, despite similar rates of evolution, the evolutionary history of the L segment appears to be more complex and can be traced back substantially longer than that for the S segment.
Arenaviruses coevolve with their natural hosts, but animal species other than the corresponding reservoir can be infected (Dalldorf 1939; Greenwood and Sanchez 2002; Salazar-Bravo et al. 2002). Information about possible arenavirus pathogenicity in their natural reservoir is rather limited, but data from LCMV infection of the mouse have shown that the same virus can behave differently depending on the host. For instance, C3H/st, BALB/WEHI, and SWR/J mice infected at birth with LCMV Armstrong, E-350, or Pasteur strains develop persistent infection, but only C3H/st mice develop growth hormone deficiency syndrome. In contrast, LCMV strains Traub and WE did not cause disease in those animals (Oldstone et al. 1984, 1985). Differential host-specific selection may be the reason why quasispecies diversity of Guanarito virus isolates from rodents in Venezuela had higher sequence variation than human isolates. One rodent isolate included a mixture of two phylogenetically distinct genotypes, suggesting a dual infection (Weaver et al. 2000). Likewise, the characterization by pyro-sequencing of the genetic stability and complexity of the Lassa vaccine candidate ML29 in vaccinated rhesus monkeys, marmosets, and mice (Zapata and Salvato 2013) revealed accumulation over time of some host-specific mutations. Protein prediction analysis showed that those mutations, at the amino acid level, induced potential structural changes in GPC and NP.
4.2 Contribution of Viral Quasispecies to Arenavirus Pathogenesis
RNA viruses within hosts exist as complex distribution of genetically closely related mutants, termed quasispecies, subjected to a process of genetic variation, competition, and selection (Domingo 1992; Domingo et al. 2001). Foremost, this swarm of mutants is possible because of the low copy-fidelity of viral RdRp. Studies in cultured cells found mutation frequencies of 1 × 10−4 to 5.7 × 10−4 substitutions per nucleotide for the Z, NP, GP, and L regions analyzed of LCMV (Grande-Perez et al. 2002, 2005a; Martin et al. 2010; Moreno et al. 2011), which are consistent with previous findings on genetic heterogeneity of LCMV (Sevilla et al. 2002) and other arenaviruses (Bowen et al. 1996, 1997; Bowen et al. 2000; Charrel and de Lamballerie 2003; Fulhorst et al. 2001; Garcia et al. 2000; Weaver et al. 2000, 2001) and also within the range commonly observed for riboviruses (10−3 to 10−5 substitutions per nucleotide) (Domingo et al. 1988; Drake and Holland 1999) (see Sect. 3.2.1).
Error-prone replication might contribute to spatial and temporal heterogeneities in RNA genome populations, favoring their rapid evolution and adaptation to different environments. The entire virus cloud contributes to the characteristics of the virus strain and will be the target of selection instead of individual variants (Perales et al. 2005). Cooperation and complementation events between variants of the quasispecies that permit the mutants carrying lethal mutations can be maintained within the population and even compete and interfere with fitter variants (de la Torre and Holland 1990; Gonzalez-Lopez et al. 2004; Martin et al. 2010; Mas et al. 2010; Perales et al. 2007; Zapata and Salvato 2013).
In addition to an error-prone polymerase, RNA recombination (Charrel et al. 2001; Fulhorst et al. 1996) and genome reassortment (Riviere 1987; Riviere et al. 1986; Riviere and Oldstone 1986) can also contribute to arenavirus genetic diversity. However, both recombination and reassortment events appear to be extremely rare in nature and only between phylogenetically closely related strains (Archer and Rico-Hesse 2002; Charrel et al. 2002; Emonet et al. 2006; Jay et al. 2005). The ability of the arenavirus to infect a wide variety of hosts could also contribute to their genetic variability (Zapata and Salvato 2013). The genetic diversity within and between arenavirus groups suggests that the spatial heterogeneity may be reflected in host range and pathogenicity (Blasdell et al. 2008). Biological functions of RNA virus distributions remain poorly understood, but evidence suggests that different arenavirus strains may exhibit different biological properties and pathogenesis attributable to differences in their quasispecies (Sevilla et al. 2002). Moreover, the genetic complexity of a quasispecies structure provides RNA viruses, including arenaviruses, with a great capacity to adapt to changing host environments (Novella et al. 1995). The quasispecies plasticity may facilitate that arenaviruses can overcome barriers to its spread, both within the host and between species (Oldstone and Campbell 2010; Sevilla et al. 2002). Viral variants, initially present at low frequency in the quasispecies, that become dominant under a selective pressure have been documented for different RNA viruses including rabies (Morimoto et al. 1998), coxsackie viruses (Beaucourt et al. 2011; Domingo et al. 2008), poliovirus (PV) (Pfeiffer and Kirkegaard 2005; Vignuzzi et al. 2006), foot-and-mouth disease virus (FMDV) (Haydon et al. 2001; Martin and Domingo 2008), and human parainfluenza virus (Prince et al. 2001). Arenaviruses are no exception, and selection of organ-specific LCMV variants with different pathogenic properties has been documented (Ahmed and Oldstone 1988; Ahmed et al. 1984). Likewise, as with other RNA viruses (Hotchin 1973; Hotchin et al. 1971; Hotchin and Sikora 1973), serial passages in cultured cells can yield LCMV variants that differ in pathogenicity with respect to the original isolate (Hotchin and Sikora 1973; Lukashevich 1992; Pulkkinen and Pfau 1970), further emphasizing the contribution of the quasispecies’ plasticity to arenavirus pathogenesis. These studies in cell culture provide strong support for the view that specific environmental conditions can influence virus–host interactions and promote selection of specific RNA species with distinct phenotypic features within the genetically complex quasispecies. The application of New Generation Sequencing to the field of virology is allowing scientists to start getting a detailed view of the genetic complexity and dynamic of RNA viral quasispecies of how they impact virus–host interactions and associated disease.
4.2.1 Selection of Immunosuppressive Variants During LCMV Persistence
Chronic infections provide investigators with an excellent experimental system for studying the contribution of virus quasispecies dynamics to virus–host interactions and associated disease as sustained virus multiplication in the persistently infected host facilitates virus evolution and adaptation resulting in selection of novel viral phenotypes that could influence the outcome of infection. Persistent infections can favor selection of viral variants with tissue-specific growth advantages that could be associated with different phenotypes and disease manifestations (Ahmed et al. 1984). Studies examining viral populations replicating in different tissues and organs of the same infected host have uncovered intra-host viral variability and selection of tissue-specific virus populations due to differences in selective pressures exerted by the different environments associated with these compartmentalized areas of the infected host (Ahmed and Oldstone 1988; Brown et al. 2011; Cabot et al. 2000; Cheng-Mayer et al. 1989; Deforges et al. 2004; Hall et al. 2001; Jelcic et al. 2004; Jridi et al. 2006; Sanjuan et al. 2004; Trivedi et al. 1994; Wright et al. 2011). LCMV can cause acute or persistent infections in mice, its natural host, and has been used as a model to study viral population dynamics in vivo (Ahmed et al. 1988; Evans et al. 1994). The age, genetics, and immune status of the host, as well as the composition of the viral quasispecies, route of infection, and dose of infecting virus, contribute to the outcome of the infection. Mice infected with LCMV within 24 h of birth do not develop an effective antiviral CTL response and become persistently infected (Matloubian et al. 1990; Salvato et al. 1991). On the other hand, iv inoculation of adult immunocompetent mice with a high (≥2 × 106 PFU) dose of Armstrong (ARM) strain of LCMV causes an acute infection that induces a protective immune response, which culminates in CD8+ CTL-mediated virus clearance. In contrast, a similar infection with the immunosuppressive clone 13 (Cl-13) strain of LCMV causes a persistent infection associated with generalized immune suppression (Ahmed et al. 1984, 1991; Ahmed and Oldstone 1988; Baranowski et al. 2000; Evans et al. 1994; Tishon et al. 1993; Wu-Hsieh et al. 1988). At early times, post-infection ARM exhibits tropism for macrophages of the red pulp of the spleen, while Cl-13 targets preferentially dendritic cells (DCs) of the spleen’s white pulp (Borrow et al. 1995; Sevilla et al. 2000). Notably, ARM and Cl-13 differ only at five nucleotide positions that involve three amino acid changes, two within GP1 [N(ARM)-176-D(Cl-13) and F(Arm)-260-L(Cl-13)] and one in the L polymerase [K(Arm)-1079-Q(C13)] (Matloubian et al. 1990, 1993; Salvato and Shimomaye 1989; Sevilla et al. 2000). Mutation F260L was found to be responsible for the strong binding affinity of Cl-13 to its receptor αDG that is expressed in DCs and facilitates over time high levels of DC infection by Cl-13 (Cao et al. 1998; Kunz et al. 2002), which is associated with Cl-13 persistence. However, at early stages of infection, ARM and Cl-13 infect similar numbers of DCs and macrophages indicating that differences between Cl-13 and ARM regarding early and yet-unknown interactions with host cell factors may determine whether LCMV persists (Cl-13) or is cleared ARM. Interestingly, mutation K1076Q in the L polymerase was found to play a critical role in the persisting phenotype of Cl-13 (Bergthaler et al. 2010; Sullivan et al. 2011).
4.2.2 Contribution of Viral Variants to LCMV-Induced Growth Hormone Deficiency Syndrome
Neonatal infection of C3He mice with certain strains of LCMV results in the development of a growth hormone disease syndrome (GHDS) characterized by growth retardation and severe hypoglycemia that result in death of the infected mice within 15–20 days of infection (Oldstone 2002; Oldstone et al. 1984, 1985). LCMV-induced GHSD was associated with decreasing levels of growth hormone (GH) mRNA that results in decreased GH protein synthesis in the anterior pituitary (Klavinskis and Oldstone 1989; Valsamakis et al. 1987). Virus-induced GHDS strictly correlated with the virus’s ability to replicate at high levels in the GH-producing cells in the anterior pituitary. Studies using reassortant viruses between strains of LCMV, which do (ARM) or do not (WE) cause GHSD, mapped the ability to cause this disorder to the S RNA (Riviere 1987). Notably, clonal analysis of the parental GHDS-nil WE population showed, as expected, that the majority (58/61) of the clones examined behaved as the parental WE clonal population and did not cause GHDS, but three clones isolated from the WE parental population did cause the characteristic GHDS (Buesa-Gomez et al. 1996). These results provided evidence that variants with the ability to cause GHDS (GHDS+) may be present within a GHDS-nil replicating WE parental population. Clonal isolates from the same parental virus population are genetically closely related. Accordingly, characterization of WEc54 (GHDS+) and WEc2.5 (GHDS−) clonal populations derived from the WE parental population identified a single amino acid at position 153 in GP1 as a critical viral genetic determinant required for induction of GHDS, illustrating how minor changes in the virus genome may have great impact in the biology of the infected host (Buesa-Gomez et al. 1996; Teng et al. 1996).
5 Implications of Arenavirus Genetic Variability for the Development of Antiviral Drugs and Vaccines to Combat Human Pathogenic Arenaviruses
5.1 Lethal Mutagenesis as a Novel Antiviral Strategy to Combat Arenavirus Infections
RNA viruses owe their potential for rapid evolution and adaptation to their genetic structure of quasispecies that is determined mainly by their low-fidelity replication machineries. Quasispecies is defined as the spectrum of closely related but nonidentical genomes (see Chaps. 1, 3, and 5). Within replicating viral quasispecies, many individual species may have the potential to exhibit distinct phenotypes including drug-resistant mutants that pose a main obstacle to the success of antiviral therapy. The high polymerase mutation rates responsible for the genetic heterogeneity displayed by RNA viruses are close to a limit, know as error threshold, beyond which the quasispecies theory predicts that biologically meaningful genetic information cannot be maintained. In the case of RNA viruses, violation of this limit is predicted to lead viral extinction, a process termed entry into error catastrophe (Eigen 2002). This model would predict that treatment of ribovirus-infected cells with mutagenic agents that increase the virus polymerase mutation rate could exert an antiviral effect by promoting viral entry into error catastrophe. Initial experimental evidence in support of this was obtained by Holland and colleagues when investigating the effect of mutagens on VSV and PV multiplication (Holland et al. 1990; Lee et al. 1997), and subsequently, the term “lethal mutagenesis ” was coined by Loeb and colleagues to describe the antiviral effect of the mutagen 5-hydrodeoxycytidine in HIV-1 infections (Loeb et al. 1999).
