
Abstract
The Ubiquitin-Proteasome System (UPS) plays an important role in the setting of the cellular response to multiple stress signals. Although the primary function of ubiquitin was initially associated with proteolysis, it is now considered as a key regulator of protein function controlling, among other functions, signalling cascades, transcription, apoptosis or oncogenesis. Failure at any level of the UPS is associated with the development of multiple pathologies including metabolic problems, immune diseases, inflammation and cancer. The successful use of the proteasome inhibitor Bortezomib (Velcade) in the treatment of multiple myeloma (MM) and mantle cell lymphoma (MCL) revealed the potential of the UPS as pharmacological target. Ten years later, new inhibitors tackling not only the proteasome but also different subsets of enzymes which conjugate or de-conjugate ubiquitin or ubiquitin-like molecules, have been developed. Most of them are excellent tools to characterize better the emerging molecular mechanisms regulating distinct critical cellular processes. Some of them have been launched already while many others are still in pre-clinical development. This chapter updates some of the most successful efforts to develop and characterize inhibitors of the UPS which tackle mechanisms involved in cancer. Particular attention has been dedicated to updating the status of the clinical trials of these inhibitors.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Proteasome
- Chemical-inhibitors
- Bortezomib
- Clinical trials
- Cancer
- Ubiquitin
- SUMO
- NEDD8
- Conjugating enzymes
- Ligases
- Isopeptidases
- Deubiquitylating enzymes
11.1 Introduction
The ubiquitin-proteasome system (UPS) is the major proteolytic system in the cytosol and nucleus of all eukaryotic cells (reviewed in Weissman et al. 2011). Most of our initial understanding of this pathway comes from biochemical approaches and genetic studies in yeast (reviewed in Weissman et al. 2011). Knowledge about the physiological roles of the UPS in mammalian cells was quite slow until cell-permeable proteasome inhibitors were developed (Palombella et al. 1994; Tsubuki et al. 1993). Proteasome inhibitors helped to demonstrate that 26S and 20S proteasomes contribute to the degradation of most short-lived and long-lived proteins that exhibit critical functions inside the cell such as transcription, apoptosis or cell cycle regulation, either through ubiquitin-dependent or ubiquitin-independent mechanisms (Glass and Gerner 1987; Tanaka et al. 1983). Consequently, UPS deregulation is linked to different human pathologies (Schwartz and Ciechanover 1999). In this chapter, we will focus on UPS inhibitors used in the treatment of cancer.
11.1.1 Ubiquitin Proteasome-System
The UPS is a complex system composed of multiple molecular machineries acting in a synchronous manner to maintain a dynamic equilibrium of their components. The UPS is often represented as two groups of separated molecular mechanisms: the first group comprises specific enzymes and cofactors acting to modify/de-modify protein substrates with a single member of the ubiquitin family; the second group of the UPS is the 26S proteasome, of approximately 2,000 kDa, which is responsible for the proteolysis of labelled substrates.
11.1.2 Protein Modification by Ubiquitin and Ubiquitin Family Members
The ubiquitin family of protein modifiers, also known as Ubiquitin-like modifiers (UbLs), is composed of at least 15 members sharing a modest primary sequence identity with ubiquitin, but preserving its compact globular β-grasp fold (Hochstrasser 2009). In mammalians, the family includes the small ubiquitin-like modifier (SUMO), the neuronal precursor cell-expressed, developmentally down-regulated protein-8 (NEDD8, also known as Rub1 in yeast), the ubiquitin cross-reactive protein (UCRP, alternatively named interferon-stimulated gene-15 ISG15), the ubiquitin-related modifier-1 (URM1), the human leukocyte antigen F-associated (FAT10), the Fau ubiquitin-like protein (FUB1), the MUB (membrane-anchored UBL), the ubiquitin fold-modifier-1 (UFM1), the ubiquitin-like protein-5 (UBL5, homologous to ubiquitin-1 [Hub1] in yeast), and the autophagy proteins ATG8 and ATG12.
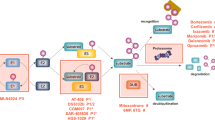
Ubiquitin and all members of the ubiquitin family are attached to protein substrates through biochemical processes which are similar but implicate distinct sets of enzymes with the capacity to act on a limited number of reactions (Fig. 11.1). All protein modifiers of the ubiquitin family are generated through the proteolytical cleavage of higher molecular weight precursors that exposes the double glycine (GG) signature, typically found in this family, and which is required for their attachment to protein substrates (reviewed in Jentsch and Pyrowolakis 2000). The attachment (or conjugation) of ubiquitin or UbL modifiers is mediated by a thiol-ester cascade of reactions that requires the action of three enzymes: an activating enzyme or E1 that will activate all molecules required for all reactions and a conjugating enzyme or E2 that, in most cases, will conjugate protein modifiers to their targets. Ubiquitin ligases or E3s are responsible for, or are required to achieve the conjugation of the modifier to its target protein (Fig. 11.1). The role that an E3 will exert depends on its capacity to form thiol-ester intermediates with protein modifiers. The ubiquitin ligases of the HECT family are typical examples of these active enzymes. Another category of E3 ligases contributes to bring together substrates and E2s in order to achieve protein conjugation without having any enzymatic activity. Among this category of E3s, we found the cullin-RING ligase (CRLs) composed of multiple subunits that will allow the specific recognition of a degradable substrate (e.g. after its phosphorylation). The contribution of E4 factors favouring chain extension has also been proposed (Hoppe 2005). Chain formation is essential to drive distinct functions. While ubiquitin chain linkages K48 and K11 have been associated with degradation, K63 chains appear to connect signalling cascades, endocytic trafficking or DNA repair. The formation of hybrid chains containing ubiquitin and UbLs such as SUMO2/3 is also possible (Aillet et al. 2012; Guzzo and Matunis 2013; Kulathu and Komander 2012) but a generic function of these chains has not been established. Deubiquitylating enzymes or DUBs recognize specific ubiquitin chains from target substrates through ubiquitin binding domains (UBDs) to know which chains should be cleaved (Eletr and Wilkinson 2014). An equivalent reaction is achieved by SUMO-specific proteases (SUSPs) or NEDD8 specific protease (NEDP1), also known as Denedylase 1 (DEN1) (Hickey et al. 2012). All isopeptidases show specificity for a modifier, however due to saturated in vitro assays, overexpression or certain stress conditions, cross-reactions have been reported thus making any conclusion on the exclusivity of these reactions difficult. Although the initial drug discovery strategies used enzymes recognizing protein substrates (E3s and isopeptidases) to develop new chemical inhibitors, nowadays all enzymes are considered as good candidates for drug development.

Available drugs in clinical trials to block the Ubiquitin-Proteasome System. All members of the ubiquitin family are generated through a high molecular weight precursor that has to be cleaved by specific proteases to generate the mature form of each modifier. The covalent attachment of ubiquitin is mediated by a thiol-ester cascade of reactions involving at least three enzymes: an activating enzyme (AE) or E1; a conjugating enzyme (CE) or E2; and a ligase (LE) or E3. Ubiquitin-like proteins such as SUMO or NEDD8 have their own set of enzymes. Demodifying enzymes such as DUBs, SUSPs or NEDP1 participate in a proofreading mechanism. Multiple chain types can be formed and the composition of the chains is essential to drive distinct functions, including proteolysis by the 26S proteasome
11.1.3 Proteasomes
The 26S proteasome is a multi-subunit complex formed by a barrel-shaped proteolytic core, the 20S core particle, and one or two regulatory 19S particles flanking the ends of the core to regulate the entry of proteins targeted for degradation (Fig. 11.1) (Bedford et al. 2010). The 20S core particles consist of four stacked heptameric ring structures that are themselves composed of two different types of subunits. The two outer rings are formed by seven alpha subunits (α1–α7) that allow the interaction with the 19S particle, while the inner rings are composed of seven beta subunits (β1–β7), three of which are responsible for the catalytic activity. The catalytic mechanism of these subunits is the same because they use the hydroxyl group of the N-terminal threonine of a mature β-subunit to perform the nucleophilic attack on the carbonyl carbon of a peptide bond (Borissenko and Groll 2007; Kisselev and Goldberg 2001; Kisselev et al. 2012; Voges et al. 1999). Each subunit reacts with specific substrates: β1 subunit possesses peptidyl-glutamyl peptide-hydrolyzing (PHGH) activity that cleaves after acid residues, β2 has trypsin-like activity that cleaves after basic residues and β5 subunit shows chymotrypsin-like activity that cleaves after hydrophobic residues. Site-directed mutagenesis studies of these catalytic residues in yeast showed that the most drastic changes occurred when β5 subunit is inactivated (Heinemeyer et al. 1997).
