
Abstract
The structural features of airways in patients with COPD are airway wall inflammation, fibrosis, muscle hypertrophy, and goblet cell metaplasia. These structural cellular changes contribute to mucus hypersecretion and destruction of the alveolar walls and a decline in forced expiratory volume in one second (FEV1). At the cellular level, macrophages, T lymphocytes, and neutrophils, driven by cytokines including interleukin-8 (IL-8), gather on the airways. The main cause of COPD inflammation is cigarette smoke. Smoke causes an increase in the secretion of matrix metalloproteinase (MMPs) and neutrophilic elastase from epithelial cells and neutrophils, which are responsible for mucin production and destruction of the lung. Initially, cigarette smoke influences the expression of pattern recognition receptors (PRRs) including Toll-like receptors (TLRs), the intracellularly located nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and receptors for advanced glycation end products (RAGE) on lung epithelial cells, endothelial cells, and leukocytes in the lung. These actions bring about the production of cytokines and activation of inflammatory cells, leading to production of MMPs and neutrophilic elastase. The inflammatory changes persist for several months and years after smoking cessation and are sometimes irreversible. Damage-associated molecular patterns (DAMPs) released from dying cells after cigarette smoking increase the number of apoptotic cells, suppress efferocytosis, induce hypoxia and oxidative stress, and prolong the inflammatory changes, even after smoking cessation. Viral and bacterial infections of the respiratory tract then fortify these inflammatory responses. Exacerbations of COPD then worsen the deterioration of COPD.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Global Initiative for Chronic Obstructive Lung Disease 2006 (GOLD 2006) states that “Smoking cessation is the single most effective and cost-effective intervention in most people to reduce the risk of developing COPD and stop its progression (Evidence A).” However, once a patient smokes and develops COPD, the inflammatory changes persist through the innate and adaptive immune systems, which are commonly activated during infection. In this chapter, we will explain why the pathogenesis of COPD progresses, even after smoking cessation, from the point of view of inflammation. The airway inflammation is related not only to the cigarette smoke itself but also viral and bacterial infections occurring before and after smoking cessation. First, the structural and cellular aspects of the inflammatory changes that are also seen with respiratory infection and that occur during COPD formation will be described in general. Then, the progression of COPD after smoking cessation will be discussed. Finally, the relationships between latent respiratory infections, COPD exacerbations, and COPD progressions will be reviewed.
2 Airway Inflammation and Changes in COPD
Clinically, most COPD patients are current or former smokers. In this chapter, we will first explain what happens in the airways of patients with COPD.
2.1 Airway Structure and Mucus Production in Patients with COPD
At the end of the peripheral airways are the alveoli, which are composed of type 1 and type 2 pneumocytes. Bronchiolar patency is stabilized by surfactant secreted from alveoli. Small airways adjacent to alveoli are lined by the epithelium. The epithelial cells are composed of two principal cell types―ciliated and secretory cells. Secretory cells are further divided into goblet and Clara cells [1]. Beneath the epithelium is the basal membrane, and further down the smooth muscle layer (Fig. 4.1) [2]. The secretory cells release mucus, which contains mucins or large glycoproteins [1]. Mucins form polymers. Two of the polymers, mucin 5 AC (MUC5AC) and MUC5B, are highly expressed in the airways [1].

Airway structure and mucus production. The epithelial cells are composed of ciliated and secretory cells. Goblet and Clara cells are secretory cells. Secretory cells release mucus, which contains mucins MUC5AC and MUC5B
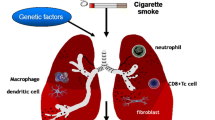
In patients with COPD, the development of airflow obstruction is associated with structural and cellular changes in both the peripheral and central airways. The structural level of peripheral changes involves airway wall inflammation, fibrosis, smooth muscle hypertrophy, goblet cell metaplasia, and lumen occlusion by mucus plugging [3]. These are all possible causes of airflow limitation (Fig. 4.2). However, despite that airway wall fibrosis can be a major contributor to the irreversible component of airflow obstruction in smokers with COPD, the presence of a precise characterization of the fibrotic tissue in peripheral airways has never been reported [4]. Goblet cell metaplasia produces an excess of mucus, which can obstruct the lumen and alter the surface tension of the fluid lining the airway, rendering the peripheral airways unstable and facilitating their closure [4]. Mucus hypersecretion from hyperplastic airway goblet cells is a hallmark of COPD [5]. A recent study showed that chronic sputum production was significantly associated with both excess of FEV1 decline and increased risk of subsequent hospitalization [4].