Because of the quasispecies genetic structure of RNA viruses, the interaction, either positive or negative, among all members of the quasispecies strongly influences the behavior of the viral population. Thereby, it is the quasispecies, rather than single independent genome species, that is subjected to selective pressures and evolution. Notably, low dose of a mutagenic agent can increase the number of mutations per genome (m) without reaching the error threshold (mc), which could result in the generation of defective-interfering genomes (DIG) that can interfere virus multiplication and production of infectious progeny, a process that has been termed “lethal defection .” This model is supported by experimental results and in silico predictions (Ahmed et al. 1984, 1991; Ahmed and Oldstone 1988; Baranowski et al. 2000; Evans et al. 1994; Gonzalez-Lopez et al. 2004; Grande-Perez et al. 2005b; Manrubia et al. 2010; Moreno et al. 2012a; Perales et al. 2007; Tishon et al. 1993; Wu-Hsieh et al. 1988). Lethal defection can explain why viral populations subjected to lethal mutagenesis can be extinguished in the absence of the loss of the master sequence, the sequence present at the highest frequency within the quasispecies, and randomization of the virus genome sequence both predicted by the quasispecies theory (Grande-Perez et al. 2005a). Disappearance of the master sequence and melting of the genetic information of the population would be observed only in situations where m increases beyond m c (Fig. 3), a situation that is highly unlikely to be observed in viral populations as the inhibitory effects exerted by DIG together with the loss of essential viral functions due to single mutations within the genome can result in viral extinction without changes in the consensus sequence of the viral population.

Lethal mutagenesis. The lethal mutagenesis concept predicts that increasing the dose of a mutagenic agent will result in an increasing number of mutations per genome (m) in the virus population. When m crosses an error threshold (m > m c), the virus population will enter into error catastrophe that is associated with the melting of the genetic information and viral extinction. The lethal defection model considers that under a moderate mutagenic dose, compatible with maintenance of the biological information, defective-interfering genomes (DIG) are generated and they interfere with the multiplication of the virus population, which could result in virus extinction in the absence of the sequence signatures of error catastrophe
Experimental evidences of lethal mutagenesis in cell culture have been achieved in several viral systems such as HIV-1 (Dapp et al. 2009, 2012; Harris et al. 2005), PV (Graci et al. 2007), or HCV (Ortega-Prieto et al. 2013). In coronavirus, the Nsp14 contains an 3′–5′ exonuclease domain that is responsible of the first described proofreading activity in riboviruses (Denison et al. 2011). Accordingly, viral mutants lacking this activity exhibit increased sensitivity to lethal mutagenesis (Smith et al. 2013). In arenaviruses, loss of infectivity and virus extinction through lethal mutagenesis have been achieved under laboratory conditions (Grande-Perez et al. 2005b; Martin et al. 2010; Moreno et al. 2011, 2012b; Ruiz-Jarabo et al. 2003) supporting the feasibility of entry into error catastrophe as a new antiviral strategy to combat arenavirus infections, a view that has been further reinforced by results from in vivo experiments (Ruiz-Jarabo et al. 2003; Sanz-Ramos et al. 2012).
Mutagenesis of LCMV with 100 µg/ml 5-FU achieved dose-dependent decreases in infectivity and systematic extinction after 48 h in one or two passages when BHK-21 cell cultures were inoculated with a moi of 0.01. However, nucleoside analogue 5-azacytidine (AZC) at 5 or 10 µg/ml was not able to eliminate LCMV after 13 passages in BHK-21 cells, not even when viral load was decreased tenfold (Grande-Perez et al. 2002). Increments in mutation frequencies in the L region reached 16.8-fold, whereas in the GP region, it was just 2.6-fold and 4.1-fold in the NP region in populations treated with a low dose (20 µg/ml) of 5-FU. However, in a preextinction population treated with 100 µg/ml of 5-FU, the mutation frequency of the polymerase increased 3.4-fold compared with the 1.6-fold found for the GP and NP regions (Grande-Perez et al. 2002). Notably, the largest increments in mutation frequencies did not correlate linearly with the lowest infectivity values (Grande-Perez et al. 2002). The type of mutations found differed between the two analogues being predominant the transitions U → C and A → G, followed by C → U in 5-FU-treated populations, whereas in AZC-treated populations, transversions C → G were the most frequent followed by C → A, G → C, and G → U. Despite the high diversity of LCMV mutagenized preextinction populations, their consensus genomic nucleotide sequences remained unaltered (Grande-Perez et al. 2005a). Furthermore, alternate virus passages in the presence and absence of 5-FU led to profound differences in fitness (measured as the capacity of LCMV to produce progeny), without modification of the consensus genomic sequence. LCMV viral populations in their way to extinction were enriched with mutants harboring a limited number of mutations called defectors (Grande-Perez et al. 2005a, b) (Fig. 3). Detailed analysis of the molecular scenario of LCMV persistent infections treated with 100 µg/ml of 5-FU showed an infectivity decrease that was paralleled by a significant increase of intracellular viral genomic L RNA. Preextinction populations recovered at different times post-infection showed specific infectivities (measured as plaque-forming units per genomic RNA molecule) lower than control populations. These experimental results together with silico simulations led to the lethal defection model of lethal mutagenesis that suggests the participation of defective-interfering genomes to explain virus extinction in cell culture when low-level mutagenesis is exerted by a mutagenic nucleoside analogue (Grande-Perez et al. 2005b). Interference caused by defectors is different to the LCMV natural defective-interfering particles, and it is produced as a consequence of 5-FU treatment (Martin et al. 2010). Supernatants of 5-FU-treated monolayers displayed interference capacity specific for LCMV, sensitive to UV irradiation and dose- and time-dependent (Martin et al. 2010). Interference did not involve significant increases of mutant spectrum complexity, as predicted by the lethal defection model (see also Chaps. 13 and 14).
Moreover, LCMV populations subjected to lethal mutagenesis exhibited a negative correlation between viral load and extinction , and infection at low moi in the presence of 5-FU was accompanied by a higher increase of mutant spectrum complexity and a higher antiviral effect than infection at high moi (Moreno et al. 2012b). This behavior was shared by VSV, another negative-stranded RNA virus, whereas the opposite was observed for the positive-stranded RNA viruses FMDV and EMCV. Increased mutagenesis caused by 5-FU treatment promoted the generation of DIG that inhibited viral RNA synthesis, which resulted in increased protection of the virus population against the mutagenic effect of 5-FU. This finding illustrates the biological consequences associated with the generation and activity of DIG. Nucleoside analogues are actively being pursued as candidate antiviral drugs that could exhibit a broad-spectrum mutagenic activity against a variety of riboviruses (Bonnac et al. 2013). The discovery of new nucleotide analogues that increase the viral mutation frequency while minimizing cell toxicity will represent a major advance for our understanding of lethal mutagenesis and its application as a novel antiviral strategy (Graci et al. 2008; Harki et al. 2002; Harris et al. 2005; Hicks et al. 2013).
5.2 Advantages of Combination Versus Single Drug Therapy to Inhibit Arenavirus Multiplication
Current antiviral drug therapy against arenavirus infections is limited to an off-label use of the nucleoside analogue ribavirin that has a limited prophylactic efficacy. In vitro and in vivo studies have documented the prophylactic and therapeutic value of RIb against several arenaviruses (Damonte and Coto 2002). Importantly, Rib reduced both morbidity and mortality in humans associated with LASV infection and experimentally in Machupo and Junin infections, if given early in the course of clinical disease (Damonte and Coto 2002). Despite its validated clinical benefits, the use of Rib has the limitations of its frequent and significant side effects, including anemia and congenital disorders, and the need of an early and intravenous administration for optimal efficacy. Several inhibitors of IMPDH (Andrei and De Clercq 1993), the S-adenosylhomocysteine (SAH) hydrolase and a variety of sulfated polysaccharides (Andrei and De Clercq 1990), phenotiazines compounds (Candurra et al. 1996), brassinosteroids (Wachsman et al. 2000), myristic acid (Cordo et al. 1999), and valproic acid (Vazquez-Calvo et al. 2013) have been reported to have antiarenaviral activity. However, most of these compounds displayed only modest and rather nonspecific effects associated with significant toxicity. Therefore, there is a pressing need for novel effective antiarenaviral drugs. In this regard, a recent high-throughput screening (HTS) using a virus-induced cytopathic effect (CPE)-based assay identified a potent small molecule inhibitor of Tacaribe virus and several other NW arenaviruses (Bolken et al. 2005). Likewise, cell-based HTS identified several small molecule inhibitors of virus cell entry mediated by LASV GP (Lee et al. 2008). These findings illustrate how complex chemical libraries used in the context of appropriate screening assays can be harnessed into a powerful tool to identify candidate antiviral drugs with highly specific activities.
Rib has been documented to be a broad-spectrum antiviral drug active against a variety of RNA viruses (Sidwell et al. 1974; Streeter et al. 1973). Several mechanisms of action have been proposed for the antiviral activity of Rib including direct incorporation into the RNA during replication, alteration of dNTP pools through IMPDH inhibition, direct inhibition of RNA synthesis, RNA capping inhibition, and immunomodulation (Graci and Cameron 2006). In LCMV-infected cells, treatment with high (100–400 µm) concentration of Rib did not increase mutation frequency values but inhibited dramatically both viral RNA synthesis and production of infectious progeny (Ruiz-Jarabo et al. 2003). However, treatment with low (20 µm) concentration of Rib increased 2.3-fold the mutation frequency of LCMV (Moreno et al. 2011), a finding that revealed a dose-dependent dual inhibitory and mutagenic activity of ribavirin for LCMV. The antiviral activity of Rib in LCMV-infected cells could be either totally or partially prevented by adding guanosine to the culture medium (Moreno et al. 2011). Inhibition of IMPDH by mycophenolic acid, which decreases intracellular GTP levels, did not increase but rather decreased LCMV mutation frequencies, thus raising some questions about whether targeting of IMPDH by Rib contributes to Rib-mediated inhibition of arenavirus multiplication. An alternative explanation for the protective effect of guanosine would be the dilution of Rib into the intracellular nucleotide pools, suggesting that Rib may be a competitive inhibitor (Moreno et al. 2011). The use of 5-FU as mutagenic agent and Rib either as inhibitor or as mutagen revealed that a treatment protocol based on a sequential inhibitor–mutagen administration exhibited the most potent antiviral activity compared to other combinations (Moreno et al. 2012a).
Rib-resistant escape mutants have been reported for several RNA viruses including PV, HCV, and FMDV (Pfeiffer and Kirkegaard 2003, 2005; Scheidel et al. 1987; Young et al. 2003) but are in general difficult to emerge. This likely reflects that Rib exerts its antiviral effect through different, mutually non-exclusive, mechanisms and thereby, a mutation conferring a virus with resistance to one mechanism may not allow viral escape from the overall antiviral activity of Rib. The observation that antiviral drugs exert their actions as a result of several mechanisms of action is not uncommon. Thus, 5-FU exerts its antiviral activity against FMDV through both its mutagenic activity and as inhibitor of initiation of viral RNA replication (Agudo et al. 2008). In coronavirus, the lack of the 3′–5′ exonuclease activity responsible of Nsp14 protein proofreading activity increases virus sensitivity to Rib and 5-FU mutagenesis, but evidence indicates that additional mechanisms of action associated with Rib treatment contribute to the overall observed reduction in virus infectivity (Smith et al. 2013). In the case of arenaviruses, Rib has been shown to exhibit a dose-dependent dual inhibitory and mutagenic activity (Moreno et al. 2011). At low concentrations (20 µm), Rib has been reported to increase the mutation frequency and mutant spectrum complexity of the population, whereas at higher concentrations (100 µm), Rib inhibited arenavirus replication without affecting the virus quasispecies complexity. The lack of a mutagenic effect of Rib when used at high concentration is not unexpected, because the corresponding strong inhibition of virus replication would limit the incorporation of Rib into the template RNA, which is required to exert its mutagenic effect.