The 19S is divided into two sub-particles, the base and the lid. The lid contains at least nine non-ATPase polypeptide chains (Rpn3, 5–9, 11, 12 and 15) that remove the polyubiquitin chains from the protein-substrates, while the base consists of four non-ATPase (Rpn1, Rpn2, and Rpn13) and six ATPase subunits (Rpt1–Rpt6) that interact directly with α-rings. These ATPases control the opening of the 20S gate that is normally locked until an unfolded substrate is recognized. Only the unfolding step involves ATP hydrolysis (Peth et al. 2010).
Apart from the proteasome, an immuno-proteasome exists in immune cells where the 11S subunit, also known as PA28 or REG, replaces the 19S subunit. The 11S is dominantly expressed in hematopoietic cells in response to pro-inflammatory signals such as interferon gamma or cytokines, and is involved in antigen processing for subsequent presentation to the MHC-I on the cell surface, allowing the initiation of the immune cell response (Rock et al. 1994; Tanaka and Kasahara 1998). Furthermore, the immune-proteasome facilitates the clearance of protein aggregates to prevent cell death produced by IFN-induced oxidative stress, (Seifert et al. 2010). All proteasome inhibitors currently used in clinical trials block the enzymatic activities of the proteasome. However, the next generation of proteasome inhibitors will block other mechanism such as gate opening or regulatory subunits.
11.2 Targeting Proteasomes
The first proteasome inhibitors made available were synthesized to specifically block the proteasome’s active sites and to understand its enzymatic mechanism. Peptide aldehydes such as MG-132 were used to develop analogues with enhanced potency, selectivity and stability. Surprisingly, several studies showed that proteasome inhibitors induced rapid and selective apoptosis in different cancer derived cell lines, leading to the idea that proteasome inhibitors could be drug candidates. This idea was confirmed by the fact that the proteasome inhibitor Bortezomib regressed tumour size of xenograft tumours and also decreased metastasis and blocked angiogenesis in patients with hematologic malignancies. Thus, Bortezomib was the first proteasome inhibitor approved by the Food and drug Association of the United States (FDA). Since its approval in 2003, it became the frontline treatment for MM, it has been accepted as second line treatment of MCL and it has also been included in hundreds of on-going clinical trials. In addition to Bortezomib, Carfilzomib, another proteasome inhibitor, has been approved for relapsed/refractory MM, and other proteasome inhibitors are in clinical and preclinical trials (Table 11.1).
11.2.1 Proteasome Inhibitors in Clinical Use
The new generation of proteasome inhibitors always aims to increase potency, specificity and stability with good bioavailability and pharmacokinetics. Conventional proteasome inhibitors efficiently block at least one of the three proteasome active sites (β1, β2 and β5) either with covalent or non-covalent, reversible or irreversible binding. Most Inhibitors and drugs in clinical trials are mimetic peptides that interact directly with the active site thus blocking the nucleophilic attack of the hydroxyl group of the proteasome N-terminal threonine active sites (Kisselev and Goldberg 2001; Orlowski and Kuhn 2008). Proteasome inhibitors are often classified according to their functional group (Table 11.1):
-
Boronic acid peptides: Peptide aldehyde analogues were synthetized substituting the aldehyde group for boronic acid (Bortezomib) (Adams et al. 1998). Bortezomib is more potent, stable and interacts more specifically with the β5 subunit, forming tetrahedral intermediates with two extra hydrogen bonds that stabilize the covalent bond (Groll et al. 2006). Bortezomib is a reversible inhibitor administrated intravenously with very low dissociation rate that behaves as an irreversible molecule. It has a dose limiting toxicity and produces multiple side effects in patients, including pain, fatigue, gastrointestinal, cardiovascular and pulmonary disorders, neutropenia, thrombocytopenia and peripheral neuropathy. Despite its potency and effectiveness, about 60 % of the treated patients develop resistance to Bortezomib after 1 year of treatment (Mujtaba et al. 2012). The resistance mechanisms are still poorly understood but have a multifactorial basis (Xolalpa et al. 2013).
-
A second-generation of proteasome inhibitors, currently in clinical trials, include the boronic acid peptides MLN2238, MLN9708 and CEP-18770. Unlike Bortezomib, these inhibitors reduce neuropathy. MLN2238 and its oral analogue MLN9708, are reversible inhibitors in phase III trials with stronger chymotrypsin-like activity inhibition in vivo and a faster dissociation rate, able to penetrate inside tissues (Kupperman et al. 2010). The oral inhibitor CEP-18770 in phase I and II trials shows encouraging results for the treatment of haematologic and solid tumours (Piva et al. 2008).
-
Epoxyketone peptides. These peptides are the most potent and specific proteasome inhibitors known. They form a morpholine adduct with the N-terminal threonine through a covalent and an irreversible bond (Groll et al. 2000). Carfilzomib (Demo et al. 2007) and ONX-0912 (Chauhan et al. 2010) belong to this class of inhibitors. They show a high chemical stability causing a stronger inhibition of the chymotrypsin-like proteasome activity than Bortezomib (Huber and Groll 2012), and overcome the problem of resistance to Bortezomib. Due to their higher specificity, neurotoxicity is reduced (Molineaux 2012) but other side effects still persist (Fostier et al. 2012). Carfilzomib is currently used for relapsed and refractory multiple myeloma and the orally bioavailable ONX-0912 is included in phase I and II trials where it has shown an improved therapeutic window.
-
β- lactones : Non-peptidic inhibitors have also been developed to improve bioavailability. The compound derived from the marine microorganism Salinispora tropica, Marizomib, is a β-lactone inhibitor in phase I trials (Fenical et al. 2009). Its β-lactone group reacts with the catalytic threonines of the proteasome active site forming an acyl-ester adduct and a tetrahydrofuran ring that stabilizes the binding leading to a highly potent, selective and orally bioactive proteasome inhibitor. Contrary to epoxyketones and peptide boronic acids, Marizomib inhibits the three activities of the proteasome irreversibly, in a stronger (more than 90 %) and longer-lasting way (Miller et al. 2011). The use of marizomib results in adverse effects such as fatigue, nausea, vomiting and dyspnea.
11.2.2 Development of New Proteasome Inhibitors
A second generation of proteasome inhibitors is being designed to specifically inhibit a subunit of interest, to reduce toxicity in normal cells and increase apoptosis in tumour cells (Parlati et al. 2009). This is the case of FV-162 that shows a better inhibition of chymotrypsin-like with an improved safety profile compared to carfilzomib, and the epoxyketone ONX 0914. ONX 0914 is a potent drug orally bioavailable for the immunoproteasome and has been developed for the treatment of autoimmune disorders.
Moreover, natural products and non-covalent proteasome inhibitors are also being developed to reduce side effects. Timosaponin AIII is an example of a natural product in preclinical development whose phases I/II in breast cancer are under preparation. Withaferin A and gambonic acid are other natural inhibitors, which have been approved by the Chinese FDA for cancer clinical trials.
To avoid resistance to proteasome inhibitors, and to limit the off-target effects, the development of compounds acting in a non-competitive way that do not directly interact with the active catalytic β subunit’s site, has been considered. These compounds could potentially be used in combination with Bortezomib to improve clinical outcomes. For example, Rapamicin and PR-39 are compounds that bind to the α7 subunits, thereby producing a disruption in the interaction between 20S and 19S regulatory subunits that blocks the entry of the substrate (Gaczynska et al. 2003). 5-amino-8-hydroxyquinoline also interacts with the α-subunits inside the proteolytic chamber; while being cytotoxic for myeloma and leukemia cells in vitro, it has also been shown to decrease tumour size in xenograft tumour growth in vivo (Li et al. 2010), and overcomes resistance to Bortezomib in cultured cell lines.
