
Abstract
In 1962, Dr. Osamu Shimomura discovered and purified green fluorescent protein (GFP) from the jellyfish Aequorea victoria. In 1992, the jellyfish GFP gene was cloned, causing a revolution in fluorescent labeling technology in cell biology. The use of genetic engineering techniques made the fluorescent labeling of cells, subcellular organelles and biomolecules possible. In 1999, new fluorescent proteins (FPs) were cloned from coral animals. At the turn of the century, genome structures of various life forms were clarified, with the players in cell workings (biomolecules) gathering together. With the demand for visualization of how biomolecules behave in live cells, expectations are high for innovations in fluorescent imaging technology and the spread of such technology. The dynamic behavior of biomolecules is crucial to understanding the mechanisms of cell proliferation, differentiation, and canceration, and the field itself has attracted the attention of the drug discovery industry, in particular. Here I describe several novel probes for cell functions that we developed in the last decade, including Fluoppi, Fucci, GEPRA, and UnaG.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


In 1962, Dr. Osamu Shimomura discovered and purified green fluorescent protein (GFP) from the jellyfish Aequorea victoria. In 1992, the jellyfish GFP gene was cloned, causing a revolution in fluorescent labeling technology in cell biology. The use of genetic engineering techniques made the fluorescent labeling of cells, subcellular organelles and biomolecules possible. In 1999, new fluorescent proteins (FPs) were cloned from coral animals. At the turn of the century, genome structures of various life forms were clarified, with the players in cell workings (biomolecules) gathering together. With the demand for visualization of how biomolecules behave in live cells, expectations are high for innovations in fluorescent imaging technology and the spread of such technology. The dynamic behavior of biomolecules is crucial to understanding the mechanisms of cell proliferation, differentiation, and canceration, and the field itself has attracted the attention of the drug discovery industry, in particular. Here I describe several novel probes for cell functions that we developed in the last decade, including Fluoppi, Fucci, GEPRA, and UnaG.
1 <Fluoppi>
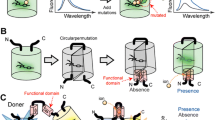
Protein-protein interactions (PPIs) play fundamental roles in cellular functions and disease development and are considered to be important targets for drug discovery. Current fluorescence-based technologies to detect PPIs, including fluorescence resonance energy transfer (FRET), bimolecular fluorescence complementation (BiFC), and translocation/aggregation-based methods, have limited dynamic range to perform robust, reversible, scalable quantitative measurements. We harnessed liquid phase transitions to develop a genetic method for optical monitoring of PPIs at the single cell level (Watanabe et al. 2017). We employed two protein modules that homo-oligomerize with PPI-dependent association to drive phase separation and condensation of crosslinked products into liquid droplets (Fig. 11.1). One module is the PB1 domain, whose concatemerizing property is crucial for phase separations. The second module is a coral-derived FP, which forms a tetrameric complex. With this system, PPI-dependent formation of liquid-phase droplets can be detected as fluorescent puncta in live cells. The method, called Fluoppi, is amenable to FP technologies and applicable to many different PPI monitoring systems that utilize high content assay (HCA) and high-throughput screening (HTS) approaches. Fluoppi is superior to other PPI-monitoring methods in contrast, sensitivity, speed, reversibility, and simplicity.

Design and validation of Fluoppi, a PPI detection system using PB1/AG tags
Left, Homo-oligomerization of the p62 PB1 domain and the green-emitting fluorescent protein AG (Azami-Green). PB1 self-associates in an equilibrium in a front-to-back topology to form a high-molecular-weight homo-oligomer. The conserved acidic/hydrophobic and lysine/arginine residues of PB1 are indicated by red and blue bars, respectively. AG forms an obligate tetramer complex to become fluorescent (green). The hydrophobic interfaces between AG subunits are indicated by thick bars on two adjacent sides. Right, Schematic representation of PPI-dependent formation of fluorescent puncta. Due to the interaction between X and Y, PB1-X and AG-Y build crosslinks, resulting in the concentration of AG fluorescence (green shading)
Dynamic phase transitions of proteins in condensed liquid compartments are emerging as a ubiquitous process in biology that underlies intracellular fluidic organization. An increasing number of membrane-less intracellular structures are known to behave like liquid-droplet phases of the cytoplasm/nucleoplasm. While many of the structures are ribonucleoprotein (RNP) granules, such as P granules and nucleoli, it was demonstrated that multivalent binding of signaling proteins is capable of assembling liquid phase droplets. Our proof-of-concept experiments revealed the rapid appearance and disappearance of cytoplasmic liquid-phase droplets in a PPI-dependent fashion (Fig. 11.2), and the stable propagation of droplets across cell division (Fig. 11.3).