Inflammatory and immune cells involved in COPD. Structural level of peripheral changes involved in airway wall inflammation, fibrosis, smooth muscle hypertrophy, goblet cell metaplasia, and destruction of the alveolar walls. Inflammatory cells include macrophages, T lymphocytes, and neutrophils in the airway lumen (From “Peter J. Barns. Nature. 2008;8:183-92”). CCL2 CC-chemokine ligand 2, CCR2 CC-chemokine receptor 2, CXCL1 CXC-chemokine ligand 1, MMP9 matrix metalloproteinase-9, Tc1 cell type 1 cytotoxic T cell, TGF-β transforming growth factor-β, T H 1 cell T helper 1 cell
2.2 Lung Destruction in COPD
Airway wall inflammatory reaction contributes not only to the mucus hypersecretion described above but also to the destruction of the alveolar walls, allowing the airway wall to deform and narrowing the airway lumen [4]. Activated inflammatory cells are thought to release elastases, which destroy the lung tissue [4]. The major sources of elastases in the lung are granulocytes and macrophages, and their products include leukocyte elastase, proteinase 3, MMPs, cysteine proteinases, and plasminogen activators [4]. Because anti-elastin antibodies are found in patients with COPD, it is thought that COPD is also an autoimmune disease of elastin [6]. MMP9 gene expression is regulated by numerous stimulatory and suppressive factors, including several cytokines and growth factors such as interleukin-1α (IL-1α), IL-2, IL-8, and interferon-γ (IFN-γ) [7]. MMPs bring about not only MUC5AC accumulation but also the destruction of the lung that leads to emphysema. MMPs probably participate in a proteolytic attack on the alveolar wall matrix [8]. MMP9 is known as gelatinase B. It has multiple potential substrates including collagens, gelatin, elastin, and pro-MMP9 and 13. It is secreted by bronchial epithelial cells, neutrophils, eosinophils, mast cells, and alveolar macrophages.
2.3 Inflammatory Cells and Cytokines in the Airway in COPD
The development of airflow obstruction is associated with an increase of macrophages and T lymphocytes in the airway wall and of neutrophils in the airway lumen [4]. Although the mechanism of neutrophil accumulation in the airway lumen of smokers with COPD is not entirely clear, it is possible that an imbalance between pro- and anti-inflammatory cytokines may play a role. For example, IL-8 is a cytokine that promotes neutrophil chemotaxis, and tumor necrosis factor (TNF-α) is a cytokine that activates an increase in adhesion molecules [4]. There is a shift in the balance of the CD4-/CD8-positive T lymphocyte ratio in favor of CD8-positive ones [4]. Indeed, the CD8-positive cytotoxic T lymphocytes infiltrate the central airways [4], peripheral airways [4], and lung parenchyma [4] suggesting a consistent inflammatory process along the entire tracheobronchial tree in smokers with COPD.
3 Airway Inflammation After Smoking Cessation
Inflammatory changes persist for several months after smoking cessation and are sometimes irreversible [3]. The association observed between smoking and the incidence of COPD is more likely to reflect an early interaction between tobacco exposure and genetic or immunologic host characteristics rather than the effect of the cumulative exposure to cigarette smoke [3].
During cessation of smoking, the number of blood leukocytes immediately falls, goblet cell hyperplasia in the airway declines remarkably, and the number of macrophages and neutrophils in bronchoalveolar lavage fluid (BALF) decreases. However, IL-8, and consequently neutrophils in the airways of ex-smokers, still remains higher than those in nonsmokers [3]. A recent cross-sectional study in which bronchial inflammation was compared between smokers and ex-smokers in patients with COPD [9] showed that the CD3+, CD4+, and plasma cell numbers were significantly higher in the ex-smokers with COPD than current smokers with COPD. Furthermore, compared with current smokers, the short-term ex-smokers showed significantly higher CD4+ and CD8+ cell numbers, whereas the long-term ex-smokers showed significantly lower CD8+ cell numbers and CD8/CD3 ratios and higher plasma cell numbers [9]. These results indicate that the inflammation persists in the airways of patients with COPD even after smoking cessation [10], though the progression of inflammation might attenuate gradually.
