
Abstract
Virus–platelet interplay is complex. Diverse virus types have been shown to associate with numerous distinct platelet receptors. This association can benefit the virus or the host, and thus the platelet is somewhat of a renegade. Evidence is accumulating to suggest that viruses are capable of entering platelets. For at least one type of RNA virus (dengue virus), the platelet has the necessary post-translational and packaging machinery required for production of replicative viral progeny. As a facilitator of immunity, the platelet also participates in eradicating the virus by direct and indirect mechanisms involving presentation of the pathogen to the innate and adaptive immune systems, thus enhancing inflammation by release of cytokines and other agonists. Virus-induced thrombocytopenia is caused by tangential imbalance of thrombopoeisis, autoimmunity, and loss of platelet function and integrity.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Respiratory Syncytial Virus
- Protein Disulfide Isomerase
- Hemorrhagic Fever
- DENV Infection
- Human Brain Microvascular Endothelial Cell
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The historical view that platelets solely participate as primary facilitators in hemostasis and thrombosis is now outdated. Reminiscent of roles played by their evolutionary counterparts in primitive organisms (Delvaeye and Conway 2009), it is now well-established that platelets directly modulate cells of the immune system through receptor-mediated cell contact with leukocytes and pathogens. In combination with these interactions, external stimuli induce secretion of cytokines and other cell modulators from platelet α-granules and dense granules to produce additional indirect effects on the cellular immune response. Moreover, in contrast to the bygone paradigms, the platelet proteome is not static. Platelets inherit from antecedent megakaryocytes a repertoire of mRNAs and the necessary post-transcriptional machinery to alter their protein composition (Schubert et al. 2014). Platelets survey their local environment, reacting dynamically to change, and are thus pivotally positioned during the defense against pathogens (Semple et al. 2011). Here, we focus on their complex interactions with viruses.
Many excellent review articles are available that recognize the importance of platelets in virology. In some cases, these highlight the effects of specific virus types (Chabert et al. 2015; Hottz et al. 2011), whereas others are more general (Assinger et al. 2014; Zapata et al. 2014). The ultimate message is that viruses trigger a complexity of biochemical and cellular events, often resulting in diminished platelet count, altered vascular permeability, and consequent bleeding diathesis. In this review, we revisit these ideas and add a temporal aspect to the emerging model. We speculate that platelets are more than immune modulators and “innocent” bystanders in the host response to viral infection and that, in the case of viruses with an RNA genome, platelets initially participate as a viral ally.
Virus Binding to Platelets
Platelets are not considered to be a primary host cellular target in virology. Therefore, to identify the receptors that bind viruses, other cell types are usually investigated. The virus–cell interaction is the key initiating step in the virus lifecycle and a logical target for development of antiviral agents. Based on the knowledge of receptors presented on typically studied cells, predictions can be made about whether platelets express the necessary receptors to facilitate specific virus interactions. To help explain platelet-related pathology as a result of infection of certain viruses, direct binding has been reported. Here we loosely divide these interactions on the basis of two principal hemostatic diseases in which direct virus–platelet interactions could logically be involved: thrombocytopenia and hemorrhagic fever (HF).
Thrombocytopenic Viruses
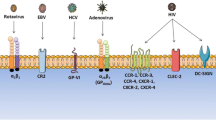
When a foreign particle, such as a virus, binds directly to the platelet surface, it is reasonable to speculate that consequent immune recognition leads to a reduction in platelet count. Thus, virus-induced thrombocytopenia has been the rationale for investigating interactions between viruses and platelets. Table 1 lists viruses that correlate with thrombocytopenia without HF. Representing at least six virus families, these viruses range broadly in structure and genome organization and, with the exception of adenoviruses, are surrounded by a lipid bilayer envelope that contains both host cell- and virus-derived elements. Therefore, an adaptive immune response as a result of viral infection of platelets may involve both virus antigens and co-epitopes originating from virus and host factors, resulting in a reduced platelet count. Receptors on the platelet that associate with viruses have been identified and include integrins α5β1, αIIbβ3, and α5β3; the lectin, dendritic cell-specific intercellular adhesion molecule-3-grapping non-integrin (DC-SIGN); Toll-like receptors (TLRs) 2 and 4; coxsackie-adenovirus receptor (CAR); complement receptor 2 (CR2); C-X-C chemokine receptor type 4 (CXCR4); C-type lectin domain family 2 (CLEC-2); chemokine C-C motif ligand (CCL); and glycoprotein (GP) VI. Co-receptor systems involving more than one virus–platelet interaction may also exist, as in human immunodeficiency virus (HIV). In some cases, the platelet receptor is unknown (e.g., SARS-CoV).
A variety of methods have been used to establish specific receptors. Early inhibition studies involving adenovirus used purified matrix proteins, adenovirus penton base proteins, and synthetic peptides. Results suggested the importance of the α5 and β1/β3 integrins for infection of several cell lines (Stevenson et al. 1997; Wickham et al. 1993). Immuno- and electron microscopy studies have shown that platelet αIIbβ3 is also important for the binding of adenovirus (Gupalo et al. 2013). Further cellular studies implicated CAR (Bergelson et al. 1997) in the virus–platelet association and identified this receptor on platelets by flow cytometry and RNA isolation (Othman et al. 2007). Additional studies using immuno-inhibition and arginyl–glycyl–aspartyl (RGD) motif peptide mimics confirmed the importance of α5 integrins in adenovirus binding to platelets; however, they failed to detect CAR expression (Shimony et al. 2009).
Hepatitis C virus (HCV) is regularly associated with thrombocytopenia (Weksler 2007). Platelet glycoprotein GPVI has been implicated in HCV–platelet interaction through peptide and immuno-inhibition studies (Pugliese et al. 2004), purified protein assays, and virus binding assays (Zahn et al. 2006) and was shown to be important for infection and dissemination (Ariede et al. 2015; Zahn et al. 2006). DC-SIGN has also been demonstrated to be involved in HCV binding.
As in infection of other cell types, HIV has been shown to associate with platelets through a variety of cell-surface receptors (Youssefian et al. 2002). Platelet DC-SIGN, as identified by flow cytometry, western blotting, and PCR, can recognize and bind pathogen-associated molecular patterns (PAMPs) on HIV because immuno-inhibition results in decreased binding (Boukour et al. 2006; Chaipan et al. 2006). Using similar immuno-inhibition and flow cytometric approaches, CLEC-2 was identified as a platelet receptor for HIV (Chaipan et al. 2006). Additional putative receptors for HIV on platelets also include CXCR4, CCL3, and CCL5 (Flaujac et al. 2010).