11.2.3 Cellular Effects of Proteasome Inhibitors
Proteasome inhibitors are successful in cancer cells with high proliferative rates. Although all mechanisms are not fully understood, it seems that they block the degradation of proapoptotic proteins and cell cycle regulators (Fig. 11.2). Among them, the tumor suppressors retinoblastome (Rb), p53 and the cyclin-dependent kinase inhibitors (CDKIs), such as p21Cip1, p27Kip1 and p57Kip2 are degraded by the proteasome (Frankland-Searby and Bhaumik 2012; Rastogi and Mishra 2012; Vlachostergios et al. 2013). Regulators of the intrinsic apoptotic pathway are also controlled by the proteasome, including members of the Bcl-2 protein family (p53-target-genes) such as the proapoptotic proteins Bax, Bid, Noxa or Bad, and anti-apoptotic members such as Bcl-XL and Bcl-2. Bortezomib treatment increases the expression of proapoptotic members such as Bax, Smac and Noxa while anti-apoptotic proteins like Bcl-2 and IAPs are down-regulated, thus favoring apoptosis in tumor cells (Crawford and Nahta 2011).

Cellular effects of proteasome inhibition. Inhibition of the proteasome stabilizes and activates critical factors that control several apoptotic pathways in cancerous cells. Most pathways affected are involved in cell cycle control, cell proliferation, oncogenic activity, apoptosis, tumour suppressor functions and includes other multiple regulatory proteins (see main text)
The mechanism to induce apoptosis of each proteasome inhibitor seems to be different, thus some combinations result in synergies. For example, Carfilzomib and Marizomib induce apoptosis mediated by caspase 8 and 9 better than Bortezomib, which is more dependent on Bax and Bak mitochondrion-mediated cell death (Kuhn et al. 2007; Miller et al. 2007).
The inhibition of NFκB by Bortezomib seems to have an important role in this drug’s mechanism of action, although it cannot explain all the anticancer effects (Fig. 11.2). NF-κB-mediated transcription plays a crucial role in tumorigenesis by controlling among others the immune and inflammatory responses; apoptosis suppression (by inducing anti-apoptotic IAPs and BCL-XL proteins); angiogenesis promotion (by inducing vascular endothelial growth factor VEGF and COX2); favouring cell proliferation (by increasing cyclin D) and increasing cell migration (by inducing metallo-proteases) (Roccaro et al. 2006). Furthermore, constitutive expression of NF-κB has been related with resistance against radiation and chemotherapy in various types of cancer (Sunwoo et al. 2001).
Disruption of the proteasome activity also leads to the aggregation of unfolded proteins, thereby producing ER stress. An extensive protein production and secretion has been found in different types of cancer, becoming more sensitive to ER stress (Hoeller and Dikic 2009). ER stress leads to up-regulation of the endoribonuclease/kinase IRE1a and the transcription factors ATF3&4 that will increase transcriptional activation of the pro-apoptotic Noxa (Armstrong et al. 2010). Autophagy is also induced by ER stress as a resistance mechanism to escape cell death through the eIF2α induced pathway, the activation of HDAC6, the IRE1-JNK pathway, by proteasomal stabilization of ATF4, the inhibition of mTOR, and by the reduced proteasomal degradation of LC3 (Belloni et al. 2010). The activation of p38 MAPK and c-Jun N-terminal kinases has been also reported after proteasome inhibition. This leads to 14-3-3 phosphorylation and consequently to cytochrome c release. The generation of reactive oxygen species (ROS) after the treatment with proteasome inhibitors results in the expression of apoptosis-related proteins (Fan et al. 2011). ROS are responsible for DNA damage and proteasome plays a crucial role in DNA repair through the activation of p53 and other pathways.
11.2.4 Experience with Clinical Trials
Bortezomib has shown promising activity and a durable response as a single agent in relapsed or refractory MCL and MM patients (Table 11.1). Trials in Phase I demonstrated an effective plateau at 65–70 % proteasome inhibition with fair tolerance levels. In phase II, 35 % of patients with refractory MM and 46.5 % of patients with pre-treated MCL responded to Bortezomib, therefore the FDA initially approved it for relapsed/refractory MM in 2003, and subsequently for relapsed/refractory MCL in 2006 (Kane et al. 2006). Moreover, survival rates of refractory MM patients using Bortezomib in phase III trials exceeded the response rates for patients treated with the previous drug choice Dexamethasone, resulting in Bortezomib being approved as a first-line therapy in 2008 (Richardson et al. 2005). However the refractory response to Bortezomib depends on the tumour cell type. Pancreatic, prostate and non-small lung tumour cells are sensitive while diffuse large B cell lymphoma, follicular lymphoma, Waldenstrom’s macroglobulinemia and some solid tumours such as sarcoma, renal cell carcinoma and glioma only provide slight and short responses (Frankland-Searby and Bhaumik 2012).
To overcome Bortezomib resistance, numerous combinations with chemotherapeutic agents, immunomodulators and/or other proteasome inhibitors are under investigation. In 2007, the FDA approved the use of a Bortezomib/Doxorubicin combination since it was better than the use of single compounds in phase III trials. Combinations with Doxorubicin/Adriamycin (ADR), Thalidomide, Melphalan, Dexamethasone, Cyclophosphamide and Myriad have also been successful. The most active combination to date includes Bortezomib, Lenalidomide and Dexamethasone, where overall responses of 100 % were obtained in phase II. Early phase I and II clinical trials using combinations of Bortezomib with new immunomodulators, radio-immunotherapy, stem cell transplantation, monoclonal antibodies or chemotherapeutic agents have been promising. In addition, targeted therapies such as histone deacetylase inhibitors, Bcl-2 inhibitors, rapamycin inhibitors, multiple kinase inhibitors (Akt, PKC, CDK inhibitors, etc.) and heat shock protein inhibitors simultaneously used with Bortezomib also show encouraging results (McBride and Ryan 2013).
MLN9708 shows synergistic effects in association with Bortezomib, vorinostat, lenalodomide and dexamethasone, in refractory MM (phase III) and first line MM patients (phase I/II) (McBride and Ryan 2013). Studies in phase I/II with Carfilzomib exhibit durable responses with acceptable toxicity in relapsed/refractory MM patients compared to Bortezomib and stem cell transplants, achieving a 24 % partial response in heavily pre-treated patients (McBride and Ryan 2013; Pautasso et al. 2013). Several phase II trials are currently on-going for haematological malignancies and solid tumours. Phase I/II trials with different compound combinations including Calfizomib/Thalidomide/Dexamethasone/Cyclosporamide are being also tested as a frontline treatment for MM, showing promising activity with high response rates in the first patients evaluated.
The last proteasome inhibitor that entered into phase I clinical trials was Marizomib, used alone or in combination with Dexamethasone for the treatment of different kinds of cancer (see Table 11.1). In preclinical trials, Marizomib is also combined with Bortezomib, since different mechanisms of action have been suggested (McBride and Ryan 2013).
11.3 Targeting NEDDylation
Together with ubiquitin and SUMO, NEDD8 is among the most studied ubiquitin-like proteins (Fig. 11.1). The best known physiological substrates of NEDDylation are the cullins, which are structurally related proteins that function as molecular scaffolds of the cullin-RING family of E3 ubiquitin-ligases (CRLs). CRLs consist of a core cullin protein bound to a RING finger protein (Rbx1/2) and an interchangeable substrate-binding adaptor protein (Bedford et al. 2011; Watson et al. 2011). Rbx1 and Rbx2 in conjunction with E2 enzymes UBC12 and UBE2F, respectively, promote NEDDylation of cullins (Huang et al. 2009). The archetypal CRLs, the so-called SCF ubiquitin ligases, contain CUL1, RBX1/2 and the adaptor protein Skp1. This core complex binds to one of the approximately 100 F-box proteins that are responsible for recruiting substrates (Bedford et al. 2011). Cullin NEDDylation has been shown to increase the ubiquitylation activity of CRLs by promoting conformational changes that increase the binding of Rbx1 to ubiquitin E2s, which results in a reduction of the distance between E2 and the substrate recognition component; it may also lead to displacement of the negative regulatory protein, CAND1, that binds to non-NEDDylated cullin (Watson et al. 2011). Specifically, the CRLs have been established to control the degradation of proteins with important biological roles, including cell cycle progression (p27, cyclin E, c-Myc), tumour suppression (p53), DNA damage (CDT1), stress responses (NRF-2, HIF-1α), and signal transduction (IκBα). In addition, other NEDD8-regulated substrates with key cancer-related functions are β-catenin, c-JUN, mTOR and MDM2. Interestingly, increasing evidence suggests that NEDD8-mediated protein turnover may be deregulated in malignant cells and could result in oncogenic transformation, disease progression, or impart a drug-resistant phenotype (Nawrocki et al. 2012). For example, disruptions in the NEDD8 pathway lead breast cancer cells to acquire anti-estrogen resistance and expression of ER alpha (Fan et al. 2003). It has also been shown that increased NEDD8 conjugation appeared to be essential for the enhancement of proliferation in several types of human carcinoma cells (Chairatvit and Ngamkitidechakul 2007).
