
Abstract
Pulmonary arterial hypertension (PAH) is characterized by endothelial cell (EC) dysfunction, loss of peripheral pulmonary arterial (PA) microvessels, and proliferation of vascular cells in proximal PAs, leading to occlusion of the lumen. Loss of function mutations in bone morphogenetic protein receptor (BMPR)2 occur in over 70% of heritable form of PAH (HPAH) patients. Intriguingly, only 20% of the mutation carriers develop clinical symptoms, suggesting that genetic variation may provide modifiers that alleviate the disease. Induced pluripotent stem cell (iPSC)-derived vascular cells provide a new opportunity to further understand the disease mechanisms in a personalized manner. In our study, we demonstrated that iPSC-derived endothelial cell (iPSC-EC) recapitulates the functional and gene expression abnormalities in native PAEC from the same PAH patients compared with healthy controls. Interestingly, PAH PAEC and iPSC-EC also respond similarly to potential PAH therapies elafin and FK506 in terms of improved angiogenesis. We then used iPSC-ECs to further understand the reduced penetrance of the BMPR2 mutation. iPSC-EC was generated from three families with unaffected mutation carriers (UMC), HPAH patients, and gender-matched controls. We identified patient-specific features of preserved function in UMC iPSC-EC attributed to the regulators of BMPR2 signaling or to cell survival gene revealed by RNA-Seq analyses. Our findings therefore highlight protective modifiers for HPAH that could help inform development of future treatment strategies. Despite the site of disease in the lung and the particular vulnerability of the PA vasculature, iPSC-EC is a good surrogate for PAH modeling and drug testing.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
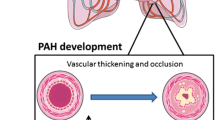
PAH is a progressive disease in which small peripheral arteries are lost or obliterated and more proximal vessels narrow and stiffen, progressively increasing resistance to flow and eventually leading to right heart failure [1, 2]. While the cause of PAH remains poorly understood, the last two decades have shed light on genetic and cellular abnormalities responsible for predisposition to vascular remodeling in PAH. In particular, it has been shown that the endothelial cells (ECs) in PAH are dysfunctional, as evidenced by susceptibility to apoptosis associated with loss of peripheral arteries [3], reduced adhesion related to impaired regeneration [4], decreased migration [5] and tube formation in angiogenesis assays [3] as well as heightened glycolysis, and emergence of apoptosis-resistant cells [6].
One of the major genetic mechanisms driving the endothelial dysfunctions is the loss of function mutations in bone-morphogenetic-protein receptor 2 (BMPR2), which is seen in 70% of patients with the heritable form of PAH (HPAH) and in 20% of sporadic cases of idiopathic PAH (IPAH). Intriguingly, in HPAH families, only 20% of the BMPR2 mutation carriers develop clinical symptoms. Various factors have been linked to the low penetrance: a TGF-β1 polymorphism [7], lower BMPR2 expression from the wild-type allele [8], and reduced expression of the estrogen metabolizing gene CYP1B1 [9]. However, due to lack of a reliable source of tissues or cells from affected and unaffected BMPR2 mutation carriers to perform in depth genetic and functional studies, our understanding of the protective modifiers or risk factors for BMPR2 mutation carriers remains incomplete.
Induced pluripotent stem cells (iPSC) reprogrammed from somatic cells provide a new resource to understand both personal and common genetic underpinnings as well as cellular dysfunction related to the pathogenesis of IPAH/HPAH. iPSC can be propagated indefinitely, differentiated into EC [10] and smooth muscle cells (SMC) [11], and used for vascular disease modeling [12] and drug testing [13] (Fig. 17.1). In our studies, we applied an iPSCs-based model to systematically study the EC abnormalities in PAH and to further understand the reduced penetrance of the BMPR2 mutation in causing the disease phenotype. By correcting the BMPR2 mutation using CRISPR/Cas9 technology, we demonstrated that in the absence of protective modifiers, the BMPR2 mutation is sufficient for EC dysfunction associated with PAH. Our studies pave the way for using iPSC-derived vascular cells, integrative “omics,” and genome editing to better understand patient-specific disease mechanisms for developing precision medicine.