Unfortunately, once mild COPD occurs, smoking cessation is not always effective in terminating the progression of COPD. The alterations in squamous metaplasia, gland size, smooth muscle mass, and fibrosis after exposure to smoking do not always recover after smoking cessation [3], though the situation is much better than not quitting smoking [11]. The hypersecretion of mucus, formation of emphysema, and fibrosis in COPD begin with the inhalation of cigarette smoke, which acts on epithelial cells, macrophages, and T lymphocytes in the airway lumen. Activation of epithelial growth factor receptor (EGFR) is responsible for mucin production after inhalation of cigarette smoke in the airways [1, 5, 7]. Meanwhile, acrolein is one of the main cigarette smoke constituents, which increases MUC5AC-positive cells, lung MMP9 transcripts, and EGFR/mitogen-activated protein kinase (MAPK) signaling. These, in turn, contribute to MUC5AC accumulation in the airways [12]. Systemic administration of acrolein causes the stress response in the endoplasmic reticulum and lung cell apoptosis, and chronic administration leads to an enlargement of the alveolar air spaces and emphysema in rats [13].
3.1 Innate Immune System
Both the innate and adaptive immune systems are involved during the progression of the pulmonary inflammation that occurs in COPD [14]. Lung epithelium is always exposed to external microbes in the air such that the innate and adaptive immune responses play important roles in reacting to those external pathogens [15]. Innate immunity is the first line of defense against foreign pathogens. Contrary to adaptive immune responses by which we can obtain protective immunity throughout our lifetime against the same pathogens once we are infected, the innate immune response occurs only once, though it can react to diverse pathogens, including the varied constituents in cigarette smoke. The role of innate immunity in the airways involves detection of pathogen-associated molecular patterns (PAMPs) or DAMPs by PRRs including TLRs and NLRs (Fig. 4.3) [14].

Innate and adaptive immune responses in the airway after smoking cessation. Cigarette smoke brings about PAMPs and DAMPs, which stimulate PRRs on the inflammatory or epithelial cells in the innate immune system and finally cause COPD. In the adaptive immune system, T cells induce notable cytokines, neutrophils, and proteinase and are also causes of COPD formation. DAMPs damage-associated molecular patterns, IL-17 interleukin-17, NLRs nucleotide-binding oligomerization domain (NOD)-like receptors, PAMPs pathogen-associated molecular patterns, PRRs pattern recognition receptors, RAGE receptors for advanced glycation end products, RLRs retinoic acid-inducible gene I (RIG-I)-like receptors, Th17 T helper 17 cell, TLRs Toll-like receptors
3.1.1 The Role of TLRs
Cells that are associated with innate immunity in the lung include macrophages, dendritic cells, monocytes, and neutrophils [15]. The recognition of microbes is first done by PRRs expressed in alveolar macrophages, dendritic cells, and epithelial cells. TLRs are a major family of PRRs, and humans have ten kinds of TLRs to recognize pathogens. The TLRs associated with COPD are TLR6 and TLR9 [16, 17]. They usually recognize bacteria or viruses, but cigarette smoke can also induce pulmonary inflammation via TLRs [17–19]. Additionally, the interaction between TLRs and EGFR increases IL-8 and vascular endothelial growth factor (VEGF) [20]. Through TLR stimulation, reactive oxygen species (ROS) activate the latent form of TNF-α-converting enzyme (TACE), which cleaves the transforming growth factor (TGF)-α proligand that activates EGFR. This results in signaling that leads to IL-8 and VEGF production [21, 22]. Indeed, it was reported that cigarette smoke augments TLR3, which stimulates IL-8 release and increases total MMP9 activity in the airways [19]. IL-8 leads to the recruitment and activation of neutrophils in the airways (see Sect. 4.3.2.2).
3.1.2 The Role of NLRs
NLRs represent a group of key sensors of infections and tissue damage in the lung [23]. Activation of most known NLRs leads to the production and release of proinflammatory cytokines and induction of cell death [23]. Ligand recognition by NOD1 and NOD2 receptors leads to signal transduction through receptor-interacting protein 2 (RIP2) kinase, with downstream activation of MAPKs and transcription factor nuclear factor-kappa B (NF-kB). This then leads to the activation of genes encoding different cytokines and chemokines such as IL-8 [23]. Inflammasomes consisting of one or two NLR proteins serve as platforms for autocatalytic caspase-1 activation, which in turn critically regulates IL-1β and IL-18 production, inducing an inflammatory form of cell death called pyroptosis. Importantly, the NLR protein 3 (NLRP3) inflammasome responds to a vast range of sterile stimuli particularly DAMPs released by dying cells such as adenosine triphosphate (ATP), uric acid metabolites, and biglycan as well as hyaluronan [23]. Experimental studies in mice suggest that activation of NLRPs by some of those DAMPs might have important functions in the pathogenesis of acute lung injury/acute respiratory distress syndrome (ALI/ARDS), COPD/emphysema, and lung fibrosis [23].
3.1.3 The Role of RLRs
Unlike other PRRs, there are almost no reports about the relationship between the components in cigarette smoke and RLRs. However, RLRs are indispensable in viral infection, and they are intimately associated with COPD progression after smoking cessation.