11.3.1 NEDDylation Inhibitors
Because many CRL substrates have tumour suppressor activity, preventing the degradation of these proteins could be an effective anticancer strategy that may also help to reduce toxicities resulting from global proteasomal inhibition (Nawrocki et al. 2012). MLN4924, a potent and selective first-in-class small molecule inhibitor developed by Millennium Pharmaceuticals, was reported as a specific inhibitor of protein NEDDylation through the inactivation of the heterodimer APPBP1 (NAE1) and UBA3 (UBE1C), also known as the NEDD8 activating enzyme NAE (Soucy et al. 2009; Wang et al. 2011). MLN4924 is an adenosine sulphamate that forms a covalent adduct with NEDD8 when bound to the NAE active site and in this way inhibits the formation of UBC12-NEDD8 thioester. The MLN4924-NEDD8 adduct mimics the NEDD8-AMP in situ which is the first intermediate in the NAE reaction cycle, but cannot be enzymatically processed further (Brownell et al. 2010; Nawrocki et al. 2012). MLN4924 inhibits CRL activity and the stabilization of their substrates by blocking NAE, with subsequent implications in cancer cell growth and survival (Bedford et al. 2011). In 2009, MLN4924 was first reported as a potent growth suppressor agent against a variety of cancer cells lines derived from solid tumours and haematological malignancies in both in vitro and in vivo xenograft models (Soucy et al. 2009; Zhao et al. 2014). During preclinical trials, tumour regression occurs after the action of different mechanisms depending on context and cell type. Induction of apoptosis is one of the reported effects mediated by MLN4924 in three different mechanisms mainly caused by the accumulation and stabilization of CRL substrates like: (1) CDT1, which trigger DNA re-replication and S phase arrest; (2) IκBα, that blocks NF-κB activation, and; (3) pro-apoptotic NOXA. In addition to apoptosis, MLN4924 also induces autophagy in a concentration- and time-dependent manner in multiple human cancer cell lines derived from carcinomas of breast, colon, liver, brain and cervix. Studies revealed that autophagy is mainly caused by inactivation of mTORC1, most likely mediated by accumulation of DEPTOR and HIF1α, followed by the activation of the HIF1-REDD1-TSC1 axis. Some studies have also shown that MLN4924 can induce irreversible senescence in multiple cancer cell lines in a p21-dependent manner. In SK-BR3 cells, p16 and p27 accumulation may also contribute to the MLN4924-induced senescence. Furthermore, MLN4924 has a potential sensitizer role in chemotherapy and radiation, involving the mechanistic accumulation of c-Jun, NOXA, CDT1, WEE1 or p21; it also affects the promotion of c-FLIP degradation, increasing the expression of Bcl-2-interacting killer (BIK), inactivating CRL3, as well as the suppression of FANCD2 monoubiquitylation and CHK1 phosphorylation, all of which results in a general increase in cellular sensitivity by enhancing DNA damage, oxidative stress, cell cycle arrest and finally apoptosis (Zhao et al. 2014). Despite this promising optimal strategy to inhibit NAE pathway, recently it has been identified that cancer cells can develop resistance to MLN4924 treatment. Resistance has been linked to heterozygous mutations in two areas of NAEβ (UBA3), the ATP binding pocket at Alanine 171 and at various residues within or close to the NEDD8-binding cleft. A point mutation in residue A171T reduced affinity for both MLN4924 and ATP. Interestingly, such resistance has been effectively bypassed by using a compound with tighter binding properties for NAE. These findings provide critical clinical aspects with respect to patient selection and consolidate efforts for the development of next-generation NAE inhibitors in order to overcome emergent mutations (Milhollen et al. 2012; Toth et al. 2012). In fact, by using a virtual screening approach, a natural product, 6,6″-biapigenin, has been identified and proposed as a second inhibitor of NAE (Leung et al. 2011). In 2012, a cyclometallated rhodium (III) complex was reported as the first metal complex to suppress the NEDDylation pathway via inhibition of NAE, which occupied the same binding pocket as MLN4924 (Zhong et al. 2012).
11.3.2 Experience with Clinical Trials
In parallel with the promising results in preclinical models, MLN4924 has been included in clinical trials for cancer therapy since 2008 (see Table 11.2). Up to now, there are a total of seven Phase I/II clinical trials for MLN4924 in patients with leukaemia, lymphoma, melanoma and several solid tumours. These trials were mainly designed to assess the safety, discomforts and risks of the inhibitor; to establish the maximum tolerated dose (MTD) that can be given to patients; to describe the pharmacokinetics (PK) parameters and pharmacodynamics (PD) effects; to evaluate disease response, and to study MLN4924 in combination with other standard treatments. In general, patients received escalating doses of MLN4924 that was administrated via intravenous infusion on different daily schedules. The pharmacokinetics of MLN4924 were measured in serial blood samples, bone marrow aspirates (BMAs), skin punch biopsies, or fine-needle tumour biopsies collected following drug dosing. The samples were analysed to measure MLN4924-NEDD8 covalent adduct and the expression of CRL substrates, such as CDT1, NRF2, and phospho-IκBα, as the biomarkers to indicate NAE inhibition in peripheral and tumour tissue (Zhao et al. 2014). The PK profiles were similar following dosing on different days, suggesting no apparent accumulation of MLN4924 in plasma with an estimated half-life of 5–15 h. In general, most clinical trials carried out thus far have concluded that MLN4924 is well tolerated in the majority of the dosing schedules studied, with evidence of target inhibition and antitumoural activity which supports continued investigation of MLN4924, both alone and in combination strategies as potential treatment for a variety of human cancers (Nawrocki et al. 2012; Zhao et al. 2014).
11.4 Targeting DUBs
The human genome encodes nearly 100 DUBs in five major classes: 60 Ubiquitin-specific proteases (USPs), 16 Otubain-domain containing proteins (OTUs), 4 Machado-Joseph Domain (Josephin domain)-containing proteins (MJD), 4 ubiquitin C-terminal hydrolases (UCHs) and 8 Jab1/MPN domain-associated metalloisopeptidases (JAMM). USPs, OTUS, MJD and UCHs use an active site cysteine as a nucleophile to attack lysine-glycine isopeptide bonds within ubiquitylated proteins (Nijman et al. 2005), whereas the fifth class of DUBs contains a JAMM zinc metalloproteinase domain (Cope and Deshaies 2003). DUBs are generally expressed as active enzymes, rather than inactive precursors. However, certain DUBs require ubiquitin binding to obtain their active conformation and prevent their uncontrolled proteolytic activity. Structural data revealed that ubiquitin-binding by DUBs is accompanied by active site rearrangements, which are necessary to induce their hydrolytic activity (Edelmann et al. 2009; Komander et al. 2009). DUB activity is also regulated through the binding of scaffold and adaptor proteins (Ventii and Wilkinson 2008), proteolytic cleavages, as well as post-translational modifications (Kessler and Edelmann 2011).
11.4.1 DUBs as Therapeutic Targets
Due to the implication of DUBs in the regulation of crucial signalling pathways such as p53 and NF-κB (Fig. 11.2), it is not surprising that their deregulation is involved in a growing number of diseases, including neurological disorders, viral infections and cancer. Indeed, some members of the DUBs family are known to contribute to neoplastic transformation, such as USP1 (in Fanconi anaemia), USP2 (in prostate cancer), DUB3 (in breast cancer), USP4 (in adenocarcinoma), USP7 (in prostate cancer and non-small-cell lung adenocarcinoma), USP9X (in both leukaemia and myelomas) and BRCC36 (in breast cancer) (Edelmann et al. 2011; Hussain et al. 2009). The genetic alteration of DUBs such as CYLD and USP6, has been associated with skin and bone marrow tumour progression, respectively. Finally, an alteration of expression of A20 (B-cell and T-cell lymphomas), USP10 (carcinomas) and BAP1 (brain, lung and testicular cancers) is observed in some cancers (Hussain et al. 2009; Nicholson et al. 2007; Sippl et al. 2011).
The high degree of substrate specificity and the well defined catalytic pocket make DUBs druggable and amenable to screening with libraries of small molecules (Eletr and Wilkinson 2014). Work carried out over the last few years has led to the identification of inhibitors with selective action against various USP targets, demonstrating the feasibility of selective targeting of DUBs. In general, the initial hits have been obtained by high throughput screening (HTS) of compound libraries, followed by further optimization using structure-activity relationships (Kramer et al. 2012). Companies like Novartis, Progenra and Hybrigenics have patented compounds inhibiting distinct DUBs (see Table 11.3). Some of them will be presented in the following section. However, to the best of our knowledge, only a few of them have, as yet, entered into clinical trials.