Disease modeling and drug screening using patient-specific iPSC in PAH. At the time of lung transplantation, both skin fibroblasts and primary PAEC were isolated from the same patient. Skin fibroblasts were then reprogramed to iPSC using Sendai virus overexpressing four Yamanaka factors. To model PAH phenotype “in a dish,” patient-specific iPSCs were further differentiated into EC and SMC. By comparing primary PAEC and iPSC-EC, we identified disease phenotypes, gene expression abnormalities, and screened for compounds that could reverse EC and SMC dysfunctions associated with PAH. IPSC also has the potential to give rise to hepatocyte, which can be used to test drug toxicity in a personalized manner. PAEC pulmonary artery endothelial cells. EC endothelial cell, SMC smooth muscle cell
2 Patient-Specific iPSC-Derived Endothelial Cells to Model PAH
The mechanism underlying PAH vary in individuals, perhaps because of genetic factors and environmental exposures. IPSC-EC that preserves the gene variants from the patient could be a good surrogate for native PAEC to understand the genotype-phenotype correlation, and to increase the opportunity to individualized therapy.
2.1 iPSC-EC Recapitulates Native Pulmonary Arterial Endothelial Cell (PAEC)
To investigate the use of iPSC-EC as a surrogate for native EC to better understand and treat PAH, we compared PAH PAEC, harvested and cultured from lungs removed at the time of transplantation, with PAH EC differentiated from iPSC (iPSC-EC) that had been reprogrammed from skin fibroblasts obtained from a small biopsy taken at the incision site of the same patients. Control fibroblasts and PAEC were obtained from unused donor lungs [13].
We found that there was marked similarity between iPSC-EC and native PAEC judged by the typical EC cobblestone morphology, acetylated low-density lipoprotein uptake, and expression of endothelial cell surface marker VE-cadherin (CD144) (Fig. 17.2). Interestingly, compared with HUVEC, iPSC-ECs from both control and PAH patients expressed increased arterial EC gene ephrin-B2 (EFNB2) and reduced venous EC gene ephrin type-B receptor 4 (EPHB4) and nuclear receptor subfamily 2 (NR2F2), possibly due to the high dose of VEGF added during the EC differentiation from iPSCs.

Characterization of iPSC-EC from healthy control. (a) iPSC-EC differentiated from skin fibroblast-derived iPSC express the endothelial surface marker CD144 (green) and CD31 (red). Nuclear staining using DAPI is in blue. (b) iPSC-ECs incorporate acetylated low-density lipoprotein (Ac-LDL, red; DAPI, Blue). (c) Angiogenesis assays: representative images of tube formation 6, 12, and 24 h after seeding cells in growth factor reduced Matrigel. iPSC-ECs start forming tubes at 6 h and will regress after 24 h
Additionally, PAH PAEC and iPSC-EC compared with those from control lungs showed reduced adhesion to laminin, decreased migration detected by wound closure scratch assay, increased vulnerability to apoptosis with serum withdrawal analyzed by caspase3/7 activity asssay, and impaired angiogenesis judged by the inability to form capillary-like tubes when seeded onto growth factor reduced Matrigel. These functional abnormalities could attribute to the impaired pSmad-ID1 pathway in both PAH PAEC and iPSC-EC compared with respective control cells. However, two abnormalities in PAH PAEC could not be recapitulated by iPSC-EC: an increase in unrepaired DNA damage judged by the “comet tails” of nuclear DNA following alkali digestion and higher mitochondrial membrane potential measured by the JC-1 assay after serum starvation in PAH PAEC compared with control subjects. This indicates that DNA damage and increased mitochondrial membrane potential in PAH EC might be related to the environment exposure in the lung, and once the cells are being reprogrammed and re-differentiated, they may lose these features.
2.2 Patient-Specific Drug Response in IPSC-EC and PAEC
Recent advances in iPSC technology also provide a new paradigm for drug screening by permitting the use of human cells with the same genetic background as the patients without the typical quantity constraints associated with patient primary cells. Thus, we compared drug response in PAH PAEC and iPSC-EC from the same group of patients using new promising treatments for PAH, FK506, and elafin. FK506 is an immunosuppressant identified as an activator of BMPR2 signaling [14], and elafin is a neutrophil elastase inhibitor that enhances BMPR2 signaling in a caveolin-1-dependent manner and improves angiogenesis in PAH [3]. In three patients with IPAH/HPAH, elafin and FK506 improved angiogenesis as judged by capillary-like tubes formed on Matrigel in both PAEC and iPSC-EC whereas in the other three patients, there was no change in either cell type. Our data indicates that PAH iPSC-EC could recapitulate the drug response in native PAEC based on in vitro EC functional assays.
3 Modeling Reduced Penetrance of BMPR2 Mutation in PAH
Due to the limited access to native PAEC from HPAH patients and no access from unaffected BMPR2 mutation carriers (UMCs), iPSC-EC could provide a unique platform to model the propensity to HPAH “in-a-dish.” The compensatory mechanisms in UMC shed light on the protective modifiers for HPAH that could help inform development of future therapeutic strategies.