RLRs, including RIG-1 and melanoma differentiation-associated gene 5 (MDA-5), are important pattern recognition receptors for viral elimination [14]. When viral RNA binds to the C-terminal regulatory domain of RIG-1 or MDA-5, it can initiate a signaling cascade. This cascade leads to activation and nuclear translocation of the transcription factors NF-kB and interferon regulatory transcription 3 (IRF-3), which are needed to turn on transcription of interferons (IFNs) [14]. Viral infection is a significant cause of COPD and acute exacerbations of COPD [14]. Up to half of COPD exacerbation cases are associated with viral infections [14]. The top four causes are rhinovirus (RV), coronavirus, influenza virus, and respiratory syncytial virus [14]. Recent studies provide evidence that MDA-5 is responsible for recognizing RV and subsequently activating signaling pathways, causing an exaggerated inflammatory response in patients with COPD. These responses include increased levels of IL-8, IL-6, CXC-chemokine ligand 1 (CXCL1), TNF-α, IL-1β, and monocyte chemotactic protein 1 (MCP-1) [14].
3.1.4 The Role of RAGE
RAGE are members of an immunoglobulin superfamily of cell surface receptors that function as pattern recognition receptors capable of signal transduction after interaction with diverse ligands [24]. RAGE is upregulated wherever its ligands accumulate in chronic conditions such as inflammation, cardiovascular disease, diabetes, cancer, and neurodegeneration [25]. RAGE expression increases in the pulmonary epithelium when tobacco smoke is present [24]. RAGE engagement activates an inflammatory signaling pathway. Smoke-induced RAGE expression mediates cytokine secretions via Ras, a GTPase that influences Raf/MAP kinase, phosphoinositide 3-kinase (PI3K), c-Jun N-terminal kinase (JNK)/p38, NF-kB [25], and the Rho proinflammatory pathway [24]. At minimum, RAGE signaling orchestrates polymorphonucleocyte recruitment and reservoirs of elastolytic enzymes including MMP9 and other mediators of emphysema regulated by RAGE signaling [24]. Recently, both soluble and circulating forms of RAGE were said to be the useful biomarkers for the presence or progression of emphysema because RAGE is generated via cleavage of full-length RAGE from the cell surface by metalloproteinases such as disintegrin, metalloproteinase domain-containing protein 10 (ADAM10), and MMP9 [25].
3.2 Adaptive Immune System
Similar to the importance of the innate immune reaction, the adaptive immune system is also a necessary element in the mechanism of COPD formation. Animal models of autoimmune emphysema not related to cigarette smoke are known [26, 27]. In this model, anti-endothelial cell antibodies (AECA) have been shown to trigger emphysema because of the reduction of the endothelium in the lung.
3.2.1 The Role of B Cells
Little is known about the role of B cells in the development of COPD [28]. The presence of B cells in lymphoid follicles has been reported in the airways and parenchyma of patients with COPD and of mice exposed to cigarette smoke [29]. In mice, the development of lymphoid follicles was progressive over time and correlated with the increase in airspace enlargement [28]. The B-cell follicles are surrounded by T cells, with the majority being CD4+ [28]. The B cells are interspaced by follicular dendritic cells that are necessary for antigen presentation and affinity maturation [28]. Follicle formation is associated with increased levels of cytokines including IL-4, IL-6, IL-8, and IL-13 [28]. These cytokines are essential for the formation and differentiation of germinal centers [28]. Overexpression of IL-13 in mice results in severe emphysema [28]. The absence of bacterial and viral products in the follicles suggests that oligoclonal B cells arise in response to lung antigens [30]. Nevertheless, viral and bacterial infections could be important in perpetuating the inflammatory process and are regarded as the main cause of the exacerbations in COPD. Barry et al. [28] hypothesized that cigarette smoke-induced breakdown products of the extracellular matrix might also be immunogenic and trigger a specific B-cell reaction [28].
3.2.2 The Role of T Cells
It is likely that antigens from necrotic and apoptotic cells in the lungs of smokers are taken up by dendritic cells and presented as antigens to CD8+ T lymphocytes [29]. Activated T cells leave the blood vessels and enter the lung parenchyma. In the lungs of smokers with COPD, CD8+ and CD4+ T cells express the tissue-specific chemokine receptors CXCR3, CXCR5, and CXCR6 [30]. These receptors correlate with the severity of the disease [29]. In both the airway and alveolar compartments, the CD8+ cytotoxic T cell is the predominant T cell in patients with COPD and causes tissue injury [29]. Any cell that displays MHC class I can be a target of CD8+ cytolytic T cells. After a cytolytic attack, target cells die of apoptosis or necrosis from the damage done by perforin, granulysin, or granzyme A or B (Fig. 4.3), all of which are proteolytic enzymes released by CD8+ T cells [15] in the lungs of patients with COPD.