11.4.2 DUBs Inhibitors
Recently, a HTS approach led to the identification of two potent and selective reversible inhibitors of the enzymatic activity of the USP1/UAF1 complex, pimozide and GW7646. USP1, one of the best-characterized DUBs, is a regulator of several important steps in the DNA damage response, mainly in Fanconi anaemia (FA), an hereditary disorder characterized by congenital abnormalities, progressive bone marrow failure, hypersensitivity to DNA cross-linking agents, genomic instability and increased susceptibility to cancer (Li et al. 2002; Reyes-Turcu et al. 2009). USP1 is frequently overexpressed in tumours like cervical and gastric cancer, melanoma and sarcoma. Importantly, USP1 inhibitors act synergistically with the chemotherapy drug cisplatin to inhibit the proliferation of cisplatin-resistant non-small cell lung cancer (NSCLC) cells (Chen et al. 2011; Garcia-Santisteban et al. 2013).
One company, Hybrigenics, has developed a variety of different inhibitors of the USP family (Table 11.3). For example, HBX41-108 is a cyano-indenopyrazine derived small molecule compound which modulates the catalytic reaction of USP7 (Colland et al. 2009). USP7, also known as herpes virus-associated USP (HAUSP), is critical in cancer progression because of its destabilizing effect on the tumour suppressor p53 (Cheon and Baek 2006; Cummins and Vogelstein 2004). However, USP7 can also modify other substrates such as claspin, FOXO4 and PTEN, suggesting that USP7 exerts both p53-dependent and -independent effects on the control of cell proliferation and apoptosis (Faustrup et al. 2009; Song et al. 2008; van der Horst et al. 2006). HBX41-108 treatments stabilize p53, activate transcription of p53-target genes without inducing genotoxic stress, and inhibit cancer cell growth. Another compound more recently developed by Hybrigenics, HBX19-818, is a second-generation of irreversible USP7 inhibitors; it is more specific than HBX41-108, and is capable of regulating USP7 substrates in cancer cells and recapitulating the USP7 knockdown phenotype (Reverdy et al. 2012). Recently, an independent screen performed by Progenra also identified compound P005091, and analogues such as P045204 and P022077, as USP7 inhibitors capable of inducing apoptosis in multiple myeloma cells resistant to both conventional and Bortezomib therapies (Chauhan et al. 2012).
Another HTS identified IU1 as a selective and reversible small-molecule inhibitor of human USP14, which is, together with UCH37 and RPN11, a DUB associated with the proteasome. IU1 binds specifically to the activated form of USP14 and enhances the degradation of several proteins involved in neurodegenerative diseases (like Tau and ataxin-3), suggesting a potential strategy to reduce levels of misfolded and aggregated proteins in cells under proteotoxic stress (Lee et al. 2010; Todi and Paulson 2011).
Inhibitors of DUBs can also be used in antiviral drug development since viruses also encode for DUBs. For example, IU1, mentioned above, is also able to inhibit replication of Dengue virus (Nag and Finley 2012). In the case of Severe Acute Respiratory Syndrome (SARS) caused by SARS-coronavirus, a DUB, called the papain-like protease (Plpro), blocks IRF3-dependent antiviral responses (Edelmann et al. 2011; Mielech et al. 2014). A non-covalent inhibitor of PLpro, GRL0617, has been synthesized. This inhibitor modifies the conformation of PLpro, inducing an inhibition of its catalytic activity, and selectively blocks SARS-Coronavirus viral replication without measurable cytotoxic effect (Ratia et al. 2008). Looking for effective therapeutics against severe acute SARS, 6-mercaptopurine (6MP) and 6-thioguanine (6TG) were found to be specific inhibitors for PLpro. The potential use of 6MP and 6TG as cellular DUB inhibitors has been further studied. The best docking score and binding energy for 6MP and 6TG is against USP14 (Chen et al. 2009; Chou et al. 2008).
11.4.3 Experience with Clinical Trials
Recently, Pimozide and GW7647 have been identified as potent inhibitors of USP1/UAF1 (Chen et al. 2011). Pimozide is an antipsychotic drug also known for its neuroleptic property in the treatment of patients with schizophrenia (Opler and Feinberg 1991), Tourette syndrome and resistant tics. In addition, pimozide inhibits the proliferation of human melanoma and breast cancer cells (Neifeld et al. 1983; Strobl 1990) and is able to reverse the chemo resistance of non-small cell lung cancer cells to the DNA cross-linker, cisplatin, further supporting USP1/UAF1 as a potential target for novel anticancer therapy (Chen et al. 2011) (Table 11.3).
6MP6 and 6TG7 are antimetabolite antineoplastic agents with immunosuppressive properties, interfering with nucleic acid synthesis by inhibiting purine metabolism. These antimetabolites, also described as PLpro and potential USP14 inhibitors, are usually used in combination with other drugs, in the treatment of or in remission-maintenance programs for leukaemia. Several clinical trials have been established or are currently in progress using 6MP6 (166 trials listed in www.clinicaltrial.gov) and 6TG7 (66 trials listed in www.clinicaltrial.gov), in combination with other chemotherapeutic drugs, for the treatment of leukaemia, brain tumour, breast and ovarian cancer, as well as immune diseases.
11.5 Targeting Ubiquitin Ligases
Several specific E3 ubiquitin ligases have been reported as deregulated or overexpressed in various diseases and cancers. Here, we will describe inhibitors of specific E3 ligases already in preclinical development or in phase I clinical trials (see Table 11.4). However, due to the importance of the E3 ligase Mdm2 in the regulation of the tumour suppressor p53, many small molecule inhibitors of this protein-protein interaction were initially developed.
11.5.1 Inhibitors of p53/Mdm2
The tumour protein, p53, is involved in a broad range of critical pathways such as cell cycle, apoptosis, senescence, DNA repair, metabolism, development and innate immunity (Vousden and Prives 2009). p53 monitors the integrity of the genome and is tightly regulated by different intracellular pathways, its main regulator being the ubiquitin E3 Hdm2 ligase (Mdm2 in mouse) that is involved in an auto-regulatory loop controlling p53 stability and activity. The transcription factor p53 has been found mutated and/or deregulated in about half of all known cancers (Soussi and Beroud 2001). In other cases, p53 remains functional but abnormally retained by overexpressed or mutated Mdm2, or its homologue MdmX/Mdm4.
In 1996, the high-resolution crystal structure of Mdm2 with p53 was published (Kussie et al. 1996) and more recently, the MdmX/p53 crystal structure was resolved (Popowicz et al. 2008). From these studies, non-peptide small molecules inhibitors have been designed to specifically target the hydrophobic pocket including three trivial amino acids (Phe19, Trp23 and Leu26) common to both Mdm2 and MdmX. Hoffmann-La Roche has been the first company to develop one of the most potent Mdm2/p53 inhibitors, namely Nutlins. There are three generations of Nutlins (Nutlin-2; Nutlin-3a; RG-7112), with each generation aiming to improve the stability, affinity and bio-availability of the inhibitor. After optimization of Nutlin-3a, RG-7112 was the first Mdm2 inhibitor used in clinical trials to treat multiple human cancers (Tovar et al. 2013). Other companies like Amgen developed a series of optimized compounds from the piperidinone class of compounds, obtaining AM-8553 that efficiently inhibits tumour regression (Rew et al. 2012). Since 2013, Amgen also developed the Mdm2 inhibitor, AMG-8735, containing a morpholinone core with a significant increase in both potency and metabolic stability compared to the piperidinone series. AMG-8735 emerges as an inhibitor with remarkable biochemical potency and pharmacokinetic properties (Gonzalez et al. 2014) (Table 11.4).
ATSP-7041, developed by Aileron Therapeutics, is the first highly potent and selective stapled α-helical peptide that functions as a dual inhibitor of Mdm2 and MdmX. ATSP-7041 binds to Mdm2 and MdmX with nanomolar affinities and restores p53 activity (Chang et al. 2013). ATSP-7041 has a potential use for the treatment of solid tumours and cancers like AML, CML, breast cancer, liposarcoma, melanoma or colon cancer. Stapled α-helical peptides emerged as a promising new modality for a wide range of therapeutic targets. Based on Nutlin-3a (Roche), MI-219 (Ascenta) and TDP 222669 (Johnson&Johnson) structures, and using an NMR spectroscopy approach as an in silico compound-selection process, Priaxon AG developed two new classes of MDM2-inhibitors, PXN-527 and PXN-523 . These inhibitors have been tested in cellular cultures for inhibition of proliferation assays and are in preclinical trials (Cheok et al. 2011).