3.1 Preserved EC Function in Unaffected BMPR2 Mutation Carrier (UMC)
To determine if the lack of clinic manifestation of the disease can be recapitulated in UMC iPSC-EC, we recruited 11 individuals from three HPAH families, with HPAH patients, UMC, and gender-matched controls [15]. We observed that cell adhesion to a wide variety of extracellular matrix substrates and cell survival measured by caspase3/7 activity either after serum withdrawal or following reoxygenation after hypoxia were preserved in UMC iPSC-EC in that values were similar to those in control cells, whereas these functions were significantly impaired in HPAH iPSC-EC. Cell migration and tube formation in Matrigel were similarly impaired in iPSC-ECs from both UMCs and HPAH patients compared with controls. However, when iPSC-EC was stimulated with BMPR2 ligand BMP4, cell migration and angiogenesis were significantly improved in UMC iPSC-EC with values similar to those in control iPSC-EC whereas these features remained impaired in iPSC-EC from HPAH patients. Our study revealed that UMC iPSC-EC showed preserved EC functions that were similar to control iPSC-EC and different from HPAH iPSC-EC.
3.2 Preserved pP38 Signaling Pathway in Unaffected BMPR2 Mutation Carrier
To determine whether compensatory BMPR2 signaling was responsible for preservation of normal UMC iPSC-EC functions, we tested both canonical and non-canonical BMP signaling pathways including pSMAD1/5-ID1, pErk, pAkt, and pP38. Interestingly, the only pathway that could distinguish UMC versus HPAH iPSC-EC is the pP38 pathway, which was consistently activated in response to BMP4 or under cell adhesion condition in iPSC-ECs from both controls and UMCs, but not in the HPAH iPSC-EC from all three families. Further studies showed that the compensatory pP38 signaling in UMC iPSC-EC was due to an increase in BMPR2 activators LRP1 and caveolin1 as well as a reduction in BMPR2 inhibitors gremlin1 and FKBP12, which could be assessed prospectively to determine whether they are biomarkers that reduce the risk of HPAH in UMC.
4 Gene Editing in PAH IPSCs
Genome editing in human iPSCs represents an opportunity to examine the contribution of pathogenic and disease-modifying mutations to molecular and cellular phenotypes [16]. Here, we summarized recent studies that utilized CRISPR/Cas9 gene editing in iPSCs to address the controversial question of whether BMPR2 mutation alone is necessary and/or sufficient for establishing the major cellular phenotypes associated with PAH.
4.1 Correction of the BMPR2 Mutation in PAH iPSCs
To determine the extent to which the BMPR2 mutation accounts for the iPSC-EC dysfunction in HPAH, we corrected the BMPR2 missense mutation in one of the HPAH iPSC lines we previously characterized using CRISPR/Cas9-mediated homology directed repair. Upon differentiation, the corrected HPAH iPSC-EC line showed control levels of cell adhesion and survival in response to either serum withdrawal or after hypoxia and reoxygenation. The pP38 signaling pathway was also rescued in the edited iPSC-EC. Tube formation and cell migration were still impaired under baseline following editing of the BMPR2 mutation; however, these were improved to control levels with BMP4 stimulation. This suggests that both protective modifiers and a corrected BMPR2 mutation are necessary to fully normalize EC function in HPAH patients.
4.2 Generation of iPSC Line with BMPR2 Mutation
To assess the effect of a BMPR2 mutation without the confounding effects of genetic differences between cell lines, Kiskin et al. generated two isogenic lines carrying either a known causal BMPR2 mutation (W9X; referred to as C2 W9X+/−) or a deletion of exon 1 (C2 ΔExon1) from a wild-type iPSC line [17]. Subsequently, these iPSCs were differentiated into SMC expressing smooth muscle actin, calponin, and myosin heavy chain 11 that had a contractile phenotype as well as iPSC-EC, which were enriched for arterial-specific EC markers. Under serum-free, chemically defined conditions, BMPR2 heterozygosity alone was sufficient to cause reduced apoptosis and increased proliferation in iPSC-SMCs, which has been shown in native PASMCs. Additionally, after serum exposure for 1 week for iPSC-ECs and serum + TNFα exposure for 1 week or serum-only for 2 weeks for iPSC-SMCs, these cells acquire inner mitochondrial membrane hyperpolarization. Although the generation of pulmonary SMC and EC from iPSC is yet to be achieved, the differentiation protocols used in this study produced cells that recapitulated key phenotypes found in diseased adult PASMCs and PAECs.