The use of monotherapy-based treatments usually facilitates the selection of drug escape-resistant mutant within the population and subsequent virus rebound and therapy failure (Domingo 1989; Nijhuis et al. 2009; Young et al. 2003). Thus, inhibitor escape mutants have been isolated from virtually any virus for which a specific inhibitor has been developed, which poses a general problem in antimicrobial therapy (Domingo 2006). Combination therapy has proven effective to minimize and delay the selection of escape mutants (Pariente et al. 2003; Tapia et al. 2005). However, the emergence of variants resistant to more than one antiviral drug is not uncommon. A novel concept of combination therapy that is based on combining antiviral drugs targeting specific steps of the virus life cycle and lethal mutagenesis could contribute to overcome this problem. Importantly, recent evidence indicates that combination therapy of inhibitors of virus multiplication together with lethal mutagenesis can be dramatically more effective, while exhibiting much less toxicity, in controlling and clearing RNA virus infections than classic combination therapy (Perales et al. 2012). Moreover, evidence indicates that mutagen-mediated increase in virus mutation rate promotes the accumulation of non-functional viral gene products that often exhibit a strong dominant negative phenotype that further contributes to inhibition of virus multiplication, which has been referred to as lethal defection model of virus extinction (Grande-Perez et al. 2005b). Therefore, modest mutagenic intensities may be sufficient to help driving a virus to extinction, which further supports a combination antiviral therapy that incorporates lethal mutagenesis.
The interactions (neutral, positive, or negative) between drugs used in combination therapy are determined by the mechanism of action and pharmacokinetics of each drug, and thereby, the implementation of combination therapy requires a detailed understanding of the mechanisms of action of each drug. It is important noting that the mutagenic effect exerted by a drug could be masked or reduced by a replication inhibitor when used in combination, whereas the sequential administration of both drugs can result in a stronger antiviral effect than the sum of both drug effects (Moreno et al. 2012a; Perales et al. 2009). Therefore, combination therapy involving the use of a mutagenic agent should employ protocols involving the sequential administration of the drugs. In this regard, theoretical predictions (Eigen 1971, 2002) support the idea that reduced viral load promotes mutagen-induced viral extinction. Studies with FMDV provided the first experimental evidence for this prediction (Perales et al. 2009; Sierra et al. 2000). Likewise, studies with LCMV showed that administration of Rib at concentrations that favored its replication inhibition activity followed (sequential treatment) by administration of the mutagen 5-FU resulted in a stronger antiviral effect than when drugs were administered simultaneously or individually (Moreno et al. 2012a). This antiviral effect correlated with a low viral progeny production, low viral RNA synthesis, and a noticeable increase in quasispecies mutant spectrum complexity. The inhibition of viral replication by Rib potentiated the mutagenic effect of the subsequently added 5-FU, promoting the generation of DIG that contributed to the loss of infectivity. None of these effects were observed when mutagenic doses of Rib were used either in combination or sequentially with 5-FU. The lesser antiviral effect observed when inhibitory concentrations of Rib were administered together with the mutagen 5-FU could be explained due to a negative interaction between mutagen and replication inhibitor (Moreno et al. 2012a). The implications of these findings for the design of protocols to treat arenavirus infections in vivo remain to be investigated.
5.3 Arenavirus Genetic Variation and Development of Broad-Spectrum Vaccines
There are not FDA-approved vaccines against HF arenaviral diseases. The JUNV live-attenuated Candid1 strain has been shown to be an effective vaccine against Argentine hemorrhagic fever (AHF) disease (Enria and Barrera Oro 2002; Enria et al. 2008). However, outside Argentina, Candid1 has only IND status and studies addressing long-term immunity and safety have not been conducted. Moreover, Candid1 does not protect against LASV. Despite significant efforts dedicated to the development of LASV vaccines, not a single LASV vaccine candidate has entered a clinical trial although the MOPV/LASV reassortant ML29, as well as recombinant VSV and vaccinia virus expressing specific LASV antigens, has shown promising results (Falzarano and Feldmann 2013). Specifically, ML29 exhibited good safety and efficacy profiles in animal models of LASV infection. However, the high prevalence of HIV within LASV endemic regions raises safety concerns about the use of VSV- or vaccinia-based platforms.
Control of LASV infection seems to be mediated mainly by cellular immune responses, and significant titers of LASV neutralizing antibodies (NAbs) are usually observed only in patients who have clinically recovered (Jahrling and Peters 1984). However, passive antibody transfer has been shown to induce protection in animal models of LF (Jahrling 1983) and in limited human studies (Monath and Casals 1975) suggesting that an ideal vaccine should induce the right combination of cellular and humoral responses.
Sequence comparison of all proteins of seven pathogenic arenaviruses revealed a high degree of genetic variation among different geographical and temporal isolates of the same virus species (Bowen et al. 2000; Fulhorst et al. 2001; Garcia et al. 2000; Weaver et al. 2000). Overall, strain variation among 54 LASV strains was found to be as high as 27 and 15 % at the nucleotide and amino acid levels, respectively (Bowen et al. 2000). The high degree of LASV genetic diversity likely contributes to underestimating its prevalence and poses also a great challenge for vaccine development. In some endemic areas, up to 32 % of anti-LASV-positive samples would not have been detected, if the IFA had only been run with LASV-Josiah (Jos) strain, which is historically used as antigen in IFA or ELISA (Emmerich et al. 2008). This underscores the need for a multivalent or “universal” LASV vaccine able to protect against infections by the related isolates of the four lineages currently identified for LASV. The rML29/Jos expresses NP and GP antigens of Josiah strain, the prototype of LASV lineage IV, the largest sublineage, containing strains isolated in Guinea, Liberia, and Sierra Leone (Bowen et al. 2000). A single dose of ML29 vaccine effectively protected guinea pigs against challenge with homologous (lineage IV) or heterologous (lineage II) strains (Carrion et al. 2012). Lineage III and IV had a sister relationship, and the protection elicited by ML29 against LASV strains from lineage II strongly suggests that ML29 will also protect against members of lineage III. In contrast, sequence differences between LP (I) and Jos (IV) are 23.6 and 20.8 % for NP and GPC genes, respectively, and only one of the three recently predicted HL2-A2-restricted CTL epitopes was conserved in GP sequences of both viruses (Botten et al. 2006). This potential problem could be overcome by the use of arenavirus reverse genetics to generate rML29 expressing NP and GP antigens from lineages I and IV to generate a blended vaccine able to provide cross-protection against members of all LASV lineages. Despite the very promising features of the rML29 platform for the development of pan-LASV vaccine, it should be noted that the mechanisms of ML29 attenuation remain poorly understood and additional mutations, including reversions, in ML29 or reassortants between ML29 and circulating virulent LASV strains, could result in viruses with enhanced virulence.
References
Abraham J, Kwong JA, Albarino CG, Lu JG, Radoshitzky SR, Salazar-Bravo J, Farzan M, Spiropoulou CF, Choe H (2009) Host-species transferrin receptor 1 orthologs are cellular receptors for nonpathogenic New World clade B arenaviruses. PLoS Pathog 5(4):e1000358. doi:10.1371/journal.ppat.1000358
Aebischer T, Moskophidis D, Rohrer UH, Zinkernagel RM, Hengartner H (1991) In vitro selection of lymphocytic choriomeningitis virus escape mutants by cytotoxic T lymphocytes. Proc Natl Acad Sci USA 88(24):11047–11051
Agudo R, Arias A, Pariente N, Perales C, Escarmis C, Jorge A, Marina A, Domingo E (2008) Molecular characterization of a dual inhibitory and mutagenic activity of 5-fluorouridine triphosphate on viral RNA synthesis: implications for lethal mutagenesis. J Mol Biol 382(3):652–666. doi:10.1016/j.jmb.2008.07.033
Ahmed R, Oldstone MB (1988) Organ-specific selection of viral variants during chronic infection. J Exp Med 167(5):1719–1724
Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB (1984) Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice: role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 160(2):521–540
Ahmed R, Simon RS, Matloubian M, Kolhekar SR, Southern PJ, Freedman DM (1988) Genetic analysis of in vivo-selected viral variants causing chronic infection: importance of mutation in the L RNA segment of lymphocytic choriomeningitis virus. J Virol 62(9):3301–3308
Ahmed R, Hahn CS, Somasundaram T, Villarete L, Matloubian M, Strauss JH (1991) Molecular basis of organ-specific selection of viral variants during chronic infection. J Virol 65(8):4242–4247
Albarino CG, Posik DM, Ghiringhelli PD, Lozano ME, Romanowski V (1998) Arenavirus phylogeny: a new insight. Virus Genes 16(1):39–46
Albarino CG, Bergeron E, Erickson BR, Khristova ML, Rollin PE, Nichol ST (2009) Efficient reverse genetics generation of infectious junin viruses differing in glycoprotein processing. J Virol 83(11):5606–5614. doi:10.1128/JVI.00276-09
Albarino CG, Palacios G, Khristova ML, Erickson BR, Carroll SA, Comer JA, Hui J, Briese T, St George K, Ksiazek TG, Lipkin WI, Nichol ST (2010) High diversity and ancient common ancestry of lymphocytic choriomeningitis virus. Emerg Infect Dis 16(7):1093–1100. doi:10.3201/eid1607.091902
Albarino CG, Bird BH, Chakrabarti AK, Dodd KA, Erickson BR, Nichol ST (2011) Efficient rescue of recombinant Lassa virus reveals the influence of S segment noncoding regions on virus replication and virulence. J Virol 85(8):4020–4024. doi:10.1128/JVI.02556-10
Andrei G, De Clercq E (1990) Inhibitory effect of selected antiviral compounds on arenavirus replication in vitro. Antiviral Res 14(4–5):287–299
Andrei G, De Clercq E (1993) Molecular approaches for the treatment of hemorrhagic fever virus infections. Antiviral Res 22(1):45–75