Platelet integrins appear to serve as the main binding partner because they contain the common RGD motif. Therefore, the presence of integrin-binding sequences in several virus families suggests that integrins are important for platelet association and signaling, with effects leading to thrombocytopenia. Viral envelope glycoproteins can serve as sources of PAMPs and facilitate virus–platelet interaction via TLRs. This is suggested as a mechanism for cytomegalovirus (CMV)-induced thrombocytopenia. Evidence implicating direct binding and consequent cell stimulation as a result of CMV-encoded glycoprotein B and glycoprotein H interactions with TLR2 (Boehme et al. 2006) on platelets or neutrophils has been obtained from immunoprecipitation and immuno-inhibition studies of co-transfected human embryonic kidney and normal fibroblast cells, respectively (Assinger et al. 2014).
Platelets also have on their surface the complement receptor type 2 (CR2), which functions as a receptor for Epstein–Barr virus (EBV), as shown by cell and immuno-inhibition techniques (Ahmad and Menezes 1997; Hutt-Fletcher 2007). Whether this also protects the virus from complement-mediated innate immune clearance is not known.
Platelets have one class of receptor for the Fc domain of antibodies, FcγRIIA. Once virus-directed antibodies are generated by the adaptive immune response, “bridged” interactions can be facilitated by platelet FcγRIIA, as demonstrated for influenza A virus (IAV) (Boilard et al. 2014). Making vaccine development difficult, some secondary viral infection mechanisms exploit antibody-dependent enhancement; examples are dengue virus (DENV) and HIV (Guzman et al. 2013). Although antibody-bridged binding to platelets has not been specifically documented for many viruses, it is reasonable to speculate that such viral immune complexes commonly form interactions with platelets. Similar to engagement of other receptors on the platelet surface, these multivalent adducts could crosslink the FcγRIIA, causing platelet activation.
Hemorrhagic Fever Viruses
Whereas the bleeding phenotype inherent to thrombocytopenia is considered to be predominantly caused by loss of platelets, the effects on hemostasis leading to virus-induced HF are typically far more complicated (Zapata et al. 2014). HF is characterized by the loss of blood homeostasis, leading to increased vascular permeability and bleeding, which can progress to shock. The causative agents of viral HF are enveloped RNA viruses from four families: Flaviviridae, Bunyaviridae, Arenaviridae, and Filoviridae (Table 2). These families contain well-known species such as Ebola and DENV that are featured as “headline news” because of the devastating and graphic illnesses these epidemic pathogens can cause. Some of the viruses are especially difficult to experimentally manipulate because of their biohazard classification level and, consequently, relatively little is known about their biochemistry. Nevertheless, receptors for viruses known to cause viral HF have been identified. Interestingly, these are similar to those receptors characterized for viruses as predominantly thrombocytopenic: β1/β3 integrins, lectins, and TLRs. Therefore, it is not surprising that HF viruses can also result in thrombocytopenia.
Using specific antibody inhibition and virological plaque-forming assays, DC-SIGN and heparan sulfate proteoglycan (HSP) were demonstrated to be important in DENV binding to platelets (Hottz et al. 2013b; Simon et al. 2015) Interestingly, additional binding sites were expressed by pretreating platelets with the agonist thrombin (Simon et al. 2015). Whether these are the same type of receptor is unknown. Employing a transduced cell model of infection, DC-SIGN has also been shown to be involved as a receptor in Ebola virus infection (Alvarez et al. 2002). Mutagenesis, binding, and RNA interference experiments have implicated Axl (Shimojima et al. 2007) and Tyro3 (Hunt et al. 2011) in Ebola’s complex cell engagement mechanism. These observations suggest that multiple receptors are occupied in the Ebola–platelet interaction. Interestingly, similar platelet surface molecules may bind Lassa virus, as indicated by the identification of DC-SIGN, Axl, and Tyro3 as receptors using cell binding and infection assays (Shimojima et al. 2012). In addition, the results of immunoblot and competition assays involving purified virus suggested that α-dystroglycan is a Lassa virus receptor (Cao et al. 1998). Neutralizing antibody experiments have implicated β3 integrins in the interaction between cells infected with hantavirus and platelets (Gavrilovskaya et al. 1999; Gavrilovskaya et al. 2010). Although cell and platelet receptors remain elusive for some HF viruses, it is possible that a particular receptor may function across an individual virus family.
Virus–Platelet Interactions with Moderate Pathology
Although virus–platelet interactions leading to life-threatening effects are well-known, there are also a number of viruses that bind to platelets without serious hematological pathology. Predominately from the Picornaviridae family, these non-enveloped RNA viruses cause enteric, throat, and nasal infections. Although not directly confirmed as analogous functional receptors for picornavirus on platelets, β1 and β3 integrins are known to facilitate interactions with receptors on other cell types. Studies following surface expression of mutated proteins in CHO cells and antibody inhibition studies have identified integrin α2β1 as a binding partner for both rotavirus and echovirus (Coulson et al. 1997; Fleming et al. 2011). Utilizing an antigenic inhibition model and CHO transfection, members of the β1 and β3 integrin families were shown to mediate parechovirus infection (Triantafilou et al. 2000). This suggests the potential for binding recognition with the integrin known to be expressed on the platelet surface. It is somewhat puzzling that thrombocytopenia is not induced because engagement of these receptors by other viruses is consistent with this outcome. Consequently, integrins may not be the only common denominator resulting in this virus-mediated hemostatic disorder.
Virus-Induced Platelet Activation
Platelets circulate in a resting state and are stimulated by ligand–receptor engagement. It is clear that protein and carbohydrate receptors on the surface of platelets bind viruses from many distinct families (Tables 1, 2, and 3). Indeed, it is not surprising that the interactions known to exist between viruses and platelet receptors can facilitate platelet activation. Interactions with DENV grown in culture have been shown to induce platelet shape changes, as monitored by atomic force and electron microscopy (Ghosh et al. 2008). DENV has also been demonstrated to cause the exposure of P-selectin and procoagulant phospholipid on the platelet surface (Hottz et al. 2013b; Simon et al. 2015), and to initiate apoptosis-like markers, including caspase activation and mitochondrial permeability changes (Hottz et al. 2013b), Similarly, HIV can induce microparticle release and other hallmarks of platelet activation (Wang et al. 2011) that are related to plasma virus levels (Mayne et al. 2012). Differential response to platelet agonists in HIV-infected individuals points to functional changes in platelets, with implications for thrombosis (Satchell et al. 2010). Interestingly, additional studies further implicated HIV treatment strategies in the development of hyper-responsive platelets (Falcinelli et al. 2013; Gresele et al. 2012). Although the direct effect of virus binding on platelet activation has not been studied extensively, outside-in signal-induced platelet changes are probably a general characteristic, regardless of the type of virus.