Visualization of Rapamycin-induced association between FRB and FKBP in HeLa cells co-expressing PB1-FKBP and FRB-AG

Liquid-like state inside Fluoppi punctate structures in CHO-K1 cells stably co-expressing PB1-p53 and AG-MDM2. Cells were time-lapse imaged. PC, phase-contrast images. AG + PC, fluorescence images merged with phase-contrast images. One cell-division event is marked by black arrows. Most large puncta (> 10 μm) were irregular in shape
We used the Fluoppi method to visualize, for the first time, ERK2-dimer formation in single living cells stimulated with EGF. Although the ERK2-dimer was originally solved by a crystal structure of phosphorylated ERK2, its in vivo existence has been debatable. Our Fluoppi method revealed not only functional in vivo dimerization but that this PPI occurs in a cell-specific wave-like manner (Fig. 11.4). Because this novel wave behavior is not synchronized among cells, our findings provide a novel caution to researchers who conventionally harvest cells for in vitro biochemical detection of ERK2-dimer formation. These discoveries could only be obtained with Fluoppi technology, which provides rapid, reversible, and quantitative PPI readouts, thus introducing a general method for discovery of novel PPIs and modulators of established PPIs.

HomoFluoppi using mAG1-PB1 tag for visualizing homo-dimerization
EGF-evoked homo-dimerization of ERK2 monitored in Cos-7 cells transiently expressing ERK2-mAG1-PB1. ERK2 homo-dimerization was quantified in 21 cells. Punctum formation was oscillatory in 9 cells and transient in 12 cells as represented by cell 1 (upper) and cell 2 (lower), respectively. Time points of image acquisition were counted after the addition of 50 ng/mL EGF. Despite heterogeneity of the temporal profile, initial punctum formation was observed 5–10 min after the addition of EGF. The time courses of P.I. normalized to the initial peak are shown (rightmost). Data points from cell 1 and cell 2 are indicated by black open circles
2 <Fucci>
Although considerable progress has been made towards understanding the mechanisms regulating cell cycle progression in single cells, little is known about the pathways coordinating the cell cycle with differentiation, morphogenesis, and cell death within multi-cellular tissues and organisms. Cell division is tightly regulated, and cells maintain tight control over the expression levels of proteins involved in the cell cycle. For example, Cdt1 and Geminin have opposite effects on DNA replication, and the amount of these proteins within cells is temporally controlled through the targeted destruction of unwanted protein. We exploited this tightly regulated process to develop a precise visual indicator of cell cycle status in living cells (Sakaue-Sawano et al. 2008). In our technique, which we call “Fucci (fluorescent ubiquitination-based cell cycle indicator)”, cells are genetically modified to express the G1 marker Cdt1 and the S/G2/M marker Geminin fused to red and green fluorescent tags, respectively. As a result, actively replicating cell nuclei in S/G2/M phases exhibit green fluorescence, while nuclei that are in G1 and not yet actively dividing fluoresce red (Fig. 11.5). Using Fucci technology, we directly observed active tumor cell growth in living animals over the course of a month. We also generated transgenic Fucci-expressing mice (Fig. 11.6), and we used these animals to perform detailed analyses of cell cycle profiles in various regions of the developing brain.

Fucci probe labels individual G1 phase nuclei in red and S/G2/M phase nuclei green