Archer AM, Rico-Hesse R (2002) High genetic divergence and recombination in Arenaviruses from the Americas. Virology 304(2):274–281
Baranowski E, Ruiz-Jarabo CM, Sevilla N, Andreu D, Beck E, Domingo E (2000) Cell recognition by foot-and-mouth disease virus that lacks the RGD integrin-binding motif: flexibility in aphthovirus receptor usage. J Virol 74(4):1641–1647
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439(7077):682–687
Beaucourt S, Borderia AV, Coffey LL, Gnadig NF, Sanz-Ramos M, Beeharry Y, Vignuzzi M (2011) Isolation of fidelity variants of RNA viruses and characterization of virus mutation frequency. J Vis Exp 52:2953–2982. doi:10.3791/2953
Bergthaler A, Flatz L, Hegazy AN, Johnson S, Horvath E, Lohning M, Pinschewer DD (2010) Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc Natl Acad Sci USA 107(50):21641–21646. doi:10.1073/pnas.1011998107
Beyer WR, Popplau D, Garten W, von Laer D, Lenz O (2003) Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J Virol 77(5):2866–2872
Blasdell KR, Becker SD, Hurst J, Begon M, Bennett M (2008) Host range and genetic diversity of arenaviruses in rodents United Kingdom. Emerg Infect Dis 14(9):1455–1458. doi:10.3201/eid1409.080209
Bodewes R, Kik MJ, Raj VS, Schapendonk CM, Haagmans BL, Smits SL, Osterhaus AD (2013) Detection of novel divergent arenaviruses in boid snakes with inclusion body disease in The Netherlands. J Gen Virol 94(Pt 6):1206–1210. doi:10.1099/vir.0.051995-0
Bolken TC, Laquerre S, Zhang Y, Bailey TR, Pevear DC, Kickner SS, Sperzel LE, Jones KF, Warren TK, Amanda Lund S, Kirkwood-Watts DL, King DS, Shurtleff AC, Guttieri MC, Deng Y, Bleam M, Hruby DE (2005) Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antiviral Res
Bonnac LF, Mansky LM, Patterson SE (2013) Structure-activity relationships and design of viral mutagens and application to lethal mutagenesis. J Med Chem 56(23):9403–9414. doi:10.1021/jm400653j
Borden KL, Campbell Dwyer EJ, Salvato MS (1998) An arenavirus RING (zinc-binding) protein binds the oncoprotein promyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J Virol 72(1):758–766
Borrow P, Evans CF, Oldstone MB (1995) Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J Virol 69(2):1059–1070
Botten J, Alexander J, Pasquetto V, Sidney J, Barrowman P, Ting J, Peters B, Southwood S, Stewart B, Rodriguez-Carreno MP, Mothe B, Whitton JL, Sette A, Buchmeier MJ (2006) Identification of protective Lassa virus epitopes that are restricted by HLA-A2. J Virol 80(17):8351–8361. doi:10.1128/JVI.00896-06 ([pii] 80/17/8351)
Bowen MD, Peters CJ, Nichol ST (1996) The phylogeny of New World (Tacaribe complex) arenaviruses. Virology 219(1):285–290. doi:10.1006/viro.1996.0248
Bowen MD, Peters CJ, Nichol ST (1997) Phylogenetic analysis of the arenaviridae: patterns of virus evolution and evidence for cospeciation between arenaviruses and their rodent hosts. Mol Phylogenet Evol 8(3):301–316
Bowen MD, Rollin PE, Ksiazek TG, Hustad HL, Bausch DG, Demby AH, Bajani MD, Peters CJ, Nichol ST (2000) Genetic diversity among Lassa virus strains. J Virol 74(15):6992–7004
Bray M (2005) Pathogenesis of viral hemorrhagic fever. Curr Opin Immunol 17(4):399–403
Brown RJ, Peters PJ, Caron C, Gonzalez-Perez MP, Stones L, Ankghuambom C, Pondei K, McClure CP, Alemnji G, Taylor S, Sharp PM, Clapham PR, Ball JK (2011) Intercompartmental recombination of HIV-1 contributes to env intrahost diversity and modulates viral tropism and sensitivity to entry inhibitors. J Virol 85(12):6024–6037. doi:10.1128/JVI.00131-11
Brunotte L, Lelke M, Hass M, Kleinsteuber K, Becker-Ziaja B, Gunther S (2011) Domain structure of Lassa virus L protein. J Virol 85(1):324–333. doi:10.1128/JVI.00721-10
Buchmeier MJ, Peters CJ, de la Torre JC (2007) Arenaviridae: the viruses and their replication. In: Knipe DM, Holey PM (eds) Fields virology, 5th edn, vol 2, pp 1792–1827
Buesa-Gomez J, Teng MN, Oldstone CE, Oldstone MB, de la Torre JC (1996) Variants able to cause growth hormone deficiency syndrome are present within the disease-nil WE strain of lymphocytic choriomeningitis virus. J Virol 70(12):8988–8992
Cabot B, Martell M, Esteban JI, Sauleda S, Otero T, Esteban R, Guardia J, Gomez J (2000) Nucleotide and amino acid complexity of hepatitis C virus quasispecies in serum and liver. J Virol 74(2):805–811
Cajimat MN, Fulhorst CF (2004) Phylogeny of the venezuelan arenaviruses. Virus Res 102(2):199–206. doi:10.1016/j.virusres.2004.01.032 ([pii] S0168170204000735)
Cajimat MN, Milazzo ML, Hess BD, Rood MP, Fulhorst CF (2007) Principal host relationships and evolutionary history of the North American arenaviruses. Virology 367(2):235–243. doi:10.1016/j.virol.2007.05.031 ([pii] S0042-6822(07)00399-6)
Cajimat MN, Milazzo ML, Mauldin MR, Bradley RD, Fulhorst CF (2013) Diversity among Tacaribe serocomplex viruses (family arenaviridae) associated with the southern plains woodrat (Neotoma micropus). Virus Res 178(2):486–494. doi:10.1016/j.virusres.2013.10.004 ([pii] S0168-1702(13)00351-1)
Campbell Dwyer EJ, Lai H, MacDonald RC, Salvato MS, Borden KL (2000) The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J Virol 74(7):3293–3300
Candurra NA, Maskin L, Damonte EB (1996) Inhibition of arenavirus multiplication in vitro by phenotiazines. Antiviral Res 31(3):149–158
Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, Nichol ST, Compans RW, Campbell KP, Oldstone MB (1998) Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 282(5396):2079–2081 (see comments)
Carrion R Jr, Bredenbeek P, Jiang X, Tretyakova I, Pushko P, Lukashevich IS (2012) Vaccine platforms to control arenaviral hemorrhagic fevers. J Vaccines Vaccin 3(7). doi:10.4172/2157-7560.1000160
Cattaneo R, Billeter MA (1992) Mutations and A/I hypermutations in measles virus persistent infections. Curr Top Microbiol Immunol 176:63–74
Charrel RN, de Lamballerie X (2003) Arenaviruses other than Lassa virus. Antiviral Res 57(1–2):89–100
Charrel RN, de Lamballerie X, Fulhorst CF (2001) The whitewater Arroyo virus: natural evidence for genetic recombination among Tacaribe serocomplex viruses (family arenaviridae). Virology 283(2):161–166. doi:10.1006/viro.2001.0874
Charrel RN, Feldmann H, Fulhorst CF, Khelifa R, de Chesse R, de Lamballerie X (2002) Phylogeny of New World arenaviruses based on the complete coding sequences of the small genomic segment identified an evolutionary lineage produced by intrasegmental recombination. Biochem Biophys Res Commun 296(5):1118–1124
Charrel RN, Lemasson JJ, Garbutt M, Khelifa R, De Micco P, Feldmann H, de Lamballerie X (2003) New insights into the evolutionary relationships between arenaviruses provided by comparative analysis of small and large segment sequences. Virology 317(2):191–196
Charrel RN, de Lamballerie X, Emonet S (2008) Phylogeny of the genus arenavirus. Curr Opin Microbiol 11(4):362–368. doi:10.1016/j.mib.2008.06.001 ([pii] S1369-5274(08)00073-8)
Chen M, Lan S, Ou R, Price GE, Jiang H, de la Torre JC, Moskophidis D (2008) Genomic and biological characterization of aggressive and docile strains of lymphocytic choriomeningitis virus rescued from a plasmid-based reverse-genetics system. J Gen Virol 89(Pt 6):1421–1433. doi:10.1099/vir.0.83464-0 ([pii] 89/6/1421)
Cheng-Mayer C, Weiss C, Seto D, Levy JA (1989) Isolates of human immunodeficiency virus type 1 from the brain may constitute a special group of the AIDS virus. Proc Natl Acad Sci USA 86(21):8575–8579
Childs JE, Peters CJ (1993) Ecology and epidemiology of arenaviruses and their hosts. In: Salvato M (ed) The arenaviridae. Plenum Press, New York, pp 331–384
Choe H, Jemielity S, Abraham J, Radoshitzky SR, Farzan M (2011) Transferrin receptor 1 in the zoonosis and pathogenesis of New World hemorrhagic fever arenaviruses. Curr Opin Microbiol 14(4):476–482. doi:10.1016/j.mib.2011.07.014 ([pii] S1369-5274(11)00098-1)
Ciurea A, Klenerman P, Hunziker L, Horvath E, Senn BM, Ochsenbein AF, Hengartner H, Zinkernagel RM (2000) Viral persistence in vivo through selection of neutralizing antibody-escape variants. Proc Natl Acad Sci USA 97(6):2749–2754. doi:10.1073/pnas.040558797
Ciurea A, Hunziker L, Zinkernagel RM, Hengartner H (2001) Viral escape from the neutralizing antibody response: the lymphocytic choriomeningitis virus model. Immunogenetics 53(3):185–189
Cordo SM, Candurra NA, Damonte EB (1999) Myristic acid analogs are inhibitors of Junin virus replication. Microbes Infect 1(8):609–614
Cornu TI, de la Torre JC (2001) RING finger Z protein of lymphocytic choriomeningitis virus (LCMV) inhibits transcription and RNA replication of an LCMV S-segment minigenome. J Virol 75(19):9415–9426
Cornu TI, de la Torre JC (2002) Characterization of the arenavirus RING finger Z protein regions required for Z-mediated inhibition of viral RNA synthesis. J Virol 76(13):6678–6688
Cornu TI, Feldmann H, de la Torre JC (2004) Cells expressing the RING finger Z protein are resistant to arenavirus infection. J Virol 78(6):2979–2983
Coulibaly-N’Golo D, Allali B, Kouassi SK, Fichet-Calvet E, Becker-Ziaja B, Rieger T, Olschlager S, Dosso H, Denys C, Ter Meulen J, Akoua-Koffi C, Gunther S (2011) Novel arenavirus sequences in Hylomyscus sp. and Mus (Nannomys) setulosus from Cote d’Ivoire: implications for evolution of arenaviruses in Africa. PLoS ONE 6(6):e20893. doi:10.1371/journal.pone.0020893 ([pii] PONE-D-11-02828)
Dalldorf G (1939) The simultaneous occurrence of the viruses of canine distemper and lymphocytic choriomeningitis : a correction of “canine distemper in the rhesus monkey”. J Exp Med 70(1):19–27
Damonte EB, Coto CE (2002) Treatment of arenavirus infections: from basic studies to the challenge of antiviral therapy. Adv Virus Res 58:125–155
Damonte EB, Mersich SE, Coto CE (1983) Response of cells persistently infected with arenaviruses to superinfection with homotypic and heterotypic viruses. Virology 129(2):474–478
Dapp MJ, Clouser CL, Patterson S, Mansky LM (2009) 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J Virol 83(22):11950–11958. doi:10.1128/JVI.01406-09
Dapp MJ, Holtz CM, Mansky LM (2012) Concomitant lethal mutagenesis of human immunodeficiency virus type 1. J Mol Biol 419(3–4):158–170. doi:10.1016/j.jmb.2012.03.003
de la Torre JC, Holland JJ (1990) RNA virus quasispecies populations can suppress vastly superior mutant progeny. J Virol 64(12):6278–6281
Deforges S, Evlashev A, Perret M, Sodoyer M, Pouzol S, Scoazec JY, Bonnaud B, Diaz O, Paranhos-Baccala G, Lotteau V, Andre P (2004) Expression of hepatitis C virus proteins in epithelial intestinal cells in vivo. J Gen Virol 85(Pt 9):2515–2523. doi:10.1099/vir.0.80071-0 ([pii] 85/9/2515)