CD4+ T cells also play an important role in COPD. The cytokine IL-17 is a 17-kDa molecule that is produced in vitro by CD4+ and CD8+ subsets of T lymphocytes from humans and mice [31]. IL-17A signaling appears to be crucial for the formation of cigarette smoking-induced emphysema [16]. The frequencies of Th17 (CD4 + IL-17+) and Tc17 (CD8 + IL-17+) cells in the lungs of smoke-exposed mice and smoke-ceased mice are positively correlated with emphysematous lesions [30].
IL-17A plays several important roles in COPD formation. First, it causes lung destruction. IL-17 induces IL-8, which is a chemoattractant for neutrophilic migration (Fig. 4.3). Activated neutrophils and macrophages cause lung destruction through the release of oxygen radicals and proteolytic enzymes such as neutrophil elastase and MMPs including MMP8, MMP9, and MMP12 (macrophage elastase) [15]. As mentioned above for “the innate immune system,” proteolytic enzymes cause cleavage of TGF-α proligands, which bind to EGFR and lead to hypersecretion of MUC5AC [20]. This results in the destruction of the extracellular matrix, tissue damage, and formation of emphysema. Second, IL-17A leads to mucus hypersecretion. In addition to recruitment and activation of the epithelial cells in the lungs, IL-17A also induces MUC5AC expression together with IL-1β, TNF-α, CXCL1, granulocyte colony-stimulating factor (G-CSF), and intracellular adhesion molecule-1 (ICAM-1) in the lungs, resulting in hypersecretion of mucus [16]. Moreover, there is evidence that both IL-6 and IL-17 mediate MUC5B expression through the extracellular signal-regulated kinase (ERK) signaling pathway [16].
Though the definition of emphysema is “non-fibrotic lesions,” fibrosis is said to play an important role in COPD, for example, pulmonary hypertension. In a recent study, IL-1β caused fibrosis in the lung. SMAD4 and NF-kB cooperate to mediate IL-1β and integrin αvβ8-dependent dendritic cell chemokine CCL20 expression [32]. Driving CCL20 expression involves integrin αvβ8-mediated activation of TGF-β [33]. Dendritic cell depletion or deficiency in the crucial dendritic cell chemokine receptor CCR6 protects cells from adenoviral IL-1β-induced adaptive T cell immune responses and fibrosis in the airways of mice [33].
3.3 COPD Deterioration After Smoking Cessation
The average duration of smoking cessation before lung volume reduction surgery is 9.2 years [10], proving that COPD deteriorates for several years after smoking cessation. The immunological changes in the lungs of COPD patients during this period are key to understanding the mechanisms involved in this pathologic progression.
3.3.1 COPD Deterioration by COPD Itself
Needless to say, cigarette smoking is not a significant cause of the progression of COPD after smoking cessation. Before explaining the relationship between COPD and infection after smoking cessation, we will first take a look at the mechanism of COPD deterioration caused by COPD itself.
First, DAMPs arise from the direct stimulation of acute exposure to cigarette smoke, but COPD itself is a cause of DAMPs. In COPD, apoptotic cells are increased because of enhanced induction of apoptosis and deficient phagocytosis of the apoptotic cells by alveolar macrophages. The increased apoptosis of lung parenchyma leads to impaired resolution of inflammation. On the other hand, apoptosis of neutrophils followed by efferocytosis (the process by which dying or dead cells are removed by phagocytic cells) not only prevents damage but also induces an anti-inflammatory macrophage phenotype (M2 or alternatively activated macrophages). In a murine model of COPD, acute cigarette exposure suppresses efferocytosis by alveolar macrophages (Fig. 4.4) [15]. Failed efferocytosis of apoptotic neutrophils leads to secondary necrosis, resulting in the release of DAMPs, neutrophil elastase, and other toxic components into the extracellular space [15]. All of these might be recognized by PRRs initially mediating the inflammatory response, eventually leading to worsening of COPD.