11.5.2 Inhibitors of Other E3 Ubiquitin Ligases
In addition to Mdm2/MdmX inhibitors, other inhibitors of ubiquitin E3 ligases are also in clinical trials. Progenra developed PO13222, a compound capable of specifically inhibiting the E3 ubiquitin ligase, MuRF1, a ligase associated with muscle wasting diseases. PO13222 specifically inhibits auto-ubiquitylation activity, without affecting ubiquitin E1 and E2 enzymes, resulting in the stabilization of muscle protein. This product is in preclinical development since 2012, the aim of which is to provide a novel therapeutic strategy to protect muscles from sarcopenia (aging) and atrophy associated with common diseases (such as cancer, AIDS and congestive heart disease) and severe burns (Eddins et al. 2011) (Table 11.4).
Progenra has also developed Synoviolin/hrd1 inhibitors for the treatment of rheumatoid arthritis (RA). Synoviolin is a RING E3 ubiquitin ligase implicated in the endoplasmic reticulum-associated degradation (ERAD) pathway. Synoviolin is highly expressed in rheumatoid synovial cells and possibly involved in the pathogenesis of RA. Using HTS, two classes of small molecules have been identified that efficiently inhibit synoviolin activity by inhibition of autoubiquitination enzymatic activity, thus blocking proliferation of synovial cells. This inhibitor has been in preclinical development since 2011 (Yagishita et al. 2012).
High-risk oncogenic human papillomavirus (HPV) types (including HPV 16 and HPV 18) are associated with 99.7 % of all cervical cancers. The E6 and E7 oncoproteins are critical factors that maintain the malignant phenotype of HPV tumour cells. By using yeast two-hybrid screening, Cancer Therapeutics CRC developed linear short peptides that specifically bind with high affinity to the HPV16 E6 oncoprotein, affecting its interaction with the HECT-E3 ubiquitin ligase E6-AP (also known as UBE3A) that degrades p53. The restoration of intracellular p53 by using these short peptides is a new strategy for the treatment of HPV cancers. This E6AP inhibitor is in preclinical stage studies since 2012 (Dymalla et al. 2009).
The E3 ubiquitin ligase Cbl-b is a key regulator of activation thresholds in mature lymphocytes, and of immunological tolerance and autoimmunity. Apeiron Biologics developed the specific Cbl-b ligase inhibitor, APN-401, that stimulates immune cells, providing a novel way to treat cancer more effectively. This compound is under preclinical development since 2012 (www.clinicaltrials.gov).
The SIAH (Seven in Absentia Homologue) family of RING-E3 ubiquitin ligases regulates cellular events that are critical for cancer development and progression. Cancer Therapeutics developed a covalent peptides-based inhibitor (Cys-trapping moiety) to disrupt SIAH interactions with adaptor proteins, and thereby block cancer progression. These small molecule SIAH inhibitors are currently being tested in a preclinical phase for patients with breast, ovarian, prostate and pancreas cancers (Stebbins et al. 2013).
11.5.3 Experience with Clinical Trials
The experience with ubiquitin E3 inhibitors in clinical trials is limited compared to proteasome inhibitors. Many of them are in phase I of clinical development and primary outcomes such as maximum tolerated dose, dose-limiting toxicities and safety are currently being evaluated. Subsequently, efficacy, pharmacokinetics and pharmacodynamics will be measured as secondary outcomes.
After the generation of the first inhibitor for Mdm2, RG-7112, Hoffmann-la Roche developed another Mdm2 inhibitor, RO-5503781, whose structure has not been published. This orally available inhibitor has been in phase I clinical trials since 2011 (NCT01462175), and is proposed to cancer patients with solid tumours and acute myelogenous advanced malignancies, except in the case of leukaemia. However, with respect to the latter, RO-5503781 has been included in another trial in combination with cytarabine, a chemotherapeutic agent used for treatment of leukaemia and non-Hodgkin lymphoma. A third trial is under consideration where RO5503781 will be used in association with cytarabine and anthracycline.
SAR-405838 is a small analogue molecule of MI-888 (small molecule inhibitors of Mdm2), developed by Ascenta Therapeutics and Sanofi, that is currently in phase I of clinical trials since 2012, either alone (Clinical trial: NCT01985191) or in combination with Pimasertib (Clinical trial: NCT01636479). This oral drug may be used for patients with malignant neoplasm (Shen et al. 2013).
CGM097 is an oral HDM2/p53 inhibitor developed by Novartis for the treatment of cancer with advanced solid tumours, or for patients that have regressed despite standard therapy, or for whom no standard therapy exists. Cells have to exhibit wild type p53 to respond to this drug. CGM097 has been in phase I clinical trials since 2012 (NCT01760525).
Merck developed the MK-8242 inhibitor that is included in phase I clinical trials for patients with advanced solid tumours when there is no other treatment available, or for recurrent acute myelogenous leukaemia and liposarcomas. Like RO5503781, MK-8242 is also being evaluated in phase I clinical trials in combination with cytarabine.
DS3032b is an oral Mdm2 inhibitor that has been developed by Daïchi Sankyo to treat patients with advanced solid tumours or lymphomas that exhibit mutated Mdm2 and a WT p53. The description of this molecule has not yet been published but it has already been included in phase I clinical trials (NCT01877382).
11.6 Conclusions and Perspectives
Even if the first pieces of the puzzle of this ATP-dependent pathway were discovered more than 30 years ago, every year new components and regulatory mechanisms controlling this system are being described. Despite this brief didactic view of the UPS classifying enzymes, cofactors and substrates, the system is a lot more complex than presented here, and there is still a distinct lack of knowledge with respect to its overall organization. For instance, it is well known that under certain circumstances several modifying/de-modifying enzymes can act on distinct members of the ubiquitin family (Leidecker et al. 2012), that proteasome subunits can be substrates of ubiquitin and ubiquitin-like molecules (Cui et al. 2014), or that the proteasome can also drive proteolysis in the absence of protein modification (Erales and Coffino 2014). Although most of the functional outcomes of recent findings are not completely understood, it becomes clear that simply targeting a single enzyme-substrate pair will be difficult. However, this should not stop the use of these inhibitors to treat distinct pathologies, as has recently occurred with proteasome inhibitors that block multiple processes, since the discovery of drugs acting at different levels will also increase the chances of clinical success. Furthermore, the experience using combinations of different inhibitors is quite positive in cases where patients do not respond or are resistant to the current treatments. Although these combinatorial drug therapies might appear to be in contradiction with the initial aim of reducing undesirable side effects, it is possible that eventually they could be more effective. A compromise between efficacy and cytotoxicity should be better defined by pharmacodynamics and pharmacokinetic studies. Finally, although we described the use of UPS inhibitors to treat some cancers, many of them have also been used to treat quite diverse diseases such as cardiac, immune and neurodegenerative disorders. The use of chemical inhibitors will contribute to a better understanding of the multiple cellular pathways regulated by the UPS. More importantly, these inhibitors will open up new perspectives for the treatment of various diseases, as has been the case for Bortezomib/Velcade (reviewed by Xolalpa et al. 2013).