Delgado S, Erickson BR, Agudo R, Blair PJ, Vallejo E, Albarino CG, Vargas J, Comer JA, Rollin PE, Ksiazek TG, Olson JG, Nichol ST (2008) Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathog 4(4):e1000047. doi:10.1371/journal.ppat.1000047
Demogines A, Abraham J, Choe H, Farzan M, Sawyer SL (2013) Dual host-virus arms races shape an essential housekeeping protein. PLoS Biol 11(5):e1001571. doi:10.1371/journal.pbio.1001571 ([pii] PBIOLOGY-D-12-04902)
Denison MR, Graham RL, Donaldson EF, Eckerle LD, Baric RS (2011) Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol 8(2):270–279
Djavani M, Rodas J, Lukashevich IS, Horejsh D, Pandolfi PP, Borden KL, Salvato MS (2001) Role of the promyelocytic leukemia protein PML in the interferon sensitivity of lymphocytic choriomeningitis virus. J Virol 75(13):6204–6208
Domingo E (1989) RNA virus evolution and the control of viral disease. Prog Drug Res Fortschritte der Arzneimittelforschung Progres des recherches pharmaceutiques 33:93–133
Domingo E (1992) Genetic variation and quasi-species. Curr Opin Genet Dev 2(1):61–63
Domingo E (2006) Quasispecies: concepts and implications for virology, vol 299., Current topics in microbiology and immunologySpringer, Berlin
Domingo E (2007) Virus evolution. In: Knipe DM, Holey PM (eds) Fields virology, 5th edn
Domingo E, Holland JJ, Ahlquist P (1988) RNA Genetics, vol I, II, III. CRC Press, Boca Ratón
Domingo E, Mas A, Yuste E, Pariente N, Sierra S, Gutierrez-Riva M, Menendez-Arias L (2001) Virus population dynamics, fitness variations and the control of viral disease: an update. Prog Drug Res 57:77–115
Domingo E, Martin V, Perales C, Escarmis C (2008) Coxsackieviruses and quasispecies theory: evolution of enteroviruses. Curr Top Microbiol Immunol 323:3–32
Downs WG, Anderson CR, Spence L, Aitken TH, Greenhall AH (1963) Tacaribe virus, a new agent isolated from artibeus bats and mosquitoes in Trinidad, West Indies. Am J Trop Med Hyg 12:640–646
Drake JW, Holland JJ (1999) Mutation rates among RNA viruses. Proc Natl Acad Sci USA 96(24):13910–13913
Eigen M (1971) Selforganization of matter and the evolution of biological macromolecules. Die Naturwissenschaften 58(10):465–523
Eigen M (2002) Error catastrophe and antiviral strategy. Proc Natl Acad Sci USA 99(21):13374–13376. doi:10.1073/pnas.212514799
Ellenberg P, Edreira M, Scolaro L (2004) Resistance to superinfection of Vero cells persistently infected with Junin virus. Arch Virol 149(3):507–522. doi:10.1007/s00705-003-0227-1
Ellenberg P, Linero FN, Scolaro LA (2007) Superinfection exclusion in BHK-21 cells persistently infected with Junin virus. J Gen Virol 88(Pt 10):2730–2739. doi:10.1099/vir.0.83041-0
Emmerich P, Gunther S, Schmitz H (2008) Strain-specific antibody response to Lassa virus in the local population of west Africa. J Clin Virol 42(1):40–44. doi:10.1016/j.jcv.2007.11.019 ([pii] S1386-6532(07)00437-4)
Emonet S, Lemasson JJ, Gonzalez JP, de Lamballerie X, Charrel RN (2006) Phylogeny and evolution of old world arenaviruses. Virology 350(2):251–257. doi:10.1016/j.virol.2006.01.026
Emonet SF, de la Torre JC, Domingo E, Sevilla N (2009a) Arenavirus genetic diversity and its biological implications. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis 9(4):417–429. doi:10.1016/j.meegid.2009.03.005
Emonet SF, Garidou L, McGavern DB, de la Torre JC (2009b) Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc Natl Acad Sci USA 106(9):3473–3478. doi:10.1073/pnas.0900088106
Emonet SE, Urata S, de la Torre JC (2011a) Arenavirus reverse genetics: new approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology 411(2):416–425. doi:10.1016/j.virol.2011.01.013 ([pii] S0042-6822(11)00018-3)
Emonet SF, Seregin AV, Yun NE, Poussard AL, Walker AG, de la Torre JC, Paessler S (2011b) Rescue from cloned cDNAs and in vivo characterization of recombinant pathogenic Romero and live-attenuated Candid #1 strains of Junin virus, the causative agent of argentine hemorrhagic fever disease. J Virol 85(4):1473–1483. doi:10.1128/JVI.02102-10
Enria DA, Barrera Oro JG (2002) Junin virus vaccines. Curr Top Microbiol Immunol 263:239–261
Enria DA, Briggiler AM, Sanchez Z (2008) Treatment of argentine hemorrhagic fever. Antiviral Res 78(1):132–139. doi:10.1016/j.antiviral.2007.10.010 ([pii] S0166-3542(07)00433-0)
Evans CF, Borrow P, de la Torre JC, Oldstone MB (1994) Virus-induced immunosuppression: kinetic analysis of the selection of a mutation associated with viral persistence. J Virol 68(11):7367–7373
Falzarano D, Feldmann H (2013) Vaccines for viral hemorrhagic fevers–progress and short comings. Curr Opin Virol 3(3):343–351. doi:10.1016/j.coviro.2013.04.007 ([pii] S1879-6257(13)00062-X)
Fichet-Calvet E, Rogers DJ (2009) Risk maps of Lassa fever in West Africa. PLoS Negl Trop Dis 3(3):e388. doi:10.1371/journal.pntd.0000388
Flanagan ML, Oldenburg J, Reignier T, Holt N, Hamilton GA, Martin VK, Cannon PM (2008) New world clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J Virol 82(2):938–948. doi:10.1128/JVI.01397-07
Flatz L, Bergthaler A, de la Torre JC, Pinschewer DD (2006) Recovery of an arenavirus entirely from RNA polymerase I/II-driven cDNA. Proc Natl Acad Sci USA 103(12):4663–4668
Freedman DO, Woodall J (1999) Emerging infectious diseases and risk to the traveler. Med Clin North Am 83(4):865–883
Fulhorst CF, Bowen MD, Ksiazek TG, Rollin PE, Nichol ST, Kosoy MY, Peters CJ (1996) Isolation and characterization of whitewater Arroyo virus, a novel North American arenavirus. Virology 224(1):114–120. doi:10.1006/viro.1996.0512
Fulhorst CF, Bowen MD, Salas RA, Duno G, Utrera A, Ksiazek TG, De Manzione NM, De Miller E, Vasquez C, Peters CJ, Tesh RB (1999) Natural rodent host associations of Guanarito and pirital viruses (family arenaviridae) in central Venezuela. Am J Trop Med Hyg 61(2):325–330
Fulhorst CF, Charrel RN, Weaver SC, Ksiazek TG, Bradley RD, Milazzo ML, Tesh RB, Bowen MD (2001) Geographic distribution and genetic diversity of whitewater Arroyo virus in the southwestern United States. Emerg Infect Dis 7(3):403–407. doi:10.3201/eid0703.010306
Fulhorst CF, Cajimat MN, Milazzo ML, Paredes H, de Manzione NM, Salas RA, Rollin PE, Ksiazek TG (2008) Genetic diversity between and within the arenavirus species indigenous to western Venezuela. Virology 378(2):205–213. doi:10.1016/j.virol.2008.05.014 ([pii] S0042-6822(08)00341-3)
Garcia JB, Morzunov SP, Levis S, Rowe J, Calderon G, Enria D, Sabattini M, Buchmeier MJ, Bowen MD, St Jeor SC (2000) Genetic diversity of the Junin virus in Argentina: geographic and temporal patterns. Virology 272(1):127–136. doi:10.1006/viro.2000.0345 ([pii] S0042682200903453)
Geisbert TW, Jahrling PB (2004) Exotic emerging viral diseases: progress and challenges. Nat Med 10(12 Suppl):S110–S121
Geleziunas R, Bour S, Wainberg MA (1994) Cell surface down-modulation of CD4 after infection by HIV-1. FASEB J 8(9):593–600
Gonzalez-Lopez C, Arias A, Pariente N, Gomez-Mariano G, Domingo E (2004) Preextinction viral RNA can interfere with infectivity. J Virol 78(7):3319–3324
Graci JD, Cameron CE (2006) Mechanisms of action of ribavirin against distinct viruses. Rev Med Virol 16(1):37–48. doi:10.1002/rmv.483
Graci JD, Harki DA, Korneeva VS, Edathil JP, Too K, Franco D, Smidansky ED, Paul AV, Peterson BR, Brown DM, Loakes D, Cameron CE (2007) Lethal mutagenesis of poliovirus mediated by a mutagenic pyrimidine analogue. J Virol 81(20):11256–11266. doi:10.1128/JVI.01028-07
Graci JD, Too K, Smidansky ED, Edathil JP, Barr EW, Harki DA, Galarraga JE, Bollinger JM Jr, Peterson BR, Loakes D, Brown DM, Cameron CE (2008) Lethal mutagenesis of picornaviruses with N-6-modified purine nucleoside analogues. Antimicrob Agents Chemother 52(3):971–979. doi:10.1128/AAC.01056-07
Grande-Perez A, Sierra S, Castro MG, Domingo E, Lowenstein PR (2002) Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc Natl Acad Sci USA 99(20):12938–12943. doi:10.1073/pnas.182426999
Grande-Perez A, Gomez-Mariano G, Lowenstein PR, Domingo E (2005a) Mutagenesis-induced, large fitness variations with an invariant arenavirus consensus genomic nucleotide sequence. J Virol 79(16):10451–10459. doi:10.1128/JVI.79.16.10451-10459.2005
Grande-Perez A, Lazaro E, Lowenstein P, Domingo E, Manrubia SC (2005b) Suppression of viral infectivity through lethal defection. Proc Natl Acad Sci USA 102(12):4448–4452
Greenwood AG, Sanchez S (2002) Serological evidence of murine pathogens in wild grey squirrels (Sciurus carolinensis) in North Wales. Vet Rec 150(17):543–546
Hall JS, French R, Hein GL, Morris TJ, Stenger DC (2001) Three distinct mechanisms facilitate genetic isolation of sympatric wheat streak mosaic virus lineages. Virology 282(2):230–236. doi:10.1006/viro.2001.0841 ([pii] S0042-6822(01)90841-4)
Harki DA, Graci JD, Korneeva VS, Ghosh SK, Hong Z, Cameron CE, Peterson BR (2002) Synthesis and antiviral evaluation of a mutagenic and non-hydrogen bonding ribonucleoside analogue: 1-beta-D-Ribofuranosyl-3-nitropyrrole. Biochemistry 41(29):9026–9033
Harris KS, Brabant W, Styrchak S, Gall A, Daifuku R (2005) KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res 67(1):1–9. doi:10.1016/j.antiviral.2005.03.004
Hass M, Golnitz U, Muller S, Becker-Ziaja B, Gunther S (2004) Replicon system for Lassa virus. J Virol 78(24):13793–13803
Hass M, Westerkofsky M, Muller S, Becker-Ziaja B, Busch C, Gunther S (2006) Mutational analysis of the lassa virus promoter. J Virol 80(24):12414–12419
Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO (2011) Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci USA 108(6):2396–2401. doi:10.1073/pnas.1016404108
Haydon DT, Bastos AD, Knowles NJ, Samuel AR (2001) Evidence for positive selection in foot-and-mouth disease virus capsid genes from field isolates. Genetics 157(1):7–15
Hetzel U, Sironen T, Laurinmaki P, Liljeroos L, Patjas A, Henttonen H, Vaheri A, Artelt A, Kipar A, Butcher SJ, Vapalahti O, Hepojoki J (2013) Isolation, identification, and characterization of novel arenaviruses, the etiological agents of boid inclusion body disease. J Virol 87(20):10918–10935. doi:10.1128/JVI.01123-13
Hicks C, Clay P, Redfield R, Lalezari J, Liporace R, Schneider S, Sension M, McRae M, Laurent JP (2013) Safety, tolerability, and efficacy of KP-1461 as monotherapy for 124 days in antiretroviral-experienced, HIV type 1-infected subjects. AIDS Res Hum Retroviruses 29(2):250–255. doi:10.1089/AID.2012.0093
Holland JJ, Domingo E, de la Torre JC, Steinhauer DA (1990) Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J Virol 64(8):3960–3962
Horga MA, Gusella GL, Greengard O, Poltoratskaia N, Porotto M, Moscona A (2000) Mechanism of interference mediated by human parainfluenza virus type 3 infection. J Virol 74(24):11792–11799