Virus Replication by Platelets
Entry
Cell binding is the first step used by all viruses to exploit the host’s cellular replication apparatus. Without question, platelets have that binding capacity. The second step in the virus lifecycle is cell entry. Two general mechanisms of cell entry can be initiated upon contact between an animal virus and a target host cell. Most viruses traverse the plasma membrane using the endocytic machinery intrinsic to the cell, whereby the virus is engulfed by a vesicular structure for intracellular transport (Ghigo 2010). Within these endosomal compartments, low pH typically induces the release of genetic contents from the viral nucleocapsid. Based on microscopic morphology and biochemistry, nearly a dozen viral endocytosis mechanisms have been discerned (Cossart and Helenius 2014; Mercer et al. 2010). A second entry mechanism is used exclusively by enveloped viruses. After receptor-mediated cell surface binding, both virus- and host-encoded (Derry et al. 2007) proteins on the virus envelope can engage to form a conformational fusion complex that merges the envelope and plasma membrane.
In contrast to the known interactions between viruses and platelets, little evidence of platelet entry has been documented. Immunogold electron microscopy revealed that purified HIV is trapped within platelet endosomal structures (Youssefian et al. 2002). Another entry mechanism was suggested by colocalization of purified adenovirus with the surface-connected channels of the platelet open canalicular system (Stone et al. 2007). Purified IAV and platelets enabled observation by electron microscopy of the surface association and uptake of virus-like particles into vacuolar structures (Danon et al. 1959). Furthermore, virus-like particles have been reported in the platelets of DENV-infected patients, although these difficult experiments lacked confirmation of virus (Noisakran et al. 2009). Electron microscopy has also been used to demonstrate encephalomyocarditis virus (EMCV) uptake by platelets in a murine model of infection (Koupenova et al. 2014). To confirm their endocytic capabilities, purified platelets have been shown to engulf Staphylococcus aureus (Youssefian et al. 2002) and liposomes that were engineered for drug delivery (Chan et al. 2015). Although viral entry mechanisms are not known, molecular details following the transfer of synthetic particles into platelets suggests that several simultaneous endocytotic pathways are involved (Chan et al. 2015). These pathways may have dynamin dependence as a common aspect and involve caveolae- or clathrin-mediated uptake (Mercer et al. 2010). Thus, observations of virus uptake by platelets are supported by the availability of entry mechanisms for other particle types.
Viral Protein Synthesis
Platelets associate with both DNA- and RNA-containing viruses. Assuming successful entry and release of genetic material into the platelet, only RNA viruses replicate because platelets are DNA transcription incompetent. The list of RNA viruses with known or suspected platelet interactions is extensive and highlighted in Tables 1, 2, and 3. Platelets contain all of the post-transcriptional apparatus necessary for potential assembly of an infectious RNA virus. To demonstrate that platelets can translate viral RNA, purified platelets inoculated with purified DENV have been shown to produce viral nonstructural protein 1 (NS1), as detected by Western blot analysis (Simon et al. 2015). Because the DENV single-stranded RNA (ssRNA) must first be translated as a polyprotein, the finding that NS1 had the predicted molecular weight of the mature protein implied that the functional DENV protease complex (NS2B/NS3) was also properly translated and processed by the platelets (Simon et al. 2015). Thus, platelets not only facilitate the initial step in the virus lifecycle, specific surface engagement (Tables 1, 2, and 3), but also have the means to allow penetration of the virus into the cytoplasm and occupation of the platelet translational mechanism.
Virus Replication
Replication of viral genomic material has been followed as a surrogate for functional virus-encoded polymerase generation. In these studies, the mRNAs of all four serotypes of purified DENV in combination with purified platelets were enhanced (Simon et al. 2015), which substantiated an earlier preliminary report (Onlamoon et al. 2010). These studies were extended using virus plaque formation assays in combination with a translation inhibitor and demonstrated that the virus is properly assembled, resulting in production of infectious progeny by platelets (Simon et al. 2015). Interestingly, platelet units stored under blood bank operating conditions also produced new DENV (Sutherland et al. 2016). A generalized model is presented in Fig. 1, highlighting the emerging concept that platelets could be a reservoir for permissive RNA viruses. The concept is based on (1) significant literature demonstrating that many types of virus bind directly to platelets; (2) several studies showing platelet entry; and (3) generation of infectious DENV progeny by platelets.

Virus replication by platelets. Examples of virus receptors present on the surface of the platelet are shown spanning the platelet membrane. Examples of an enveloped virus (DENV binding to the co-receptors DC-SIGN and HSP) and a non-enveloped virus (adenovirus binding to CAR) are depicted. This receptor engagement triggers mechanisms that can involve dynamin-dependent engulfment processes, leading to entry into the platelet as an endosomal inclusion. At least for DENV, which has an RNA genome unlike adenoviruses, evidence is accumulating that suggests viral genomic material is released from the nucleocapsid into the cytoplasm, where it is translated by platelet ribosomal complexes or used as a template for replication by the virus-encoded polymerase. Other virus-encoded genes contribute to controlling the function of the cell or post-translational modification of the viral proteins, including proteolytic maturation if the viral genome is organized to produce a polyprotein. The viral structural proteins and genome copies are then transported to the platelet Golgi apparatus, where they are assembled and delivered to the exterior by exosomal transport or possibly by cell disruption (not shown). Platelet mitochondria are available for energy-demanding aspects of the mechanism
Although evidence suggests that platelets could be recruited by RNA viruses as conspirators for replication, it is conceivable that only platelet subsets can fulfill this role. As an example, ~20 % of platelets are positive for DC-SIGN (Hottz et al. 2013b) and may account for binding to HIV, DENV, HCV, Ebola, and LASV, which are known to use DC-SIGN for cell surface attachment (Tables 1, 2, and 3). Furthermore, TLR2 is found on a subset of ~14 % of platelets and is involved in at least the CMV interaction (Boehme et al. 2006). Some platelet-interacting viruses are known to use co-receptor systems, of which candidate receptors are found on platelets. But, whether these too are distributed as subpopulations, like DC-SIGN and TLR2, has not been evaluated. Of these functionally distinct classes of platelets, only some may be permissive to entry and replication after binding.
Platelet Immune Response Against Viruses
Platelets may initially harbor and replicate certain viruses (Simon et al. 2015). But, as known facilitators and modulators of the immune response to pathogen invasion (Jenne and Kubes 2015), platelets also competently present bound viruses to leukocytes for clearance. To enhance this innate immune recognition, the association of viruses with platelets induces the release of cytokines, which causes local infiltration of immune cells. Many platelet surface molecules mediate leukocyte cross-talk and have an essential effect on viral infection. The modulation of platelet function by viruses, leading to their ultimate involvement in innate and adaptive immunity, is summarized in Fig. 2.