References
Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL (1998) Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg Med Chem Lett 8:333–338
Aillet F, Lopitz-Otsoa F, Egana I, Hjerpe R, Fraser P, Hay RT, Rodriguez MS, Lang V (2012) Heterologous SUMO-2/3-ubiquitin chains optimize IkappaBalpha degradation and NF-kappaB activity. PLoS ONE 7:e51672
Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP (2010) Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem 285:6091–6100
Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A, Le J, Ohashi PS, Sarosi I, Nishina H, Lipkowitz S, Penninger JM (2000) Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 403(6766):211–216
Bedford L, Paine S, Sheppard PW, Mayer RJ, Roelofs J (2010) Assembly, structure, and function of the 26S proteasome. Trends Cell Biol 20:391–401
Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE (2011) Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov 10:29–46
Belloni D, Veschini L, Foglieni C, Dell’Antonio G, Caligaris-Cappio F, Ferrarini M, Ferrero E (2010) Bortezomib induces autophagic death in proliferating human endothelial cells. Exp Cell Res 316:1010–1018
Borissenko L, Groll M (2007) 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev 107:687–717
Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL et al (2010) Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 37:102–111
Chairatvit K, Ngamkitidechakul C (2007) Control of cell proliferation via elevated NEDD8 conjugation in oral squamous cell carcinoma. Mol Cell Biochem 306:163–169
Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A et al (2013) Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A 110:E3445–E3454
Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarelli B, Raje N, Richardson P, Anderson KC (2010) A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116:4906–4915
Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, Weinstock J, Kingsbury WD, Hideshima T, Shah PK, Minvielle S, Altun M, Kessler BM, Orlowski R, Richardson P, Munshi N, Anderson KC (2012) A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 22(3):345–358. doi:10.1016/j.ccr.2012.08.007
Chen X, Chou CY, Chang GG (2009) Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir Chem Chemother 19:151–156
Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, Maloney DJ, Jadhav A, Simeonov A, Zhuang Z (2011) Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol 18:1390–1400
Cheok CF, Verma CS, Baselga J, Lane DP (2011) Translating p53 into the clinic. Nat Rev Clin Oncol 8:25–37
Cheon KW, Baek KH (2006) HAUSP as a therapeutic target for hematopoietic tumors (review). Int J Oncol 28:1209–1215
Chou CY, Chien CH, Han YS, Prebanda MT, Hsieh HP, Turk B, Chang GG, Chen X (2008) Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem Pharmacol 75:1601–1609
Chun-Nam L, Lai-King S, Fuli L, Chi-Ming C (2011) Activation of autophagy of aggregation-prone ubiquitylated proteins by timosaponin A-III. J Biol Chem 286(36):31684–31696
Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J et al (2009) Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther 8:2286–2295
Cope GA, Deshaies RJ (2003) COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell 114:663–671
Crawford A, Nahta R (2011) Targeting Bcl-2 in herceptin-resistant breast cancer cell lines. Curr Pharmacogenomics Personal Med 9:184–190
Cui Z, Scruggs SB, Gilda JE, Ping P, Gomes AV (2014) Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J Mol Cell Cardiol 71:32–42. doi:10.1016/j.yjmcc.2013.10.008, Epub 2013 Oct 17
Cummins JM, Vogelstein B (2004) HAUSP is required for p53 destabilization. Cell Cycle 3:689–692
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F et al (2007) Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 67:6383–6391
Dymalla S, Scheffner M, Weber E, Sehr P, Lohrey C, Hoppe-Seyler F, Hoppe-Seyler K (2009) A novel peptide motif binding to and blocking the intracellular activity of the human papillomavirus E6 oncoprotein. J Mol Med (Berl) 87:321–331
Eddins MJ, Marblestone JG, Suresh Kumar KG, Leach CA, Sterner DE, Mattern MR, Nicholson B (2011) Targeting the ubiquitin E3 ligase MuRF1 to inhibit muscle atrophy. Cell Biochem Biophys 60:113–118
Edelmann MJ, Iphofer A, Akutsu M, Altun M, di Gleria K, Kramer HB, Fiebiger E, Dhe-Paganon S, Kessler BM (2009) Structural basis and specificity of human otubain 1-mediated deubiquitination. Biochem J 418:379–390
Edelmann MJ, Nicholson B, Kessler BM (2011) Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev Mol Med 13:e35
Eletr ZM, Wilkinson KD (2014) Regulation of proteolysis by human deubiquitinating enzymes. Biochim Biophys Acta 1843:114–128
Erales J, Coffino P (2014) Ubiquitin-independent proteasomal degradation. Biochim Biophys Acta 1843:216–221
Fan M, Bigsby RM, Nephew KP (2003) The NEDD8 pathway is required for proteasome-mediated degradation of human estrogen receptor (ER)-alpha and essential for the antiproliferative activity of ICI 182,780 in ERalpha-positive breast cancer cells. Mol Endocrinol 17:356–365
Fan WH, Hou Y, Meng FK, Wang XF, Luo YN, Ge PF (2011) Proteasome inhibitor MG-132 induces C6 glioma cell apoptosis via oxidative stress. Acta Pharmacol Sin 32:619–625
Faustrup H, Bekker-Jensen S, Bartek J, Lukas J, Mailand N (2009) USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. J Cell Biol 184:13–19
Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC (2009) Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg Med Chem 17:2175–2180
Fostier K, De Becker A, Schots R (2012) Carfilzomib: a novel treatment in relapsed and refractory multiple myeloma. OncoTargets Ther 5:237–244
Frankland-Searby S, Bhaumik SR (2012) The 26S proteasome complex: an attractive target for cancer therapy. Biochim Biophys Acta 1825:64–76
Gaczynska M, Osmulski PA, Gao Y, Post MJ, Simons M (2003) Proline- and arginine-rich peptides constitute a novel class of allosteric inhibitors of proteasome activity. Biochemistry 42:8663–8670
Garcia-Santisteban I, Peters GJ, Giovannetti E, Rodriguez JA (2013) USP1 deubiquitinase: cellular functions, regulatory mechanisms and emerging potential as target in cancer therapy. Mol Cancer 12:91
Glass JR, Gerner EW (1987) Spermidine mediates degradation of ornithine decarboxylase by a non-lysosomal, ubiquitin-independent mechanism. J Cell Physiol 130:133–141
Gonzalez AZ, Eksterowicz J, Bartberger MD, Beck HP, Canon J, Chen A, Chow D, Duquette J, Fox BM, Fu J et al (2014) Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J Med Chem 57:2472–2488
Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D (2000) A gated channel into the proteasome core particle. Nat Struct Biol 7:1062–1067
Groll M, Huber R, Potts BC (2006) Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc 128:5136–5141
Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK, Lindow S, Kaiser M, Dudler RA (2008) Plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 452(7188):755–758
Guzzo CM, Matunis MJ (2013) Expanding SUMO and ubiquitin-mediated signaling through hybrid SUMO-ubiquitin chains and their receptors. Cell Cycle 12:1015–1017
Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH (1997) The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem 272:25200–25209
Hickey CM, Wilson NR, Hochstrasser M (2012) Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13:755–766
Hochstrasser M (2009) Origin and function of ubiquitin-like proteins. Nature 458:422–429
Hoeller D, Dikic I (2009) Targeting the ubiquitin system in cancer therapy. Nature 458:438–444
Hoppe T (2005) Multiubiquitylation by E4 enzymes: ‘one size’ doesn’t fit all. Trends Biochem Sci 30:183–187
Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF et al (2009) E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell 33:483–495
Huber EM, Groll M (2012) Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew Chem Int Ed Engl 51:8708–8720
Hussain S, Zhang Y, Galardy PJ (2009) DUBs and cancer: the role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 8:1688–1697
Jentsch S, Pyrowolakis G (2000) Ubiquitin and its kin: how close are the family ties? Trends Cell Biol 10:335–342
Kane RC, Farrell AT, Sridhara R, Pazdur R (2006) United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res 12:2955–2960
Kessler BM, Edelmann MJ (2011) PTMs in conversation: activity and function of deubiquitinating enzymes regulated via post-translational modifications. Cell Biochem Biophys 60:21–38
Kisselev AF, Goldberg AL (2001) Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8:739–758
Kisselev AF, van der Linden WA, Overkleeft HS (2012) Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 19:99–115
Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D (2009) Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep 10:466–473
Kramer HB, Nicholson B, Kessler BM, Altun M (2012) Detection of ubiquitin-proteasome enzymatic activities in cells: application of activity-based probes to inhibitor development. Biochim Biophys Acta 1823:2029–2037
Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA et al (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110:3281–3290
Kulathu Y, Komander D (2012) Atypical ubiquitylation – the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol 13:508–523
Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F et al (2010) Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 70:1970–1980
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274:948–953
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP et al (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467:179–184
Leidecker O, Matic I, Mahata B, Pion E, Xirodimas DP (2012) The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle 11:1142–1150