Hotchin J (1973) Transient virus infection: spontaneous recovery mechanism of lymphocytic choriomeningitis virrus-infected cells. Nat New Biol 241(113):270–272
Hotchin J, Sikora E (1973) Low-pathogenicity variant of lymphocytic choriomeningitis virus. Infect Immun 7(5):825–826
Hotchin J, Kinch W, Benson L (1971) Lytic and turbid plaque-type mutants of lymphocytic choriomeningitis virus as a cause of neurological disease or persistent infection. Infect Immun 4(3):281–286
Huang IC, Li W, Sui J, Marasco W, Choe H, Farzan M (2008) Influenza A virus neuraminidase limits viral superinfection. J Virol 82(10):4834–4843. doi:10.1128/JVI.00079-08
Huggins JW (1989) Prospects for treatment of viral hemorrhagic fevers with ribavirin, a broad-spectrum antiviral drug. Rev Infect Dis 11(Suppl 4):S750–S761
Hugot JP, Gonzalez JP, Denys C (2001) Evolution of the Old World arenaviridae and their rodent hosts: generalized host-transfer or association by descent? Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis 1(1):13–20 ([pii] S156713480100003X)
Hundley HA, Bass BL (2010) ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci 35(7):377–383. doi:10.1016/j.tibs.2010.02.008 ([pii] S0968-0004(10)00029-0)
Irwin NR, Bayerlova M, Missa O, Martinkova N (2012) Complex patterns of host switching in New World arenaviruses. Mol Ecol 21(16):4137–4150. doi:10.1111/j.1365-294X.2012.05663.x
Isaacson M (2001) Viral hemorrhagic fever hazards for travelers in Africa. Clin Infect Dis 33(10):1707–1712
Ishii A, Thomas Y, Moonga L, Nakamura I, Ohnuma A, Hang’ombe BM, Takada A, Mweene AS, Sawa H (2012) Molecular surveillance and phylogenetic analysis of Old World arenaviruses in Zambia. J Gen Virol 93(Pt 10):2247–2251. doi:10.1099/vir.0.044099-0
Jackson AP, Charleston MA (2004) A cophylogenetic perspective of RNA-virus evolution. Mol Biol Evol 21(1):45–57. doi:10.1093/molbev/msg232
Jahrling PB (1983) Protection of Lassa virus-infected guinea pigs with Lassa-immune plasma of guinea pig, primate, and human origin. J Med Virol 12(2):93–102
Jahrling PB, Peters CJ (1984) Passive antibody therapy of Lassa fever in cynomolgus monkeys: importance of neutralizing antibody and Lassa virus strain. Infect Immun 44(2):528–533
Jahrling PB, Peters CJ (1992) Lymphocytic choriomeningitis virus A neglected pathogen of man. Arch Pathol Lab Med 116(5):486–488
Jay MT, Glaser C, Fulhorst CF (2005) The arenaviruses. J Am Vet Med Assoc 227(6):904–915
Jelcic I, Hotz-Wagenblatt A, Hunziker A, Zur Hausen H, de Villiers EM (2004) Isolation of multiple TT virus genotypes from spleen biopsy tissue from a Hodgkin’s disease patient: genome reorganization and diversity in the hypervariable region. J Virol 78(14):7498–7507. doi:10.1128/JVI.78.14.7498-7507.2004 ([pii] 78/14/7498)
Jridi C, Martin JF, Marie-Jeanne V, Labonne G, Blanc S (2006) Distinct viral populations differentiate and evolve independently in a single perennial host plant. J Virol 80(5):2349–2357. doi:10.1128/JVI.80.5.2349-2357.2006 ([pii] 80/5/2349)
Kilgore PE, Peters CJ, Mills JN, Rollin PE, Armstrong L, Khan AS, Ksiazek TG (1995) Prospects for the control of Bolivian hemorrhagic fever. Emerg Infect Dis 1(3):97–100
Kilgore PE, Ksiazek TG, Rollin PE, Mills JN, Villagra MR, Montenegro MJ, Costales MA, Paredes LC, Peters CJ (1997) Treatment of Bolivian hemorrhagic fever with intravenous ribavirin. Clin Infect Dis 24(4):718–722
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (2012) Ninth report of the international committee on taxonomy of viruses. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy: classification and nomenclature of viruses. Elsevier, San Diego
Klavinskis LS, Oldstone MB (1989) Lymphocytic choriomeningitis virus selectively alters differentiated but not housekeeping functions: block in expression of growth hormone gene is at the level of transcriptional initiation. Virology 168(2):232–235
Kranzusch PJ, Whelan SP (2011) Arenavirus Z protein controls viral RNA synthesis by locking a polymerase-promoter complex. Proc Natl Acad Sci USA 108(49):19743–19748. doi:10.1073/pnas.1112742108
Kunz S, Borrow P, Oldstone MB (2002) Receptor structure, binding, and cell entry of arenaviruses. Curr Top Microbiol Immunol 262:111–137
Kunz S, Edelmann KH, de la Torre J-C, Gorney R, Oldstone MBA (2003a) Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology (in press)
Kunz S, Edelmann KH, de la Torre JC, Gorney R, Oldstone MB (2003b) Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology 314(1):168–178 ([pii] S0042682203004215)
Kunz S, Sevilla N, Rojek JM, Oldstone MB (2004) Use of alternative receptors different than alpha-dystroglycan by selected isolates of lymphocytic choriomeningitis virus. Virology 325(2):432–445
Kunz S, Rojek JM, Kanagawa M, Spiropoulou CF, Barresi R, Campbell KP, Oldstone MB (2005) Posttranslational modification of alpha-dystroglycan, the cellular receptor for arenaviruses, by the glycosyltransferase LARGE is critical for virus binding. J Virol 79(22):14282–14296. doi:10.1128/JVI.79.22.14282-14296.2005 ([pii] 79/22/14282)
Lan S, McLay Schelde L, Wang J, Kumar N, Ly H, Liang Y (2009) Development of infectious clones for virulent and avirulent pichinde viruses: a model virus to study arenavirus-induced hemorrhagic fevers. J Virol 83(13):6357–6362. doi:10.1128/JVI.00019-09
Lecompte E, ter Meulen J, Emonet S, Daffis S, Charrel RN (2007) Genetic identification of Kodoko virus, a novel arenavirus of the African pigmy mouse (Mus Nannomys minutoides) in West Africa. Virology 364(1):178–183. doi:10.1016/j.virol.2007.02.008 ([pii] S0042-6822(07)00118-3)
Lee CH, Gilbertson DL, Novella IS, Huerta R, Domingo E, Holland JJ (1997) Negative effects of chemical mutagenesis on the adaptive behavior of vesicular stomatitis virus. J Virol 71(5):3636–3640
Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC (2000) NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J Virol 74(8):3470–3477
Lee AM, Rojek JM, Spiropoulou CF, Gundersen AT, Jin W, Shaginian A, York J, Nunberg JH, Boger DL, Oldstone MB, Kunz S (2008) Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J Biol Chem 283(27):18734–18742. doi:10.1074/jbc.M802089200
Lelke M, Brunotte L, Busch C, Gunther S (2010) An N-terminal region of Lassa virus L protein plays a critical role in transcription but not replication of the virus genome. J Virol 84(4):1934–1944. doi:10.1128/JVI.01657-09
Lenz O, ter Meulen J, Klenk HD, Seidah NG, Garten W (2001) The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc Natl Acad Sci USA 98(22):12701–12705. doi:10.1073/pnas.221447598
Lewicki H, Tishon A, Borrow P, Evans CF, Gairin JE, Hahn KM, Jewell DA, Wilson IA, Oldstone MB (1995) CTL escape viral variants I: generation and molecular characterization. Virology 210(1):29–40. doi:10.1006/viro.1995.1314 ([pii] S0042-6822(85)71314-1)
Liang Y, Lan S, Ly H (2009) Molecular determinants of Pichinde virus infection of guinea pigs–a small animal model system for arenaviral hemorrhagic fevers. Ann N Y Acad Sci 1171(Suppl 1):E65–E74. doi:10.1111/j.1749-6632.2009.05051.x ([pii] NYAS5051)
Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI (1999) Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc Natl Acad Sci USA 96(4):1492–1497
Lopez N, Jacamo R, Franze-Fernandez MT (2001) Transcription and RNA replication of tacaribe virus genome and antigenome analogs require N and L proteins: z protein is an inhibitor of these processes. J Virol 75(24):12241–12251
Loureiro ME, Wilda M, Levingston Macleod JM, D’Antuono A, Foscaldi S, Marino Buslje C, Lopez N (2011) Molecular determinants of arenavirus Z protein homo-oligomerization and L polymerase binding. J Virol 85(23):12304–12314. doi:10.1128/JVI.05691-11
Lukashevich IS (1992) Generation of reassortants between African arenaviruses. Virology 188(2):600–605
Lukashevich IS (2012) Advanced vaccine candidates for Lassa fever. Viruses 4(11):2514–2557. doi:10.3390/v4112514
Lukashevich IS, Patterson J, Carrion R, Moshkoff D, Ticer A, Zapata J, Brasky K, Geiger R, Hubbard GB, Bryant J, Salvato MS (2005) A live attenuated vaccine for Lassa fever made by reassortment of Lassa and Mopeia viruses. J Virol 79(22):13934–13942. doi:10.1128/JVI.79.22.13934-13942.2005
Mallela A, Nishikura K (2012) A-to-I editing of protein coding and noncoding RNAs. Crit Rev Biochem Mol Biol 47(6):493–501. doi:10.3109/10409238.2012.714350
Manrubia SC, Domingo E, Lazaro E (2010) Pathways to extinction: beyond the error threshold. Philos Trans R Soc Lond B Biol Sci 365(1548):1943–1952. doi:10.1098/rstb.2010.0076
Martin V, Domingo E (2008) Influence of the mutant spectrum in viral evolution: focused selection of antigenic variants in a reconstructed viral quasispecies. Mol Biol Evol 25(8):1544–1554. doi:10.1093/molbev/msn099
Martin V, Abia D, Domingo E, Grande-Perez A (2010) An interfering activity against lymphocytic choriomeningitis virus replication associated with enhanced mutagenesis. J Gen Virol 91(Pt 4):990–1003. doi:10.1099/vir.0.017053-0
Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC (2006) Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 80(18):9192–9199
Martinez-Sobrido L, Giannakas P, Cubitt B, Garcia-Sastre A, de la Torre JC (2007) Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J Virol 81(22):12696–12703
Martinez-Sobrido L, Emonet S, Giannakas P, Cubitt B, Garcia-Sastre A, de la Torre JC (2009) Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 83(21):11330–11340. doi:10.1128/JVI.00763-09
Mas A, Lopez-Galindez C, Cacho I, Gomez J, Martinez MA (2010) Unfinished stories on viral quasispecies and Darwinian views of evolution. J Mol Biol 397(4):865–877. doi:10.1016/j.jmb.2010.02.005 ([pii] S0022-2836(10)00155-5)
Matloubian M, Somasundaram T, Kolhekar SR, Selvakumar R, Ahmed R (1990) Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. J Exp Med 172(4):1043–1048
Matloubian M, Kolhekar SR, Somasundaram T, Ahmed R (1993) Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J Virol 67(12):7340–7349
McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, Johnson KM, Elliott LH, Belmont-Williams R (1986) Lassa fever: effective therapy with ribavirin. N Engl J Med 314(1):20–26
McKee KT Jr, Huggins JW, Trahan CJ, Mahlandt BG (1988) Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob Agents Chemother 32(9):1304–1309
Merkler D, Horvath E, Bruck W, Zinkernagel RM, Del la Torre JC, Pinschewer DD (2006) “Viral deja vu” elicits organ-specific immune disease independent of reactivity to self. J Clin Invest 116(5):1254–1263
Mets MB, Barton LL, Khan AS, Ksiazek TG (2000) Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am J Ophthalmol 130(2):209–215
Meyer BJ, Southern PJ (1994) Sequence heterogeneity in the termini of lymphocytic choriomeningitis virus genomic and antigenomic RNAs. J Virol 68(11):7659–7664