Virus-induced platelet modulation leading to immune clearance. Platelet receptor binding or possibly entry of the virus induces outside-in platelet signaling, with morphological and biochemical effects to the platelet. Typical platelet “activation” markers are expressed, such as exposure of procoagulant phospholipid (proPL) as measured by annexin V (Anx5) binding and P-selectin (CD62P). Platelet microparticles may be released. The proPL surface can propagate the hypercoagulable state often induced by some of these viruses by enabling assembly of clotting enzyme complexes, where neighboring platelet surface PARs can be activated by thrombin (FIIa) in a feedback-amplified manner to further promote platelet modulation. Virus–platelet engagement has been shown to induce caspase activation and mitochondrial membrane potential changes indicative of an apoptotic state, which similarly occurs for megakaryocytes (not shown) with consequent reduction in thrombopoeisis. Virus-induced platelet stimulation causes the release of α-granule and dense-granule contents. This has profound stimulatory effects on all cells of the immune system, orchestrating localized innate and adaptive responses against virus invasion
Toll-Like Receptors
TLRs (Cognasse et al. 2015) are a family of innate immune regulators that recognize pathogen-associated molecular patterns (PAMPs), which are markers associated with viruses, bacteria, and fungi that lead to neutrophil-mediated pathogen destruction. Known to recognize ssRNA, typical of many platelet-interacting viruses (Tables 1, 2, and 3), platelet TLR7 was identified as vital for EMCV clearance by platelets (Koupenova et al. 2014). Interestingly, this receptor is expressed within endosomes and its functional involvement implies endocytosis of the virus by the platelet. Penetrance of EMCV was TLR7 dependent, with activation of TLR7 resulting in the release of the α-granules that house proinflammatory cytokines, leading to neutrophil aggregation, endothelial cell adhesion, and inflammation. The TLR7 platelet response was shown to contribute to host survival, as EMCV levels decreased and there were no observable prothrombotic events as a consequence of potential platelet activation (Koupenova et al. 2014).
Unlike TLR7, TLR2 is expressed on the platelet surface. CMV was found to associate predominately with the TLR2-positive platelet subpopulation (Boehme et al. 2006). As seen using flow cytometry, CMV induces rapid surface expression of P-selectin, leading to the release of proinflammatory CD40 ligand and interleukin-1β cytokine from the platelets (Assinger et al. 2014). CMV-induced TLR2 activation was confirmed by antigenic inhibition and could be blocked by inhibiting phosphoinositide 3-kinase signaling. Vascular endothelial-derived growth factor (VEGF) is a proangiogenic cytokine that is released by TLR2-induced platelet activation and involved in endothelial migration, proliferation, and increased vascular permeability (Assinger et al. 2014). To add further to the many links reported between CMV and vascular disease (Al-Ghamdi 2012; Bruggeman 2000), this TLR2-mediated VEGF release can allow leukocyte recruitment to an atherosclerotic plaque, promoting growth (Holm et al. 2009).
Integrins
Consisting of a broad group of homologous heterodimeric proteins (Bennett et al. 2009), cell surface integrins are fundamental to the important platelet–leukocyte connection. In addition to the role of integrins in facilitating numerous direct platelet–virus interactions (Tables 1, 2, and 3), they are also important in processes leading to immune clearance of blood-borne viruses. One example is through the recruitment of dendritic cells by the interaction of platelet surface junctional adhesion molecule-C (JAM-C) and dendritic cell integrin αMβ2 (Langer et al. 2007). Trafficking of cytotoxic T lymphocytes (CTLs) to sites of infection can be mediated by platelet activation (Iannacone et al. 2005). In the case of hepatitis B virus (HBV), CTLs enter the liver parenchyma and accumulate where HBV may reside and replicate. CTL activity lowers the virus count, but unfortunately contributes to progression of liver damage (Iannacone et al. 2005).
The HIV-1 transactivator of transcription protein (Tat) directly interacts with platelets, resulting in their activation and degranulation (Wang et al. 2011). This mechanism requires the β3 integrin and chemokine receptor CCR3 to be expressed on the platelet surface. This involvement of integrins was unambiguously demonstrated using a β3 knockout mouse model (Wang et al. 2011). Virus-induced platelet degranulation involves the release of proinflammatory CD40 ligand, which promotes platelet–monocyte complex formation via platelet P-selectin and monocyte P-selectin glycoprotein ligand-1 (PSGL-1) (Singh et al. 2014). This was demonstrated by injecting wild-type mice with recombinant soluble CD40 ligand and analyzing cell associations by flow cytometry. The platelet–monocyte complexes derived from HIV-1 infected donors exhibited enhanced adhesion to human brain microvascular endothelial cells, suggesting a role for platelets in HIV-associated neuroinflammation (Singh et al. 2014).
Selectins
Although not as extensive as integrins, selectins are also a family of cellular adhesion receptors. Selectins recognize various carbohydrate structures found on opposing surfaces. Platelets constitutively store P-selectin (i.e., CD62P) in α-granules. In response to stimulus-induced activation, P-selectin is transported to the platelet surface where it mediates tethering to numerous cell types. Important to the role of platelets in orchestrating the immune response, platelet surface P-selectin mediates adhesion through association with PSGL-1 found on the neutrophil surface. P-selectin on platelets also interacts with PSGL-1 on a subset of Th1 leukocytes. Thus, when binding of viruses to the platelet surface triggers platelet activation and P-selectin expression on the surface, innate immune clearance of the pathogen is facilitated.
Protease-Activated Receptors
Hemostasis and inflammation are regulated and linked by protease-activated receptors (PARs) (Rothmeier and Ruf 2012). Four PAR types have been identified and are implicated both in virus replication (Aerts et al. 2013; Khoufache et al. 2013; Sutherland et al. 2012) and in the host innate response to viral infection (Antoniak et al. 2013). Enveloped viruses can assemble and activate coagulation enzyme complexes directly on their surface, which mediate PAR stimulation. Through these pathways, PAR1 and PAR2 on human umbilical vein endothelial cells have been shown to enhance HSV1 infection in vitro (Pryzdial et al. 2014). A similar mechanism may stimulate virus-bound platelets, which express high levels of PAR1. A more recent study showed that IAV induced platelet activation and aggregation through platelet-surface PAR, which exacerbated acute lung injury (Le et al. 2015). This injury was attributed to the resulting massive inflammation through platelet-induced recruitment of neutrophils to the lung.
Chemokine Receptors
Chemokine receptors are members of the G-protein-coupled receptor family, whose major functions include cellular recruitment via chemokine recognition. Several chemokine receptors (CCR1, CCR3, CCR4, CXCR1, and CXCR4) bind to select ligands, resulting in enhancement but not initiation of inflammatory pathways, platelet aggregation, hemostasis, and thrombosis (Zarbock et al. 2007). Activated platelets also secrete numerous chemokines, such as CXCL7 and CXCL8. CXCL7 promotes chemotaxis, adhesion to endothelial cells, and degranulation of neutrophils (Schenk et al. 2002), whereas CXCL8 is important in recruitment of neutrophils (Baggiolini et al. 1994). Active CD40L is secreted by platelets in response to stimulation and binds endothelial CD40, eliciting chemokine secretion and increasing the expression of adhesion molecules on the endothelium (Henn et al. 1998).