COPD deterioration by COPD itself. In COPD, apoptotic cells are increased, while phagocytosis and efferocytosis are deficient. This condition is thought to cause activation of pattern recognition receptors (PRRs) and makes COPD even worse
Second, hypoxia is another factor involved in COPD deterioration. Recently, a close relationship between hypoxia and inflammation was identified [34]. Hypoxia could perpetuate inflammatory changes in COPD [15]. Hypoxia seen in COPD also promotes prolonged COPD deterioration. Under hypoxic conditions, hypoxia-inducible factor 1α (HIF1α) translocates from the cytoplasm to the nucleus, inducing transcription of multiple genes, including those of TLRs and nuclear factor-kB, which are the influential factors in COPD progression. HIF1α also prolongs the life span of neutrophils by inhibiting apoptosis and promoting COPD progression [34]. On the other hand, the decrease of HIF1α is reported to bring about emphysema or COPD by reducing VEGF [35]. More research is necessary to clarify the impact of HIF1α on COPD or emphysema.
Other factors include oxidative stress, which is discussed in another chapter.
3.3.2 COPD Deterioration by Latent Respiratory Infection
While smoking is a very important determinant for adult lung function and COPD, there is a wide variation in adult lung function that is not related to smoking [36]. An association between lower respiratory infections and adult lung function impairment is reasonably well documented (Fig. 4.5) [36]. Viral infection plays an important role. Recently, the subject of viral infection and COPD was taken up mainly in the topic of COPD exacerbation (mainly rhinovirus), but the groundwork of COPD could already be set by respiratory viral infections during childhood, and adult lung function could be determined early in life [36]. The estimates for individual childhood disadvantage factors such as maternal asthma, paternal asthma, childhood asthma, and severe respiratory infection were comparable to or larger than the estimate for smoking 10–19 cigarettes daily [36]. One of the most well-known viruses in relation with COPD is adenovirus. Latent adenoviral infection may amplify inflammation and predispose to COPD [10]. Double-stranded DNA viruses have the ability to persist in the airway epithelial cells long after the acute infection has cleared. The expression of the adenoviral trans-activating protein has been demonstrated in the airway epithelium and is associated with an amplification of the cigarette smoke-induced inflammatory response and emphysema. In vitro, human airway epithelial cells with viral genes upregulate the expression of intercellular adhesion molecule 1 (ICAM-1) and IL-8 in these cells [37]. Human rhinovirus (HRV) is also a main virus of respiratory infection and closely related with COPD. IL-17A synergistically enhances HRV type 16 (HRV-16)-induced epithelial production of neutrophil chemoattractant of IL-8 [38].

COPD deterioration by respiratory infection and COPD exacerbation. Viral and bacterial infections of the airways bring about COPD exacerbation and accelerate COPD progression
Bacterial infection is also reported to be the cause of COPD. Tuberculosis is an important matter. The results of a nationwide survey of adults in South Africa suggested that the strongest predictor of COPD was a history of pulmonary tuberculosis [36], but other pathogenic bacteria are, of course, important. Viral infections yield a negative chain of succeeding bacterial infections. Preceding rhinovirus infections precipitate secondary bacterial infections in 60 % of COPD patients by the production of rhinovirus-induced neutrophil elastase and reductions in antimicrobial molecules [39]. In COPD, impaired innate lung defenses predispose patients to microbial colonization and infection of the lower respiratory tract, which causes additional airway epithelial injury. This cyclical sequence of events amplifies chronic inflammation and perpetuates microbial infections. The frequency of chronic bacterial colonization and infection increases progressively with disease severity [15]. In a study of bacterial colonization of the lower respiratory tract, potentially pathogenic bacteria were recovered in 34.6 % of patients with COPD compared with 6.7 % in ex-smokers without COPD and nonsmokers by means of BAL culture. Neutrophilia and decreases in macrophages were seen among subjects with COPD after smoking cessation. Additionally, patients with COPD demonstrated increased levels of IL-8 in the BAL. COPD patients also had increased levels of proteinase and proteinase inhibitors, including neutrophil elastase/antineutrophil elastase (NE-A1AT) complex, MMP9, and tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) [40]. In short, bacterial colonization in the lower respiratory tract is related to IL-8 and neutrophils and could contribute to airway inflammation and progressive airway destruction and obstruction in COPD [40].
On the contrary, COPD prolongs bacterial infections. The alveolar macrophage is a key defense against inhaled particulate matter and pathogens. Indeed, the phagocytosis of bacteria such as Haemophilus influenzae and Streptococcus pneumoniae by alveolar macrophages is impaired in COPD [15, 41]. Histone deacetylase (HDAC) is a key molecule in the repression of proinflammatory cytokine production in alveolar macrophages, and its expression is diminished in COPD patients [42]. The decreased TLR2 expression on alveolar macrophages has also been observed in patients with COPD, and the decrements in TLR4 expression in nasal and tracheal epithelium have been noted in severe COPD [41]. Additional important factors of attacking infections in COPD include the impairment of mucociliary clearance because the airway surface liquid is rich in endogenous antimicrobial polypeptides, including cationic polypeptides and collectins. Several of these molecules also have important immunoregulatory functions [41].