5 Future Directions and Clinical Implications
The studies summarized here demonstrate that iPSC-EC and PAEC from the same group of PAH patients showed similar functional impairment, reduced BMPR2 signaling, and subgroup responsiveness to potential PAH therapy, suggesting that despite the site of disease in the lung and the particular vulnerability of the PA vasculature, iPSC-EC are good surrogates for native PAEC to study the fundamental pathobiology in PAH. The results of these studies also suggest the potential of using of iPSC-derived vascular cells to model other vascular diseases where the phenotype may be variable, e.g., Marfan, Williams, or Alagille syndromes.
Because patient-specific iPSC has the potential to give rise to unlimited number of all the cell types in the vasculature including EC, SMC and fibroblasts, it holds great promise for screening compounds that reverse the cellular phenotype associated with PAH in a high-throughput manner, which may ultimately change the current paradigm of treating PAH with “one size fits all” strategy to precision medicine.
Additionally, since PAH iPSC-EC does not recapitulate all the phenotypes in native EC from the lung such as increased DNA damage and higher mitochondrial membrane potential, the next step would be to generate tissue-specific, functionally distinct endothelial cell subtypes from iPSCs. This will allow us to study fundamental questions about lineage specification during development and provide a scalable cell-specific source for tissue engineering, cell therapy, disease modeling, and drug discovery.
References
Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–13. https://doi.org/10.1172/JCI60658.
Tuder RM, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D4–12. https://doi.org/10.1016/j.jacc.2013.10.025.
Nickel NP, et al. Elafin Reverses Pulmonary Hypertension via Caveolin-1-Dependent Bone Morphogenetic Protein Signaling. Am J Respir Crit Care Med. 2015;191:1273–86. https://doi.org/10.1164/rccm.201412-2291OC.
de Jesus Perez VA, et al. Loss of adenomatous poliposis coli-alpha3 integrin interaction promotes endothelial apoptosis in mice and humans. Circ Res. 2012;111:1551–64. https://doi.org/10.1161/CIRCRESAHA.112.267849.
Rhodes CJ, et al. RNA sequencing analysis detection of a novel pathway of endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:356–66. https://doi.org/10.1164/rccm.201408-1528OC.
Masri FA, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–54. https://doi.org/10.1152/ajplung.00428.2006.
Phillips JA 3rd, et al. Synergistic heterozygosity for TGFbeta1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension. Genet Med. 2008;10:359–65. https://doi.org/10.1097/GIM.0b013e318172dcdf.
Hamid R, Cogan JD, Hedges LK, Austin E, Phillips JA 3rd, Newman JH, Loyd JE. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. 2009;30:649–54. https://doi.org/10.1002/humu.20922.
West J, et al. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics. 2008;1:45. https://doi.org/10.1186/1755-8794-1-45.
Gu M. Efficient differentiation of human pluripotent stem cells to endothelial cells. Curr Protoc Hum Genet. 2018;6:e64. https://doi.org/10.1002/cphg.64.
Patsch C, et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol. 2015;17:994–1003. https://doi.org/10.1038/ncb3205.
Theodoris CV, et al. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell. 2015;160:1072–86. https://doi.org/10.1016/j.cell.2015.02.035.
Sa S, et al. Induced pluripotent stem cell model of pulmonary arterial hypertension reveals novel gene expression and patient specificity. Am J Respir Crit Care Med. 2017;195:930–41. https://doi.org/10.1164/rccm.201606-1200OC.
Spiekerkoetter E, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–13. https://doi.org/10.1172/JCI65592.
Gu M, et al. Patient-specific iPSC-derived endothelial cells uncover pathways that protect against pulmonary hypertension in bmpr2 mutation carriers. Cell stem cell. 2017;20:490–504e495. https://doi.org/10.1016/j.stem.2016.08.019.
Hockemeyer D, Jaenisch R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell. 2016;18:573–86. https://doi.org/10.1016/j.stem.2016.04.013.
Kiskin FN, et al. Contributions of BMPR2 mutations and extrinsic factors to cellular phenotypes of pulmonary arterial hypertension revealed by induced pluripotent stem cell modeling. Am J Respir Crit Care Med. 2018;198:271–5. https://doi.org/10.1164/rccm.201801-0049LE.
Acknowledgment
This work was supported by funding from NIH HL135258 (M.G.)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Gu, M. (2020). Using Patient-Specific Induced Pluripotent Stem Cells to Understand and Treat Pulmonary Arterial Hypertension. In: Nakanishi, T., Baldwin, H., Fineman, J., Yamagishi, H. (eds) Molecular Mechanism of Congenital Heart Disease and Pulmonary Hypertension. Springer, Singapore. https://doi.org/10.1007/978-981-15-1185-1_17
Download citation
DOI: https://doi.org/10.1007/978-981-15-1185-1_17
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-1184-4
Online ISBN: 978-981-15-1185-1
eBook Packages: MedicineMedicine (R0)