A coronal section of an E13 Fucci transgenic embryo, containing the bran, olfactory system, and vomeronasal system. Red and green fluorescence signals are merged. Scale bar, 0.1 mm
While our original paper represented a significant step forward in the understanding of cell cycle regulation, many challenges remain, and foremost among these is the development of a reliable and multifaceted screening technique for the discovery and characterization of novel anticancer drugs. We identify several significant variations in the cell cycle that cells exposed to chemotherapeutic agents can undergo. The variations would be missed by conventional cytometry analysis that only quantifies dye-incorporation to examine DNA content. By combining cytometry analysis with the Fucci probe, we have developed a novel assay that fully integrates the complexity of cell cycle regulation into drug discovery screens (Sakaue-Sawano et al. 2011). We believe that this assay system represents a powerful drug-discovery tool for the development of the next generation of anti-cancer therapies.
Since 2008, we have received many requests for Fucci-related products, including cDNAs, cell lines, and transgenic mouse lines. Importantly, quite a few people have also contacted us with inquiries about the performance of Fucci in non-mammalian animal model systems, such as zebrafish and fruit flies. In order to compile dynamic profiles of cell proliferation in the whole embryo, we turned to zebrafish, a powerful vertebrate model whose external embryonic development and transparency provide good access to almost all stages of embryogenesis for in vivo imaging. Thus, we examined whether the original Fucci based on human Cdt1 and Geminin operates effectively in fish cells. Unfortunately, we found that the original Fucci does not fully function in fish cells because the ubiquitin-mediated degradation of Cdt1 differs between mammals and fish. In 2009, however, we reported the development of zebrafish Fucci (zFucci), a time- and work-intensive project that involved the generation of DNA constructs using the zebrafish homologs of Cdt1 and Geminin, the characterization of these constructs using cultured fish cells, and the construction of transgenic zebrafish, Cecyil (Cell cycle illuminated) (Sugiyama et al. 2009). The transgenic lines, Cecyil and Cecyil2, which express zFucci and zFucci-S/G2/M(NC), respectively, exhibit normal development, confirming that the indicators do not affect normal cell cycle progression of fish cells. As the developers of this tool, we were privileged to be the first to use fish embryos to reveal the intricate and dynamic patterns of cell cycle progression in several parts of the body. Two organs were highlighted for their cell-cycle related morphogenesis. One was the retina, which has already been very well characterized, and the other was the notochord, which is a less explored embryonic structure in vertebrates in terms of cell proliferation/differentiation. Remarkably, we discovered two waves of cell-cycle transitions traveling from the anterior to the posterior of the notochord (Fig. 11.7) (Sugiyama et al. 2014).

Cell-cycle transition waves in the differentiating notochord. Confocal images of the posterior region of the notochord at 19, 20, and 21 hpf (hours post-fertilization)
3 <GEPRA>
Owing to the combined efforts of molecular biologists and geneticists in the last half century, our understanding of the spatiotemporal regulation of embryonic development has greatly advanced, particularly by the discovery of morphogens, diffusible molecules that impart positional information to cells within the embryo. Nevertheless, it remains unclear how a morphogen is spatially distributed along various axes of the body. Even though a source-sink model of morphogen dynamics was proposed by Francis Crick in 1970 predicting that a local source and a local sink can generate a linear concentration gradient of a morphogen based on simple diffusion, no one had yet succeeded in real-time monitoring of endogenous morphogen concentrations in a live embryo. Generally, morphogen gradients are surmised based on data that map the regions of enzymes that synthesize or degrade morphogen molecules in fixed samples. This is also the case with retinoic acid (RA). Despite the importance of RA in vertebrate development, the presence of its gradient in the zone flanked by synthesis (raldh2 enzyme) and degradation (cyp26 enzyme) regions is still controversial. Some researchers think that there is no RA gradient but a uniform or rectangular distribution. Moreover, assuming that a RA gradient exists, it is unknown whether it is linear or not. Due to its non-peptidic nature it is not possible to obtain even static images of RA distribution.
We developed three genetically encoded probes for RA (GEPRAs) with different binding affinities, for quantitative real-time imaging of the intracellular concentration of RA ([RA]i) in live zebrafish embryos (Fig. 11.8) (Shimozono et al. 2013). By imaging both [RA]i and morphology over an extended period and combining the imaging with pharmacological and genetic manipulations, we were able to gain a comprehensive understanding of [RA]i dynamics along the anterior-posterior axis in relation to hindbrain formation and somitogenesis. The [RA]i distribution was demonstrated to be linear which definitively resolves the historical controversy.