3.3.3 COPD Deterioration by Exacerbation
Lastly, we look at the relationship between acute exacerbations and subsequent COPD progression (Fig. 4.5). There is a considerable new evidence that infection is the predominant cause of exacerbations and likely contributes to the pathogenesis of COPD [41]. Viruses can be detected in 10–15 % of sputum samples from patients with COPD and 30–60 % of those with COPD exacerbations, depending on the sensitivity of the detection method [15]. Exacerbations of COPD result in elevated levels of circulating fragments of structural proteins. This suggests that patients with COPD have accelerated extracellular matrix turnover during exacerbations that is related to disease severity [43]. The worsening of airway obstruction is also caused by viral infection in COPD exacerbation. The CD4+ T cells of BALF from patients with COPD showed a mixed Th1 and Th2 cell cytokine phenotype during acute rhinovirus infection [15]. Though Th1-biased immunity has previously been linked to the immunopathology of COPD and emphysema, Th2 as well as Th1 bias is seen in acute exacerbations of COPD [44]. HRV-encoded proteinase 2A induces strong Th1 and Th2 immune responses from CD4+ T cells, implicating this microbial proteinase as an adjuvant factor during respiratory tract infections in patients with COPD. Th2 induces the production of cytokines such as IL-13, IL-4, IFN-γ, and IL-2, which causes an asthma-like attack [44]. This mechanism of the worsening obstruction in acute exacerbations of COPD triggered by viral infection could accumulate the inflammatory microbes and particles, such as the bacteria themselves and the related DAMPs and PAMPs, in the respiratory airways, eventually accelerating COPD deterioration.
4 Conclusion
COPD deterioration is not just the result of exposure to substances in cigarette smoke. The progression of COPD is associated with delayed immunological reactions caused by neutrophils, macrophages, and lymphatic cells and the cytokines secreted by these cells. The viral and bacterial infections accelerate pathological processes in the respiratory tract, even several years after smoking cessation. In view of these, airway inflammation persists after smoking cessation.
References
Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–47.
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–53.
Willemse BW, Postma DS, Timens W, et al. The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation. Eur Respir J. 2004;23(3):464–76.
Saetta M, Turato G, Maestrelli P, et al. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(6):1304–9.
Takeyama K, Jung B, Shim JJ, et al. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 2001;280(1):L165–72.
Kasahara Y, Tuder RM, Taraseviciene-Stewart L, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106(11):1311–9.
Deshmukh HS, Shaver C, et al. Acrolein-activated matrix metalloproteinase 9 contributes to persistent mucin production. Am J Respir Cell Mol Biol. 2008;38(4):446–54.
Churg A, Zhou S, Wright JL. Series “matrix metalloproteinases in lung health and disease”: matrix metalloproteinases in COPD. Eur Respir J. 2012;39(1):197–209.
Gamble E, Grootendorst DC, Hattotuwa K, O’Shaughnessy T, Ram FS, Qiu Y, Zhu J, Vignola AM, Kroegel C, Morell F, Pavord ID, Rabe KF, Jeffery PK, Barnes NC. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J. 2007;30(3):467–71. Epub 2007 May 15.
Shapiro SD. End-stage chronic obstructive pulmonary disease: the cigarette is burned out but inflammation rages on. Am J Respir Crit Care Med. 2001;164(3):339–40.
Vlies BH, Walker PP. Improving the diagnosis and management of COPD. Practitioner. 2015;259(1787):15–9, 2.
Haswell LE, Hewitt K, Thorne D, et al. Cigarette smoke total particulate matter increases mucous secreting cell numbers in vitro: a potential model of goblet cell hyperplasia. Toxicol In Vitro. 2010;24(3):981–7.
Kitaguchi Y, Taraseviciene-Stewart L, Hanaoka M, et al. Acrolein induces endoplasmic reticulum stress and causes airspace enlargement. PLoS One. 2012;7(5), e38038.
Zhang Q, Wan H, Huang S, et al. Critical role of RIG-I-like receptors in inflammation in chronic obstructive pulmonary disease. Clin Respir J. 2014;3:22–31.
Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–26.
Nyunoya T, Mebratu Y, Contreras A, et al. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am J Respir Cell Mol Biol. 2014;50(3):471–82.
Doz E, Noulin N, Boichot E, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. 2008;180(2):1169–78.