Platelet α-granule contents have been shown to limit the spread of HIV-1 in co-cultured T cells (Solomon et al. 2013). CXCL4 (platelet activating factor) and CXCL7 are the most abundant chemokines in the α-granules of platelets (Blair and Flaumenhaft 2009). In particular, CXCL4 released by activated platelets binds to HIV-1 major viral envelope glycoprotein, gp120, proximal to the essential CD4-binding site (Auerbach et al. 2012). The resulting steric inhibition reduced HIV-1 infection by 80 % compared with non-activated platelets using a HIV-1-sensitive cell line that uses reporter gene constructs to quantify infection (Solomon et al. 2013). CXCL4 stimulates neutrophil–endothelial cell attachment and also acts as a co-stimulator of TNF in the release of neutrophil secondary granules (Kasper and Petersen 2011), as further means of enhancing localized immune cell influx.
The proinflammatory cytokine interleukin (IL)-1β is synthesized by platelets as a precursor protein and cleaved by caspase-1 to produce an active form that is released in microparticles (Hottz et al. 2013b). DENV2 was shown to induce IL-1β synthesis directly and secretion from platelets by activating the assembly of a nucleotide-binding domain leucine-rich repeat-containing protein (NLRP3) inflammasome, which controls caspase-1 activity (Hottz et al. 2013a). Generally, IL-1β is important in the acute-phase response, where proteins such as C-reactive protein, complement components, and fibrinogen are produced to destroy or contain microbes (Morrell et al. 2014). Although aiding in the immune response, the IL-1β released from platelets during DENV infection contributes to increased endothelial permeability, thrombosis, and dysregulated hemostasis (Hottz et al. 2013a).
Defensins
Defensins are cationic antimicrobial peptides that are key elements in the innate immune system. They act on bacteria, enveloped viruses, and non-enveloped viruses. There are many forms of these small 4–5 kDa peptides, with human platelets expressing β-defensins (hBD) 1, 2, and 3 (Kraemer et al. 2011; Tohidnezhad et al. 2011; Tohidnezhad et al. 2012). Immunofluorescence studies in vitro showed that the inclusion of a selective agonist of neutrophils (phorbol 12-myristate 13-acetate) or platelet PAR1 (thrombin receptor agonist peptide 1) induced secretion of an adhesive complex from neutrophils, identified as a pathogen “snare.” The molecular networks were identified as being composed of long uncoiled strands of DNA and were named neutrophil extracellular traps (NETs) (Brinkmann et al. 2004). NET release is induced by β-defensin 1 secreted from activated platelets (Kraemer et al. 2011). Although most NET involvement in pathogen defense has been elucidated using bacteria, super-resolution structured illumination microscopy revealed that HIV-1 virus particles are also captured by NETs (Saitoh et al. 2012). When these entrapped virus particles were extracted, their infectivity was reduced as a result of highly enriched levels of α-defensin and myeloperoxidase within the NETs.
In addition to HIV-1, hBD-2 is also known to inhibit human respiratory syncytial virus (RSV) entry into human lung epithelial cells, as followed by 35S-labeled RSV uptake (Kota et al. 2008). It was also shown through electron microscopy and buoyant density profiles that hBD-2, but not hBD-1, disrupted the viral envelope, possibly because the cationic nature of the peptide led to lipid destabilization (Kota et al. 2008). HIV-1 induced hBD-2 and hBD-3 expression in human oral epithelial cells, which associated directly with HIV-1 and neutralized infection in vitro (Quinones-Mateu et al. 2003). IAV hemagglutinin and hBD-3 binding also resulted in inhibition of epithelial cell infection (Leikina et al. 2005). hBD-3 was shown to alter fusion between IAV, sindbis virus, baculovirus, and synthetic target membrane by crosslinking virus surface proteins (Leikina et al. 2005). Although reasonable to speculate, it is not known whether platelet-derived hBD-2 and hBD-3 mediate similar direct antiviral effects.
Other Secreted Platelet Components
Human donor platelet concentrates contain unknown antiviral activity that reduced the viral titer of poliovirus 1, adenovirus 5, and vaccinia virus by approximately 2.63 ± 0.5 to 5.6 ± 0.9 log units (Maurice et al. 2002). The same group also observed platelet activation in all virus–platelet co-cultures with epithelial monolayers (Vero cells); recent knowledge (Flaujac et al. 2010) suggests that platelet releasate is the antimicrobial factor(s) in these studies.
Defensins and cytokines are stored in platelet α-granules. However, dense granule constituents such as adenosine diphosphate (ADP) (Packham and Rand 2011), polyphosphates (polyP) (Smith and Morrissey 2014), and serotonin (Jedlitschky et al. 2012) also have immune-modulating properties (Morrell et al. 2014). These have not been as well characterized for a role in viral innate immunity as those contained in α-granules. Nevertheless, the release of platelet dense granule contents by viruses has been investigated in CMV infection. This was mediated by platelet surface TLR2 occupation. Furthermore, inhibition of the ADP receptor, P2Y12, identified ADP release as an important trigger for secondary platelet activation (Assinger et al. 2014). Dendritic cells also express P2Y12 and their stimulation increases antigen endocytosis and processing (Vanderstocken et al. 2010), which could involve localized platelet response to pathogen interactions. PolyP induces proinflammatory responses by acting on the nuclear factor κB (NF-κB) pathway in vascular endothelial cells (Bae et al. 2012). PolyP can also interact with chromatin-associated nuclear proteins such as high mobility group box 1 (HMGB1) to amplify proinflammatory responses (Dinarvand et al. 2014). Because HMGB1 is also secreted by platelets (Maugeri et al. 2012), polyP warrants continued study as a bridge between platelets and innate immunity, especially in the context of complement, which is suppressed by polyP (Wat et al. 2014) and could be a viral survival mechanism.
Virus-Induced Thrombocytopenia
According to some estimates, approximately two-thirds of acute thrombocytopenia cases are preceded by viral infection (Rand and Wright 1998). This strikingly high correlation suggests mechanisms directly linking the virus to the platelet, such as receptor-mediated binding (Table 1). In contrast to viral thrombocytopenia, viral HF (Table 2) is thought to be driven by severe suppression of innate immunity and the resulting cytokine flood that counters systemic virus replication. HF can also result in a reduced platelet count, but through mechanisms indirectly involving platelets, as detailed in excellent reviews elsewhere (Feldmann and Geisbert 2011; Messaoudi and Basler 2015; Zapata et al. 2014). Here, we overview five primary pathways that simultaneously contribute to viral thrombocytopenia in the absence of HF: decreased thrombopoeisis, direct and indirect virus interactions with platelets and megakaryocytes (tipping the intricate balance between thrombopoeisis and platelet clearance), altered platelet function, and virus-induced immune response against platelets.
Decreased Thrombopoeisis
Viral infection results in production of interferon, which has antiviral activity. However, type I interferons can also inhibit megakaryocytes, resulting in impaired platelet production (Rivadeneyra et al. 2015). Megakaryocyte growth and differentiation is stimulated by thrombopoietin (TPO), which is predominantly produced in the liver (Giannini et al. 2002). Impaired liver function is consequently detrimental to TPO production and the resulting thrombocytopenia often suffered by chronic HCV patients correlates with low TPO and attenuated thrombopoeisis (Giannini et al. 2002; Wenzel et al. 2010).