A ratiometric [RA]i image in a bud-stage embryo expressing GEPRA
4 <UnaG>
The discovery and utilization of FPs has catalyzed rapid advances in many fields of biomedical research. While new variants continue to be genetically-engineered, nature remains the best source of novel fluorescent sensors. In 2009, researchers at Kagoshima University in Japan described a green fluorescence in the muscle of the Japanese eel (Hayashi and Toda 2009), a traditional part of Japanese cuisine but suffering from population decline. In 2013, we reported a comprehensive characterization of the protein that is responsible for the fluorescence, named ‘UnaG,’ including its molecular cloning, mechanism, structure and clinical use (Kumagai et al. 2013).
The mechanism of UnaG fluorescence is unexpected: we found that it binds non-covalently to the heme catabolite molecule bilirubin that serves as a fluorogenic chromophore. This is in marked contrast to the autocatalytic but oxygen-dependent chromophore formation of GFP and GFP-like proteins. The binding of bilirubin to UnaG is strong (K d = 98 pM) and highly specific to the lipophilic bilirubin species of medical importance. We resolved the crystal structure of holoUnaG at considerably high resolution (1.2 Å), and defined the binding pocket as co-planar non-covalent interactions (Fig. 11.9). All of known bilin (linear tetrapyrrole)-binding proteins found in the plant/protozoan kingdom and their derivatives contain covalently bound bilin chromophores (Miyawaki 2016). However, UnaG is the first bilin-inducible FP, the first bilin-binding FP that belongs to the fatty-acid binding protein (FABP) family, and the first FP from the vertebrate subphylum. In many respects, therefore, UnaG can be regarded as a novel FP.

A crystal structure of holo-UnaG (complex with bilirubin). The bilirubin chromophore is shown as a stick representation with atoms colored (carbon, green; oxygen, red; nitrogen, blue)
The accumulation of bilirubin in the human body causes the diseases jaundice and kernicterus, and bilirubin itself is a global health indicator of liver function. Until our molecular cloning of UnaG, however, humankind had not obtained a direct sensor for this substance. On the whole, there have been significant problems related to current measurements of bilirubin in clinical laboratories. One of the problems stems from the principle of bilirubin detection that uses light absorption; absorptiometric methods are insensitive and affected by many factors. With UnaG, in contrast, we have developed a much superior fluorescence measurement assay that will revolutionize the ability to monitor bilirubin in human samples (Fig. 11.10). Our study also provides new insight on the role of a fluorescent protein system in the muscle of migratory eels.

A schematic showing the simple procedure for bilirubin level determination in a sample using lyophilized apoUnaG
References
Hayashi S, Toda Y (2009) A novel fluorescent protein purified from eel muscle. Fish Sci 75(6):1461–1469
Kumagai A, Ando R, Miyatake H, Greimel P, Kobayashi T, Hirabayashi Y, Shimogori T, Miyawaki A (2013) A bilirubin-inducible fluorescent protein from eel muscle. Cell 153(7):1602–1611
Miyawaki A (2016) Exploiting the cyanobacterial light-harvesting machinery for developing fluorescent probes. Nat Methods 13(9):729–730
Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A (2008) Visualizing spatiotemporal dynamics of multicellular cell cycle progression. Cell 132(3):487–498
Sakaue-Sawano A, Kobayashi T, Ohtawa K, Miyawaki A (2011) Drug-induced cell cycle modulation leading to cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC Cell Biol 12:2
Shimozono S, Iimura T, Kitaguchi T, Higashijima SI, Miyawaki A (2013) Visualization of an endogenous retinoic acid gradient across embryonic development. Nature 496(7445):363–366
Sugiyama M, Sakaue-Sawano A, Iimura T, Fukami K, Kitaguchi T, Kawakami K, Okamoto H, Higashijima S, Miyawaki A (2009) Illuminating cell-cycle progression in the developing zebrafish embryo. Proc Natl Acad Sci U S A 106(49):20812–20817
Sugiyama M, Saitou T, Kurokawa H, Sakaue-Sawano A, Imamura T, Miyawaki A, Iimura T (2014) Live imaging-based model selection reveals periodic regulation of the stochastic g1/s phase transition in vertebrate axial development. PLoS Comput Biol 10(12):e1003957
Watanabe T, Seki T, Fukano T, Sakaue-Sawano A, Karasawa S, Kubota M, Kurokawa H, Inoue K, Akatsuka J, Miyawaki A (2017) Genetic visualization of protein interactions harnessing liquid phase transitions. Sci Rep 7:46380
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Supplementary Electronic Material(s)
(MP4 748925 kb)
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Miyawaki, A., Sakurai, H. (2020). Comprehensive Approaches Using Luminescence to Studies of Cellular Functions. In: Toyama, Y., Miyawaki, A., Nakamura, M., Jinzaki, M. (eds) Make Life Visible. Springer, Singapore. https://doi.org/10.1007/978-981-13-7908-6_11
Download citation
DOI: https://doi.org/10.1007/978-981-13-7908-6_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7907-9
Online ISBN: 978-981-13-7908-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)