Todt JC, Freeman CM, Brown JP, et al. Smoking decreases the response of human lung macrophages to double-stranded RNA by reducing TLR3 expression. Respir Res. 2013;14:33.
Koarai A, Yanagisawa S, Sugiura H, Ichikawa T, Akamatsu K, Hirano T, Nakanishi M, Matsunaga K, Minakata Y, Ichinose M. Cigarette smoke augments the expression and responses of toll-like receptor 3 in human macrophages. Respirology. 2012;17(6):1018–25.
Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1068–75.
Nakanaga T, Nadel JA, Ueki IF, Koff JL, Shao MX. Regulation of interleukin-8 via an airway epithelial signaling cascade. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1289–96.
Richter A, O’Donnell RA, Powell RM, Sanders MW, Holgate ST, Djukanović R, Davies DE. Autocrine ligands for the epidermal growth factor receptor mediate interleukin-8 release from bronchial epithelial cells in response to cigarette smoke. Am J Respir Cell Mol Biol. 2002;27(1):85–90.
Chaput C, Sander LE, Suttorp N, Opitz B. NOD-like receptors in lung diseases. Front Immunol. 2013;4:393.
Stogsdill MP, Stogsdill JA, Bodine BG, et al. Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am J Respir Cell Mol Biol. 2013;49(1):128–34.
Yonchuk JG, Silverman EK, Bowler RP, Agustí A, Lomas DA, Miller BE, Tal-Singer R, Mayer RJ. Circulating soluble receptor for advanced glycation end products(sRAGE) as a biomarker of emphysema and the RAGE axis in the lung. Am J Respir Crit Care Med. 2015;192(7):785–92.
Taraseviciene-Stewart L, Scerbavicius R, Voelkel NF. An animal model of autoimmune emphysema. Am J Respir Crit Care Med. 2005;171(7):734–42.
Hanaoka M, Nicolls MR, Taraseviciene-Stewart L, et al. Immunomodulatory strategies prevent the development of autoimmune emphysema. Respir Res. 2010;11:179.
van der Strate BW, Postma DS, Brandsma CA, Melgert BN, et al. Cigarette smoke-induced emphysema: a role for the B cell? Am J Respir Crit Care Med. 2006;173(7):751–8.
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–54.
Duan MC, Tang HJ, Zhong XN, et al. Persistence of Th17/Tc17 cell expression upon smoking cessation in mice with cigarette smoke-induced emphysema. Clin Dev Immunol. 2013;2013:350727.
Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278(19):17036–43.
Brand OJ, Somanath S, Moermans C, et al. Transforming growth factor-β and Interleukin-1β signaling pathways converge on the Chemokine CCL20 promoter. J Biol Chem. 2015;290(23):14717–28.
Hashimoto M, Yanagisawa H, Minagawa S, Sen D, Goodsell A, Ma R, Moermans C, McKnelly KJ, Baron JL, Krummel MF, Nishimura SL. A critical role for dendritic cells in the evolution of IL-1β-mediated murine airway disease. J Immunol. 2015;194(8):3962–9.
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–65.
Yasuo M, Mizuno S, Kraskauskas D, et al. Hypoxia inducible factor-1α in human emphysema lung tissue. Eur Respir J. 2011;37(4):775–83.
Svanes C, Sunyer J, Plana E, et al. Early life origins of chronic obstructive pulmonary disease. Thorax. 2010;65(1):14–20.
Hogg JC. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med. 2001;164(10 Pt 2):S71–5.
Wiehler S, Proud D. Interleukin-17A modulates human airway epithelial responses to human rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L505–15.
Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(11):1117–24.
Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(9):991–8.
Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359(22):2355–65.
Ito K, Ito M, Elliott WM, Cosio B, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352(19):1967–76.
Sand JM, Knox AJ, Lange P, et al. Accelerated extracellular matrix turnover during exacerbations of COPD. Respir Res. 2015;16:69.
Singh M, Lee SH, Porter P, et al. Human rhinovirus proteinase 2A induces TH1 and TH2 immunity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2010;125(6):1369–78.e2.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Kato, A., Hanaoka, M. (2017). Pathogenesis of COPD (Persistence of Airway Inflammation): Why Does Airway Inflammation Persist After Cessation of Smoking?. In: Nakamura, H., Aoshiba, K. (eds) Chronic Obstructive Pulmonary Disease. Respiratory Disease Series: Diagnostic Tools and Disease Managements. Springer, Singapore. https://doi.org/10.1007/978-981-10-0839-9_4
Download citation
DOI: https://doi.org/10.1007/978-981-10-0839-9_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-0838-2
Online ISBN: 978-981-10-0839-9
eBook Packages: MedicineMedicine (R0)