Direct impairment of platelet production as a result of viral replication in megakaryocytes was observed in vitro for HIV, HCV, DENV, and CMV (Basu et al. 2008; Crapnell et al. 2000; Li et al. 1999; Sato et al. 2000; Sridharan et al. 2013). An increase in cell death via apoptosis and decreased megakaryocyte differentiation are probable reasons for reduced platelet production. For example, HIV infection of megakaryocytes resulted in downregulation of the TPO receptor c-Mpl (Gibellini et al. 2013), causing reduced sensitivity to thrombopoeisis induction. To further investigate this mechanism, umbilical cord blood hematopoietic progenitor cells that were induced by TPO toward a megakaryocytic lineage in vitro have been used as a model for HIV infection. The virus surface glycoprotein gp120 interacts with CD4 on these cells (Gibellini et al. 2007), resulting in specific protein and mRNA changes that elevate TGF-β1 levels and decrease tumor necrosis factor (ligand) superfamily, member 13 (TNFSF13) levels. Both of these effects contribute to inhibition of megakaryocytic proliferation and promote apoptosis (Gibellini et al. 2007). Further evidence supporting the concept that viral thrombocytopenia involves attenuated thrombopoiesis comes from an elegant humanized mouse model showing that DENV infection reduces platelet production (Sridharan et al. 2013). Thus, viral infection has the capacity to reduce thrombopoeisis by direct effects on the megakaryocyte, conceivably involving the same surface receptors for specific viruses that have been identified on platelets and other cells (Tables 1, 2, and 3).
Direct Virus–Platelet Associations
Activation of platelets, degranulation, and recruitment of neutrophils and dendritic cells contribute to phagocytosis of platelet fragments by leukocytes. The final clearance of these potentially virus-laden particles is in the spleen and liver (Bondanza et al. 2001; Grozovsky et al. 2010; Koupenova et al. 2014). CMV- or EMCV-mediated activation of platelets via TLR2 or TLR7, respectively, enhanced the interaction between platelets and neutrophils, resulting in platelet clearance (Assinger et al. 2014; Koupenova et al. 2014). In a similar manner, hantavirus also induced platelet activation and clearance by binding to platelet surface integrins αvβ3 and αIIbβ3 (Gavrilovskaya et al. 2010). The interaction of hantavirus with platelets also contributed to viral dissemination and activation of endothelial cell functions, thereby increasing vascular permeability (Feldmann and Geisbert 2011; Gavrilovskaya et al. 2010).
Adenovirus correspondingly induces thrombocytopenia, as seen when adenovirus gene therapy vectors are intravenously administered to rhesus macaques and mice (Othman et al. 2007; Wolins et al. 2003). Current literature indicates that CAR mediates the binding of adenovirus to platelets, enabling subsequent entry (Othman et al. 2007; Shimony et al. 2009). The virus–platelet interaction is predominantly localized to sites of intercellular complex formation, implying that CAR expression is enhanced in response to platelet activation (Gupalo et al. 2011). Similar to the effects reported for other viruses, platelet activation is probable upon the initial association of adenovirus and platelet-surface CAR.
The IAV envelope has neuraminidase activity that cleaves sialic acid on the surface of platelets (Madoff et al. 1964). Removal of more than 15 % of total sialic acid on rabbit platelets caused complete platelet clearance within 1 h of administering 51Cr-labeled platelets into rabbits (Greenberg et al. 1975). The removal of circulating platelets was presumably caused by recognition of exposed terminal galactose residues by scavenger cells expressing the asialoglycoprotein receptor (Sorensen et al. 2009). This adds to the repertoire of virus-clearance mechanisms that can be facilitated through direct virus–platelet association.
Indirect Virus–Platelet Associations
Platelets express Fc receptors that allow recognition of immune complexes or aggregated immunoglobulin. In addition to PAR-1 activation by thrombin, IAV can activate platelets through low-affinity FcγRIIA signaling (Boilard et al. 2014). For this to occur, IAV must be decorated with anti-IAV IgG. Crossreactive antibodies resulting from immune recognition of different IAV strains (H1N1 versus H3N2) was sufficient to produce this effect. When wild-type mice (which do not express FcγRIIA) and transgenic mice expressing human FcγRIIA were challenged intravenously with a sublethal dose of H1N1, there was a drop in circulating platelet count in only the transgenic mice. This indicates that platelet homeostasis is affected by the accessibility of Fc receptors on platelets, supporting the link to pathogenic thrombocytopenia (Boilard et al. 2014).
Activation of endothelial cells has been implicated in adenovirus-induced thrombocytopenia. In these studies, the release of ultralarge von Willebrand factor (vWF) from the Weibel–Palade bodies (Gupalo et al. 2011) was evaluated using a murine model. Adenovirus induced thrombocytopenia in wild-type animals but, in sharp contrast, vWF knockout mice were protected from a reduced platelet count (Gupalo et al. 2011). Thus, endothelial vWF could contribute in an indirect manner to virus-induced thrombocytopenia by supporting platelet aggregation through interactions with platelet surface GP1b, (the vWF receptor). Clearance of these platelet aggregates is subsequently facilitated by splenic macrophages or Kupffer cells in the liver (Othman et al. 2007).
Systemic inflammation as a result of viral infection can result in platelet interactions and increased clearance, as demonstrated during IAV, rhinovirus, and CMV infections (Bouwman et al. 2002). The mechanism involves mononuclear leukocytic release of CXCL4. DENV infection was used in a subsequent study and resulted in increased inflammation, vascular permeability, and platelet aggregation and activation (Yang et al. 1995).
Altered Platelet Function
Thrombocytopenia can result from virus activation of platelets or endothelial cells, which induces cell–cell adhesion processes via expression of integrin and selectin function (Zapata et al. 2014). The flip-side is that inhibition of platelet aggregation can also result in impaired platelet function. DENV infections have been shown to stimulate platelet-directed IgM autoantibodies that inhibit ADP-induced platelet aggregation (Lin et al. 2001). Although these autoantibodies have an effect on immune clearance of platelets, they also affect the development of HF. For hantavirus, this can include renal syndrome resulting from defective platelet aggregation (Cosgriff et al. 1991). Other HF viruses such as Lassa virus, Junin virus, and Ebola virus have inhibitors of platelet aggregation, but these have not been identified (Cummins et al. 1989; Cummins et al. 1990, Feldmann and Klenk 1996). Additionally, vaccinia virus causes impairment of platelet aggregation induced by ADP, collagen, or thrombin (Bik et al. 1982). Ebola virus infection also results in increased levels of type I interferons, which downregulate platelet production and function (Rivadeneyra et al. 2015; Villinger et al. 1999).