Meyer BJ, de la Torre JC, Southern PJ (2002) Arenaviruses: genomic RNAs, transcription, and replication. Curr Top Microbiol Immunol 262:139–149
Mills JN, Barrera Oro JG, Bressler DS, Childs JE, Tesh RB, Smith JF, Enria DA, Geisbert TW, McKee KT Jr, Bowen MD, Peters CJ, Jahrling PB (1996) Characterization of Oliveros virus, a new member of the Tacaribe complex (arenaviridae: arenavirus). Am J Trop Med Hyg 54(4):399–404
Monath TP, Casals J (1975) Diagnosis of Lassa fever and the isolation and management of patients. Bull World Health Organ 52(4–6):707–715
Moreno H, Gallego I, Sevilla N, de la Torre JC, Domingo E, Martin V (2011) Ribavirin can be mutagenic for arenaviruses. J Virol 85(14):7246–7255. doi:10.1128/JVI.00614-11
Moreno H, Grande-Perez A, Domingo E, Martin V (2012a) Arenaviruses and lethal mutagenesis: prospects for new ribavirin-based interventions. Viruses 4(11):2786–2805. doi:10.3390/v4112786
Moreno H, Tejero H, de la Torre JC, Domingo E, Martin V (2012b) Mutagenesis-mediated virus extinction: virus-dependent effect of viral load on sensitivity to lethal defection. PLoS ONE 7(3):e32550. doi:10.1371/journal.pone.0032550
Morimoto K, Hooper DC, Carbaugh H, Fu ZF, Koprowski H, Dietzschold B (1998) Rabies virus quasispecies: implications for pathogenesis. Proc Natl Acad Sci USA 95(6):3152–3156
Morin B, Coutard B, Lelke M, Ferron F, Kerber R, Jamal S, Frangeul A, Baronti C, Charrel R, de Lamballerie X, Vonrhein C, Lescar J, Bricogne G, Gunther S, Canard B (2010) The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog 6(9):e1001038. doi:10.1371/journal.ppat.1001038
Morrison TG, McGinnes LW (1989) Avian cells expressing the Newcastle disease virus hemagglutinin-neuraminidase protein are resistant to Newcastle disease virus infection. Virology 171(1):10–17
Moshkoff DA, Salvato MS, Lukashevich IS (2007) Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes 34(2):169–176. doi:10.1007/s11262-006-0050-3
Muller G, Bruns M, Martinez Peralta L, Lehmann-Grube F (1983) Lymphocytic choriomeningitis virus. IV. Electron microscopic investigation of the virion. Arch Virol 75(4):229–242
Murphy DG, Dimock K, Kang CY (1991) Numerous transitions in human parainfluenza virus 3 RNA recovered from persistently infected cells. Virology 181(2):760–763
Neuman BW, Adair BD, Burns JW, Milligan RA, Buchmeier MJ, Yeager M (2005) Complementarity in the supramolecular design of arenaviruses and retroviruses revealed by electron cryomicroscopy and image analysis. J Virol 79(6):3822–3830
Neumann G, Kawaoka Y (2004) Reverse genetics systems for the generation of segmented negative-sense RNA viruses entirely from cloned cDNA. Curr Top Microbiol Immunol 283:43–60
Nijhuis M, van Maarseveen NM, Boucher CA (2009) Antiviral resistance and impact on viral replication capacity: evolution of viruses under antiviral pressure occurs in three phases. Handb Exp Pharmacol 189:299–320. doi:10.1007/978-3-540-79086-0_11
Nishikura K (2010) Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79:321–349. doi:10.1146/annurev-biochem-060208-105251
Novella IS, Duarte EA, Elena SF, Moya A, Domingo E, Holland JJ (1995) Exponential increases of RNA virus fitness during large population transmissions. Proc Natl Acad Sci USA 92(13):5841–5844
Oldstone MB (2002) Biology and pathogenesis of lymphocytic choriomeningitis virus infection. In: Oldstone MB (ed) Arenaviruses, vol 263, pp 83–118
Oldstone MB, Campbell KP (2010) Decoding arenavirus pathogenesis: essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology 411(2):170–179. doi:10.1016/j.virol.2010.11.023 ([pii] S0042-6822(10)00741-5)
Oldstone MB, Rodriguez M, Daughaday WH, Lampert PW (1984) Viral perturbation of endocrine function: disordered cell function leads to disturbed homeostasis and disease. Nature 307(5948):278–281
Oldstone MB, Ahmed R, Buchmeier MJ, Blount P, Tishon A (1985) Perturbation of differentiated functions during viral infection in vivo I: relationship of lymphocytic choriomeningitis virus and host strains to growth hormone deficiency. Virology 142(1):158–174
Ortega-Prieto AM, Sheldon J, Grande-Perez A, Tejero H, Gregori J, Quer J, Esteban JI, Domingo E, Perales C (2013) Extinction of hepatitis C virus by ribavirin in hepatoma cells involves lethal mutagenesis. PLoS ONE 8(8):e71039. doi:10.1371/journal.pone.0071039
Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI (2008) A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med 358(10):991–998
Pariente N, Airaksinen A, Domingo E (2003) Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J Virol 77(12):7131–7138
Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S (2011) Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog 7(9):e1002232. doi:10.1371/journal.ppat.1002232 ([pii] 10-PLPA-RA-4139)
Perales C, Martin V, Ruiz-Jarabo CM, Domingo E (2005) Monitoring sequence space as a test for the target of selection in viruses. J Mol Biol 345(3):451–459. doi:10.1016/j.jmb.2004.10.066 ([pii] S0022-2836(04)01372-5)
Perales C, Mateo R, Mateu MG, Domingo E (2007) Insights into RNA virus mutant spectrum and lethal mutagenesis events: replicative interference and complementation by multiple point mutants. J Mol Biol 369(4):985–1000. doi:10.1016/j.jmb.2007.03.074
Perales C, Agudo R, Tejero H, Manrubia SC, Domingo E (2009) Potential benefits of sequential inhibitor-mutagen treatments of RNA virus infections. PLoS Pathog 5(11):e1000658. doi:10.1371/journal.ppat.1000658
Perales C, Iranzo J, Manrubia SC, Domingo E (2012) The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol 20(12):595–603. doi:10.1016/j.tim.2012.08.010 ([pii] S0966-842X(12)00155-2)
Perez M, de la Torre JC (2002) Characterization of the genomic promoter of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). J Virol (in Press)
Perez M, Craven RC, de la Torre JC (2003) The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc Natl Acad Sci USA 100(22):12978–12983
Perez M, Greenwald DL, de la Torre JC (2004) Myristoylation of the RING finger Z protein is essential for arenavirus budding. J Virol 78(20):11443–11448
Peters CJ (2002) Human Infection with Arenaviruses in the Americas. In: Oldstone MB (ed) Arenaviruses i, vol 262., Current topics in microbiology and immunologySpringer, Berlin, pp 65–74
Peters CJ (2006) Lymphocytic choriomeningitis virus—an old enemy up to new tricks. N Engl J Med 354(21):2208–2211
Pfeiffer JK, Kirkegaard K (2003) A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci USA 100(12):7289–7294. doi:10.1073/pnas.1232294100
Pfeiffer JK, Kirkegaard K (2005) Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog 1(2):e11. doi:10.1371/journal.ppat.0010011
Pinschewer DD, Perez M, Sanchez AB, de la Torre JC (2003) Recombinant lymphocytic choriomeningitis virus expressing vesicular stomatitis virus glycoprotein. Proc Natl Acad Sci USA 100(13):7895–7900
Pinschewer DD, Perez M, de la Torre JC (2005) Dual role of the lymphocytic choriomeningitis virus intergenic region in transcription termination and virus propagation. J Virol 79(7):4519–4526
Pinschewer DD, Flatz L, Steinborn R, Horvath E, Fernandez M, Lutz H, Suter M, Bergthaler A (2010) Innate and adaptive immune control of genetically engineered live-attenuated arenavirus vaccine prototypes. Int Immunol 22(9):749–756. doi:10.1093/intimm/dxq061
Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel RM (1990) Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature 346(6285):629–633. doi:10.1038/346629a0
Poch O, Sauvaget I, Delarue M, Tordo N (1989) Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J 8(12):3867–3874
Polson AG, Bass BL (1994) Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J 13(23):5701–5711
Prince GA, Ottolini MG, Moscona A (2001) Contribution of the human parainfluenza virus type 3 HN-receptor interaction to pathogenesis in vivo. J Virol 75(24):12446–12451. doi:10.1128/JVI.75.24.12446-12451.2001
Pulkkinen AJ, Pfau CJ (1970) Plaque size heterogeneity: a genetic trait of lymphocytic choriomeningitis virus. Appl Microbiol 20(1):123–128
Qi X, Lan S, Wang W, Schelde LM, Dong H, Wallat GD, Ly H, Liang Y, Dong C (2010) Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468(7325):779–783. doi:10.1038/nature09605
Radoshitzky SR, Abraham J, Spiropoulou CF, Kuhn JH, Nguyen D, Li W, Nagel J, Schmidt PJ, Nunberg JH, Andrews NC, Farzan M, Choe H (2007) Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446(7131):92–96 (Epub 2007 Feb 2007)
Radoshitzky SR, Kuhn JH, Spiropoulou CF, Albarino CG, Nguyen DP, Salazar-Bravo J, Dorfman T, Lee AS, Wang E, Ross SR, Choe H, Farzan M (2008) Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. Proc Natl Acad Sci USA 105(7):2664–2669. doi:10.1073/pnas.0709254105
Richmond JK, Baglole DJ (2003) Lassa fever: epidemiology, clinical features, and social consequences. BMJ 327(7426):1271–1275. doi:10.1136/bmj.327.7426.1271
Rima BK, Gatherer D, Young DF, Norsted H, Randall RE, Davison AJ (2014) Stability of the parainfluenza virus 5 genome revealed by deep sequencing of strains isolated from different hosts and following passage in cell culture. J Virol. doi:10.1128/JVI.03351-13
Riviere Y (1987) Mapping arenavirus genes causing virulence. Curr Top Microbiol Immunol 133:59–65
Riviere Y, Oldstone MB (1986) Genetic reassortants of lymphocytic choriomeningitis virus: unexpected disease and mechanism of pathogenesis. J Virol 59(2):363–368
Riviere Y, Ahmed R, Oldstone MB (1986) The use of lymphocytic choriomeningitis virus reassortants to map viral genes causing virulence. Med Microbiol Immunol 175(2–3):191–192
Rodrigo WW, de la Torre JC, Martinez-Sobrido L (2011) Use of single-cycle infectious lymphocytic choriomeningitis virus to study hemorrhagic fever arenaviruses. J Virol 85(4):1684–1695. doi:10.1128/JVI.02229-10
Rojek JM, Kunz S (2008) Cell entry by human pathogenic arenaviruses. Cell Microbiol 10(4):828–835
Rojek JM, Perez M, Kunz S (2008a) Cellular entry of lymphocytic choriomeningitis virus. J Virol 82(3):1505–1517
Rojek JM, Sanchez AB, Thao NN, de la Torre JC, Kunz S (2008b) Different mechanisms of cell entry by human pathogenic Old World and New World arenaviruses. J Virol
Rojek JM, Pasqual G, Sanchez AB, Nguyen NT, de la Torre JC, Kunz S (2010) Targeting the proteolytic processing of the viral glycoprotein precursor is a promising novel antiviral strategy against arenaviruses. J Virol 84(1):573–584. doi:10.1128/JVI.01697-09
Rueda P, Garcia-Barreno B, Melero JA (1994) Loss of conserved cysteine residues in the attachment (G) glycoprotein of two human respiratory syncytial virus escape mutants that contain multiple A-G substitutions (hypermutations). Virology 198(2):653–662 ([pii] S0042682284710774)
Ruiz-Jarabo CM, Ly C, Domingo E, de la Torre JC (2003) Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 308(1):37–47