Leung CH, Chan DS, Yang H, Abagyan R, Lee SM, Zhu GY, Fong WF, Ma DL (2011) A natural product-like inhibitor of NEDD8-activating enzyme. Chem Commun (Camb) 47:2511–2513
Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416:648–653
Li X, Wood TE, Sprangers R, Jansen G, Franke NE, Mao X, Wang X, Zhang Y, Verbrugge SE, Adomat H et al (2010) Effect of noncompetitive proteasome inhibition on bortezomib resistance. J Natl Cancer Inst 102:1069–1082
McBride A, Ryan PY (2013) Proteasome inhibitors in the treatment of multiple myeloma. Expert Rev Anticancer Ther 13:339–358
Mielech AM, Chen Y, Mesecar AD, Baker SC (2014) Nidovirus papain-like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. pii:S0168–1702(14)00040-9. doi:10.1016/j.virusres.2014.01.025. [Epub ahead of print]
Milhollen MA, Thomas MP, Narayanan U, Traore T, Riceberg J, Amidon BS, Bence NF, Bolen JB, Brownell J, Dick LR et al (2012) Treatment-emergent mutations in NAEbeta confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell 21:388–401
Miller CP, Ban K, Dujka ME, McConkey DJ, Munsell M, Palladino M, Chandra J (2007) NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 110:267–277
Miller CP, Manton CA, Hale R, Debose L, Macherla VR, Potts BC, Palladino MA, Chandra J (2011) Specific and prolonged proteasome inhibition dictates apoptosis induction by marizomib and its analogs. Chem Biol Interact 194:58–68
Molineaux SM (2012) Molecular pathways: targeting proteasomal protein degradation in cancer. Clin Cancer Res 18:15–20
Mujtaba T, Kanwar J, Wan SB, Chan TH, Dou QP (2012) Sensitizing human multiple myeloma cells to the proteasome inhibitor bortezomib by novel curcumin analogs. Int J Mol Med 29:102–106
Nag DK, Finley D (2012) A small-molecule inhibitor of deubiquitinating enzyme USP14 inhibits Dengue virus replication. Virus Res 165:103–106
Nawrocki ST, Griffin P, Kelly KR, Carew JS (2012) MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs 21:1563–1573
Neifeld JP, Tormey DC, Baker MA, Meyskens FL Jr, Taub RN (1983) Phase II trial of the dopaminergic inhibitor pimozide in previously treated melanoma patients. Cancer Treat Rep 67:155–157
Nicholson B, Marblestone JG, Butt TR, Mattern MR (2007) Deubiquitinating enzymes as novel anticancer targets. Future Oncol 3:191–199
Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123:773–786
Opler LA, Feinberg SS (1991) The role of pimozide in clinical psychiatry: a review. J Clin Psychiatry 52:221–233
Orlowski RZ, Kuhn DJ (2008) Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 14:1649–1657
Palombella VJ, Rando OJ, Goldberg AL, Maniatis T (1994) The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78:773–785
Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y et al (2009) Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 114:3439–3447
Pautasso C, Bringhen S, Cerrato C, Magarotto V, Palumbo A (2013) The mechanism of action, pharmacokinetics, and clinical efficacy of carfilzomib for the treatment of multiple myeloma. Expert Opin Drug Metab Toxicol 9:1371–1379
Peth A, Uchiki T, Goldberg AL (2010) ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol Cell 40:671–681
Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, Giai V, Coscia M, Peola S, Massaia M et al (2008) CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 111:2765–2775
Popowicz GM, Czarna A, Holak TA (2008) Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle 7:2441–2443
Rastogi N, Mishra DP (2012) Therapeutic targeting of cancer cell cycle using proteasome inhibitors. Cell Div 7:26
Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME et al (2008) A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci U S A 105:16119–16124
Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, Harpon J, Battaglia V, Vivat V, Sippl W et al (2012) Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem Biol 19:467–477
Rew Y, Sun D, Gonzalez-Lopez De Turiso F, Bartberger MD, Beck HP, Canon J, Chen A, Chow D, Deignan J, Fox BM et al (2012) Structure-based design of novel inhibitors of the MDM2-p53 interaction. J Med Chem 55:4936–4954
Reyes-Turcu FE, Ventii KH, Wilkinson KD (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78:363–397
Richardson PG, Schlossman R, Hideshima T, Anderson KC (2005) New treatments for multiple myeloma. Oncology (Williston Park) 19:1781–1792; discussion 1792, 1795–1787
Roccaro AM, Vacca A, Ribatti D (2006) Bortezomib in the treatment of cancer. Recent Pat Anticancer Drug Discov 1:397–403
Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL (1994) Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78:761–771
Schwartz AL, Ciechanover A (1999) The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med 50:57–74
Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, Prozorovski T, Lange N, Steffen J, Rieger M et al (2010) Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 142:613–624
Shen M, Schmitt S, Buac D, Dou QP (2013) Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets 17:1091–1108
Sippl W, Collura V, Colland F (2011) Ubiquitin-specific proteases as cancer drug targets. Future Oncol 7:619–632
Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP (2008) The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455:813–817
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S et al (2009) An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458:732–736
Soussi T, Beroud C (2001) Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer 1:233–240
Stebbins JL, Santelli E, Feng Y, De SK, Purves A, Motamedchaboki K, Wu B, Ronai ZA, Liddington RC, Pellecchia M (2013) Structure-based design of covalent Siah inhibitors. Chem Biol 20:973–982
Strobl R (1990) The schizophrenic world view–psychopathologic aspects of ontogenetic regression. Fortschr Neurol Psychiatr 58:1–6
Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C (2001) Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res 7:1419–1428
Tanaka K, Kasahara M (1998) The MHC class I ligand-generating system: roles of immunoproteasomes and the interferon-gamma-inducible proteasome activator PA28. Immunol Rev 163:161–176
Tanaka K, Waxman L, Goldberg AL (1983) ATP serves two distinct roles in protein degradation in reticulocytes, one requiring and one independent of ubiquitin. J Cell Biol 96:1580–1585
Todi SV, Paulson HL (2011) Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci 34:370–382
Toth JI, Yang L, Dahl R, Petroski MD (2012) A gatekeeper residue for NEDD8-activating enzyme inhibition by MLN4924. Cell Rep 1:309–316
Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K et al (2013) MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res 73:2587–2597
Tsubuki S, Kawasaki H, Saito Y, Miyashita N, Inomata M, Kawashima S (1993) Purification and characterization of a Z-Leu-Leu-Leu-MCA degrading protease expected to regulate neurite formation: a novel catalytic activity in proteasome. Biochem Biophys Res Commun 196:1195–1201
van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BM (2006) FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol 8:1064–1073
Ventii KH, Wilkinson KD (2008) Protein partners of deubiquitinating enzymes. Biochem J 414:161–175
Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, Oerlemans R, Chan ET, Kirk CJ, Peters GJ, van der Heijden JW, de Gruijl TD, Scheper RJ, Jansen G (2012) Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno) proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 1:174–182
Vlachostergios PJ, Voutsadakis IA, Papandreou CN (2013) Mechanisms of proteasome inhibitor-induced cytotoxicity in malignant glioma. Cell Biol Toxicol 29:199–211
Voges D, Zwickl P, Baumeister W (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68:1015–1068
Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137:413–431
Wang M, Medeiros BC, Erba HP, DeAngelo DJ, Giles FJ, Swords RT (2011) Targeting protein neddylation: a novel therapeutic strategy for the treatment of cancer. Expert Opin Ther Targets 15:253–264
Watson IR, Irwin MS, Ohh M (2011) NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell 19:168–176
Weissman AM, Shabek N, Ciechanover A (2011) The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol 12:605–620
Xolalpa W, Perez-Galan P, Rodriguez MS, Roue G (2013) Targeting the ubiquitin proteasome system: beyond proteasome inhibition. Curr Pharm Des 19:4053–4093
Yagishita N, Aratani S, Leach C, Amano T, Yamano Y, Nakatani K, Nishioka K, Nakajima T (2012) RING-finger type E3 ubiquitin ligase inhibitors as novel candidates for the treatment of rheumatoid arthritis. Int J Mol Med 30:1281–1286
Zhang W, Sidhu SS (2014) Development of inhibitors in the ubiquitination cascade. FEBS Lett 588(2):356–367. doi:10.1016/j.febslet.2013.11.003, Epub 2013 Nov 12. Review
Zhao Y, Morgan MA, Sun Y (2014) Targeting neddylation pathways to inactivate Cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. [Epub ahead of print]
Zhong HJ, Yang H, Chan DS, Leung CH, Wang HM, Ma DL (2012) A metal-based inhibitor of NEDD8-activating enzyme. PLoS ONE 7:e49574
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Mata-Cantero, L., Lobato-Gil, S., Aillet, F., Lang, V., Rodriguez, M.S. (2015). The Ubiquitin-Proteasome System (UPS) as a Cancer Drug Target: Emerging Mechanisms and Therapeutics. In: Wondrak, G. (eds) Stress Response Pathways in Cancer. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9421-3_11
Download citation
DOI: https://doi.org/10.1007/978-94-017-9421-3_11
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9420-6
Online ISBN: 978-94-017-9421-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)