Virus-Induced Immune Response Against Platelets
The molecular relationship between autoantibodies against platelet proteins and viral infection is complex and not yet clearly understood. Virus-induced thrombocytopenia typically worsens as damage to the liver progresses and can deteriorate into a more severe clinical complication (Aref et al. 2009). Anti-platelet autoantibodies are linked to immune thrombocytopenic purpura (ITP) (Liebman 2008). Secondary ITP can result from vaccines such as measles-mumps-rubella (MMR) (incidence of 1 in 40,000 administrations) or infections with homologous herpes family viruses, hepatitis C, HIV, hantavirus, and severe acute respiratory syndrome coronavirus (Goeijenbier et al. 2012; Liebman 2008).
Non-AIDS early HIV-1 infections can result in ITP induced by autoimmune antibodies. Affinity purification of circulating serum immune complexes in HIV patients identified an anti-HIV IgG1 antibody that recognizes amino acid residues 49–66 of the integrin β3 subunit that induces platelet vessiculation (Nardi et al. 2001). The complement pathway was ruled out in this reaction because neither the F(ab′)2 fragment of the antibody raised against integrin β3 residues 49–66 in wild-type mice nor treatment with full-length antibody in C3-deficient mice could affect platelet microparticle formation (Nardi et al. 2001). Peroxide generation was monitored through the use of an intracellular fluorescent probe and revealed a novel mechanism by which the autoantibody induced damage in platelets through a NADPH oxidase peroxide-generating pathway (Nardi et al. 2001). Similar outside-in signaling events could be generated by other virus-induced platelet autoantibodies.
ITP occurs in 20 % of HCV patients and is potentially attributed to the presence of antibodies that are crossreactive with HCV core envelope 1 protein and platelet β3 integrin (Rajan et al. 2005; Zhang et al. 2009). The crossreactivity was found by using the antibody specific for integrin β3 residues 49–66 to screen a phage-display peptide library (Zhang et al. 2009). The recognized peptides were then aligned with the viral genome to define similarities. The matched peptides were rationally designed as tools to inhibit the binding of autoantibodies to platelets or to produce platelet antibodies with functional effects (Li et al. 2005; Zhang et al. 2009). This approach was similarly used to discover molecular mimics in the HIV-1-encoded negative regulatory factor (nef) protein (Li et al. 2005), both leading toward therapeutic design in virus-induced thrombocytopenia.
Antibody crossreactivity between antiviral antigens and platelet antigens has been demonstrated during DENV infection (Cheng et al. 2009). Pairwise sequence alignment analysis programs were used to annotate homologous peptide sequences between DENV NS1 and platelet protein disulfide isomerase (PDI) (Cheng et al. 2009). This tool identified several sequence homologies, of which amino acid residues 311–330 (P311–330) was the most dominant epitope recognized by both anti-NS1 and anti-PDI, as determined by ELISA (Cheng et al. 2009). The P311–330 antibodies generated from hyperimmunized mice bound platelet PDI and inhibited both thiol isomerase activity and platelet aggregation induced by ADP (Cheng et al. 2009). The presence of anti-platelet autoantibodies with anti-DENV activity in DENV patient sera is associated with thrombocytopenia and the severity of the disease during the acute phase of secondary DENV infection (Saito et al. 2004). However, the direct implication of these crossreactive antibodies in DENV pathogenesis has not yet been elucidated. The discovery that platelets can translate and express NS1 (Simon et al. 2015) suggests that viral antibodies might not be crossreactive with platelets, but actually recognize the virus-encoded gene product expressed on the platelet, further complicating vaccine development.
Conclusion
Associations between viruses and platelets can lead to pathology. Viral infection often precedes thrombocytopenia and, therefore, an understanding of the mechanisms that facilitate virus–platelet interactions, their direct effect on platelets, and indirect effects on the cellular environment can lead to therapeutic control. This is a difficult challenge because platelets also help to eradicate viruses by steering innate and adaptive immune responses. Thus, ideal therapeutic control of virus-induced thrombocytopenia can discretely manage both the detrimental and positive involvement of platelets in viral infection.
Take Home Messages
-
Diverse virus families can bind to platelets, resulting in mild to severe clinical outcomes.
-
Virus–platelet interplay results in changes to innate and adaptive immunity.
-
Platelets replicate the RNA genome of permissive viruses, which can contribute to pathology.
References
Aerts L, Hamelin M-E, Rheaume C, Lavigne S, Couture C, Kim W, Susan-Resiga D, Prat A, Seidah NG, Vergnolle N, Riteau B, Boivin G (2013) Modulation of protease activated receptor 1 influences human metapneumovirus disease severity in a mouse model. PLoS One 8:1–13
Agbanyo FR, Wasi S (1994) Human cytomegalovirus interaction with platelets and adhesive glycoproteins: significance in viral pathogenesis. J Infect Dis 170:1120–1127
Ahmad A, Menezes J (1997) Binding of the Epstein-Barr virus to human platelets causes the release of transforming growth factor-beta. J Immunol 159(8):3984–3988
Al-Ghamdi A (2012) Role of herpes simplex virus-1, cytomegalovirus and Epstein-Barr virus in atherosclerosis. Pak J Pharm Sci 25(1):89–97
Alvarez CP, Lasala F, Carrillo J, Muniz O, Corbi AL, Delgado R (2002) C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J Virol 76(13):6841–6844
Antoniak S, Owens AP, Baunacke M, Williams JC, Lee RD, Weithauser A, Sheridan PA, Malz R, Luyendyk JP, Esserman DA, Trejo J, Kirchhofer D, Blaxall BC, Pawlinski R, Beck MA, Rauch U, Mackman N (2013) PAR-1 contributes to the inate immune response during viral infection. J Clin Invest 123:1310–1322
Aref S, Sleem T, El MN, Ebrahiem L, Abdella D, Fouda M, Samara NA, Menessy A, Abdel-Ghaffar H, Bassam A, Abdel WM (2009) Antiplatelet antibodies contribute to thrombocytopenia associated with chronic hepatitis C virus infection. Hematology 14(5):277–281
Ariede JR, Pardini MI, Silva GF, Grotto RM (2015) Platelets can be a biological compartment for the Hepatitis C Virus. Braz J Microbiol 46(2):627–629
Assinger A, Kral JB, Yaiw KC, Schrottmaier WC, Kurzejamska E, Wang Y, Mohammad AA, Religa P, Rahbar A, Schabbauer G, Butler LM, Soderberg-Naucler C (2014) Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler Thromb Vasc Biol 34(4):801–809