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Sun W, Wang H, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe’er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Sham PC, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Johnson TA, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Yakub I, Birren BW, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L’Archeveque P, Bellemare G, Saeki K, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J (2007) Genome-wide detection and characterization of positive selection in human populations. Nature 449(7164):913–918. doi:10.1038/nature06250
Salazar-Bravo J, Ruedas LA, Yates TL (2002) Mammalian reservoirs of arenaviruses. In: Oldstone MBA (ed) Arenaviruses I: the epidemiology, molecular and cell biology of arenaviruses, vol 262., Current topics in microbiology and immunologySpringer, Berlin, pp 25–63
Salvato MS, Shimomaye EM (1989) The completed sequence of lymphocytic choriomeningitis virus reveals a unique RNA structure and a gene for a zinc finger protein. Virology 173(1):1–10
Salvato M, Borrow P, Shimomaye E, Oldstone MB (1991) Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J Virol 65(4):1863–1869
Sanchez AB, de la Torre JC (2005) Genetic and biochemical evidence for an oligomeric structure of the functional L polymerase of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 79(11):7262–7268
Sanchez AB, de la Torre JC (2006) Rescue of the prototypic Arenavirus LCMV entirely from plasmid. Virology 350(2):370–380
Sanjuan R, Codoner FM, Moya A, Elena SF (2004) Natural selection and the organ-specific differentiation of HIV-1 V3 hypervariable region. Evolution 58(6):1185–1194
Sanz-Ramos M, Rodriguez-Calvo T, Sevilla N (2012) Mutagenesis-mediated decrease of pathogenicity as a feature of the mutant spectrum of a viral population. PLoS ONE 7(6):e39941. doi:10.1371/journal.pone.0039941
Saunders AA, Ting JP, Meisner J, Neuman BW, Perez M, de la Torre JC, Buchmeier MJ (2007) Mapping the landscape of the lymphocytic choriomeningitis virus stable signal peptide reveals novel functional domains. J Virol 81(11):5649–5657. doi:10.1128/JVI.02759-06
Scheidel LM, Durbin RK, Stollar V (1987) Sindbis virus mutants resistant to mycophenolic acid and ribavirin. Virology 158(1):1–7
Sevilla N, de la Torre JC (2006) Arenavirus diversity and evolution: quasispecies in vivo. In: Domingo E (ed) Quasispecies: concepts and implications for virology, current topics in microbiology and immunology, vol 299, pp 315–335
Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, Campbell KP, de La Torre JC, Oldstone MB (2000) Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J Exp Med 192(9):1249–1260
Sevilla N, Domingo E, de la Torre JC (2002) Contribution of LCMV towards deciphering biology of quasispecies in vivo. Curr Top Microbiol Immunol 263:197–220
Sidwell RW, Huffman JH, Allen LB, Meyer RB Jr, Shuman DA, Simon LN, Robins RK (1974) In vitro antiviral activity of 6-substituted 9-beta-D-ribofuranosylpurine 3′, 5′-cyclic phosphates. Antimicrob Agents Chemother 5(6):652–657
Sierra S, Davila M, Lowenstein PR, Domingo E (2000) Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J Virol 74(18):8316–8323
Smith EC, Blanc H, Vignuzzi M, Denison MR (2013) Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog 9(8):e1003565. doi:10.1371/journal.ppat.1003565
Sogoba N, Feldmann H, Safronetz D (2012) Lassa fever in West Africa: evidence for an expanded region of endemicity. Zoonoses Public Health 59(Suppl 2):43–47. doi:10.1111/j.1863-2378.2012.01469.x
Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, Reavill DR, Dunker F, Derisi JL (2012) Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. MBio 3(4):e00180–e00112. doi:10.1128/mBio.00180-12
Strecker T, Eichler R, Meulen J, Weissenhorn W, Dieter Klenk H, Garten W, Lenz O (2003) Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles. J Virol 77(19):10700–10705 (corrected)
Strecker T, Maisa A, Daffis S, Eichler R, Lenz O, Garten W (2006) The role of myristoylation in the membrane association of the Lassa virus matrix protein Z. Virol J 3:93. doi:10.1186/1743-422X-3-93
Streeter DG, Witkowski JT, Khare GP, Sidwell RW, Bauer RJ, Robins RK, Simon LN (1973) Mechanism of action of 1-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc Natl Acad Sci USA 70(4):1174–1178
Subbarao K, Katz JM (2004) Influenza vaccines generated by reverse genetics. Curr Top Microbiol Immunol 283:313–342
Sullivan BM, Emonet SF, Welch MJ, Lee AM, Campbell KP, de la Torre JC, Oldstone MB (2011) Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc Natl Acad Sci USA 108(7):2969–2974. doi:10.1073/pnas.1019304108
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729. doi:10.1093/molbev/mst197
Tapia N, Fernandez G, Parera M, Gomez-Mariano G, Clotet B, Quinones-Mateu M, Domingo E, Martinez MA (2005) Combination of a mutagenic agent with a reverse transcriptase inhibitor results in systematic inhibition of HIV-1 infection. Virology 338(1):1–8. doi:10.1016/j.virol.2005.05.008
Teng MN, Borrow P, Oldstone MB, de la Torre JC (1996) A single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with the ability to cause growth hormone deficiency syndrome. J Virol 70(12):8438–8443
Tishon A, Eddleston M, de la Torre JC, Oldstone MB (1993) Cytotoxic T lymphocytes cleanse viral gene products from individually infected neurons and lymphocytes in mice persistently infected with lymphocytic choriomeningitis virus. Virology 197(1):463–467 ([pii] S0042682283716132)
Tortorici MA, Albarino CG, Posik DM, Ghiringhelli PD, Lozano ME, Rivera Pomar R, Romanowski V (2001) Arenavirus nucleocapsid protein displays a transcriptional antitermination activity in vivo. Virus Res 73(1):41–55 ([pii] S0168-1702(00)00222-7)
Trivedi P, Meyer KK, Streblow DN, Preuninger BL, Schultz KT, Pauza CD (1994) Selective amplification of simian immunodeficiency virus genotypes after intrarectal inoculation of rhesus monkeys. J Virol 68(11):7649–7653
Urata S, Noda T, Kawaoka Y, Yokosawa H, Yasuda J (2006) Cellular factors required for Lassa virus budding. J Virol 80(8):4191–4195
Urata S, Yun N, Pasquato A, Paessler S, Kunz S, de la Torre JC (2011) Antiviral activity of a small-molecule inhibitor of arenavirus glycoprotein processing by the cellular site 1 protease. J Virol 85(2):795–803. doi:10.1128/JVI.02019-10
Valsamakis A, Riviere Y, Oldstone MB (1987) Perturbation of differentiated functions in vivo during persistent viral infection III: decreased growth hormone mRNA. Virology 156(2):214–220
Vazquez-Calvo A, Martin-Acebes MA, Saiz JC, Ngo N, Sobrino F, de la Torre JC (2013) Inhibition of multiplication of the prototypic arenavirus LCMV by valproic acid. Antiviral Res 99(2):172–179. doi:10.1016/j.antiviral.2013.05.012
Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R (2006) Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439(7074):344–348. doi:10.1038/nature04388
Volpon L, Osborne MJ, Capul AA, de la Torre JC, Borden KL (2010) Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc Natl Acad Sci USA 107(12):5441–5446. doi:10.1073/pnas.0909877107
von Messling V, Cattaneo R (2004) Toward novel vaccines and therapies based on negative-strand RNA viruses. Curr Top Microbiol Immunol 283:281–312
Wachsman MB, Lopez EM, Ramirez JA, Galagovsky LR, Coto CE (2000) Antiviral effect of brassinosteroids against herpes virus and arenaviruses. Antivir Chem Chemother 11(1):71–77
Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB (2011) RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci USA 108(1):331–336. doi:10.1073/pnas.1017241108
Weaver SC, Salas RA, de Manzione N, Fulhorst CF, Duno G, Utrera A, Mills JN, Ksiazek TG, Tovar D, Tesh RB (2000) Guanarito virus (arenaviridae) isolates from endemic and outlying localities in Venezuela: sequence comparisons among and within strains isolated from Venezuelan hemorrhagic fever patients and rodents. Virology 266(1):189–195. doi:10.1006/viro.1999.0067 ([pii] S0042-6822(99)90067-3)
Weaver SC, Salas RA, de Manzione N, Fulhorst CF, Travasos da Rosa AP, Duno G, Utrera A, Mills JN, Ksiazek TG, Tovar D, Guzman H, Kang W, Tesh RB (2001) Extreme genetic diversity among Pirital virus (Arenaviridae) isolates from western Venezuela. Virology 285(1):110–118. doi:10.1006/viro.2001.0954 ([pii] S0042-6822(01)90954-7)
Wright CF, Morelli MJ, Thebaud G, Knowles NJ, Herzyk P, Paton DJ, Haydon DT, King DP (2011) Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J Virol 85(5):2266–2275. doi:10.1128/JVI.01396-10
Wu-Hsieh B, Howard DH, Ahmed R (1988) Virus-induced immunosuppression: a murine model of susceptibility to opportunistic infection. J Infect Dis 158(1):232–235
York J, Nunberg JH (2006) Role of the stable signal peptide of Junin arenavirus envelope glycoprotein in pH-dependent membrane fusion. J Virol 80(15):7775–7780. doi:10.1128/JVI.00642-06 ([pii] 80/15/7775)
York J, Nunberg JH (2007) Distinct requirements for signal peptidase processing and function in the stable signal peptide subunit of the Junin virus envelope glycoprotein. Virology 359(1):72–81. doi:10.1016/j.virol.2006.08.048 ([pii] S0042-6822(06)00620-9)
York J, Romanowski V, Lu M, Nunberg JH (2004) The signal peptide of the Junin arenavirus envelope glycoprotein is myristoylated and forms an essential subunit of the mature G1-G2 complex. J Virol 78(19):10783–10792. doi:10.1128/JVI.78.19.10783-10792.2004 ([pii] 78/19/10783)
Young PR, Howard CR (1983) Fine structure analysis of Pichinde virus nucleocapsids. J Gen Virol 64(Pt 4):833–842
Young KC, Lindsay KL, Lee KJ, Liu WC, He JW, Milstein SL, Lai MM (2003) Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 38(4):869–878. doi:10.1053/jhep.2003.50445
Zahn RC, Schelp I, Utermohlen O, von Laer D (2007) A-to-G hypermutation in the genome of lymphocytic choriomeningitis virus. J Virol 81(2):457–464. doi:10.1128/JVI.00067-06
Zapata JC, Salvato MS (2013) Arenavirus variations due to host-specific adaptation. Viruses 5(1):241–278. doi:10.3390/v5010241
Zinkernagel RM (2002) Lymphocytic choriomeningitis virus and immunology. Curr Top Microbiol Immunol 263:1–5
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Grande-Pérez, A., Martin, V., Moreno, H., de la Torre, J.C. (2015). Arenavirus Quasispecies and Their Biological Implications. In: Domingo, E., Schuster, P. (eds) Quasispecies: From Theory to Experimental Systems. Current Topics in Microbiology and Immunology, vol 392. Springer, Cham. https://doi.org/10.1007/82_2015_468
Download citation
DOI: https://doi.org/10.1007/82_2015_468
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23897-5
Online ISBN: 978-3-319-23898-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)