Auerbach DJ, Lin Y, Miao H, Cimbro R, Difiore MJ, Gianolini ME, Furci L, Biswas P, Fauci AS, Lusso P (2012) Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc Natl Acad Sci U S A 109(24):9569–9574
Bae JS, Lee W, Rezaie AR (2012) Polyphosphate elicits pro-inflammatory responses that are counteracted by activated protein C in both cellular and animal models. J Thromb Haemost 10(6):1145–1151
Baggiolini M, Dewald B, Moser B (1994) Interleukin-8 and related chemotactic cytokines—Cxc and Cc chemokines. Adv Immunol 55(55):97–179
Basu A, Jain P, Gangodkar SV, Shetty S, Ghosh K (2008) Dengue 2 virus inhibits in vitro megakaryocytic colony formation and induces apoptosis in thrombopoietin-inducible megakaryocytic differentiation from cord blood CD34+ cells. FEMS Immunol Med Microbiol 53(1):46–51
Bennett JS, Berger BW, Billings PC (2009) The structure and function of platelet integrins. J Thromb Haemost 7:200–205
Bergelson JM, Cunningham JA, Droguett G, KurtJones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW (1997) Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275(5304):1320–1323
Bik T, Sarov I, Livne A (1982) Interaction between vaccinia virus and human blood platelets. Blood 59:482–487
Blair P, Flaumenhaft R (2009) Platelet alpha-granules: basic biology and clinical correlates. Blood Rev 23(4):177–189
Boehme KW, Guerrero M, Compton T (2006) Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol 177(10):7094–7102
Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, Borgeat P, Flamand L (2014) Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood 123(18):2854–2863
Bondanza A, Manfredi AA, Zimmermann VS, Iannacone M, Tincani A, Balestrieri G, Sabbadini MG, Querini PR (2001) Anti-beta2 glycoprotein I antibodies cause inflammation and recruit dendritic cells in platelet clearance. Thromb Haemost 86(5):1257–1263
Boukour S, Masse JM, Benit L, Dubart-Kupperschmitt A, Cramer EM (2006) Lentivirus degradation and DC-SIGN expression by human platelets and megakaryocytes. J Thromb Haemost 4(2):426–435
Bouwman JJ, Visseren FL, Bosch MC, Bouter KP, Diepersloot RJ (2002) Procoagulant and inflammatory response of virus-infected monocytes. Eur J Clin Invest 32(10):759–766
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535
Bruggeman CA (2000) Does cytomegalovirus play a role in atherosclerosis? Herpes 7(2):51–54
Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, Nichol ST, Compans RW, Campbell KP, Oldstone MBA (1998) Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and lassa fever virus. Science 282(5396):2079–2081
Caviness AC, Demmler GJ, Selwyn BJ (2008) Clinical and laboratory features of neonatal herpes simplex virus infection—A case-control study. Pediatr Infect Dis J 27(5):425–430
Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, Garraud O (2015) Human platelets and their capacity of binding viruses: meaning and challenges? BMC Immunol 16:26
Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, Geier M, Stewart EA, Eisemann J, Steinkasserer A, Suzuki-Inoue K, Fuller GL, Pearce AC, Watson SP, Hoxie JA, Baribaud F, Pohlmann S (2006) DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J Virol 80(18):8951–8960
Chan V, Novakowski SK, Law S, Klein-Bosgoed C, Kastrup CJ (2015) Controlled transcription of exogenous mRNA in platelets using protocells. Angew Chem Int Ed 54(46):13590–13593
Cheng HJ, Lei HY, Lin CF, Luo YH, Wan SW, Liu HS, Yeh TM, Lin YS (2009) Anti-dengue virus nonstructural protein 1 antibodies recognize protein disulfide isomerase on platelets and inhibit platelet aggregation. Mol Immunol 47(2–3):398–406
Cognasse F, Nguyen KA, Damien P, Mcnicol A, Pozzetto B, Hamzeh-Cognasse H, Garraud O (2015) The inflammatory role of platelets via their TLRs and Siglec receptors. Front Immunol 6:83
Cosgriff TM, Lee HW, See AF, Parrish DB, Moon JS, Kim DJ, Lewis RM (1991) Platelet dysfunction contributes to the haemostatic defect in haemorrhagic fever with renal syndrome. Trans R Soc Trop Med Hyg 85(5):660–663
Cossart P, Helenius A (2014) Endocytosis of viruses and bacteria. Cold Spring Harb PerspectBiol 6(8)
Coulson BS, Londrigan SL, Lee DJ (1997) Rotavirus contains integrin ligand sequences and a disintegrin-like domain that are implicated in virus entry into cells. Proc Natl Acad Sci U S A 94(10):5389–5394
Crapnell K, Zanjani ED, Chaudhuri A, Ascensao JL, St JS, Maciejewski JP (2000) In vitro infection of megakaryocytes and their precursors by human cytomegalovirus. Blood 95(2):487–493
Cummins D, Fisher-Hoch SP, Walshe KJ, Mackie IJ, McCormick JB, Bennett D, Perez G, Farrar B, Machin SJ (1989) A plasma inhibitor of platelet aggregation in patients with Lassa fever. Br J Haematol 72(4):543–548
Cummins D, Molinas FC, Lerer G, Maiztegui JI, Faint R, Machin SJ (1990) A plasma inhibitor of platelet aggregation in patients with Argentine hemorrhagic fever. Am J Trop Med Hyg 42(5):470–475
Danon D, Jerushalmy Z, Devries A (1959) Incorporation of influenza virus in human blood platelets invitro—electron microscopical observation. Virology 9(4):719–722
de Almeidal AJ, Campos-De-Magalhdes M, Brandao-Mello CE, de Oliveira RV, Yoshida CFT, Lampe E (2007) Detection of hepatitis C virus in platelets: evaluating its relationship to viral and host factors. Hepato-Gastroenterol 54(75):964–968
de Boer SM, Kortekaas J, de Haan CAM, Rottier PJM, Moormann RJM, Bosch BJ (2012) Heparan sulfate facilitates rift valley fever virus entry into the cell. J Virol 86(24):13767–13771
Delvaeye M, Conway EM (2009) Coagulation and innate immune responses: can we view them separately? Blood 114(12):2367–2374
Derry MC, Sutherland MR, Restall CM, Waisman DM, Pryzdial ELG (2007) Annexin 2-mediated enhancement of cytomegalovirus infection opposes inhibition by annexin 1 or annexin 5. J Gen Virol 88:19–27
Dinarvand P, Hassanian SM, Qureshi SH, Manithody C, Eissenberg JC, Yang LK, Rezaie AR (2014) Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y(1) purinergic receptor. Blood 123(6):935–945
Falcinelli E, Francisci D, Belfiori B, Petito E, Guglielmini G, Malincarne L, Mezzasoma A, Sebastiano M, Conti V, Giannini S, Bonora S, Baldelli F, Gresele P (2013) In vivo platelet activation and platelet hyperreactivity in abacavir-treated HIV-infected patients. Thromb Haemost 110(2):349–357
Feldmann H, Geisbert TW (2011) Ebola haemorrhagic fever. Lancet 377(9768):849–862
Feldmann H, Klenk HD (1996) Filoviruses. In: Baron S (ed) Medical microbiology (4th edn). University of Texas Medical Branch at Galveston, Galveston, Chap. 72. Available from https://www.ncbi.nlm.nih.gov/books/NBK8129/
Flaujac C, Boukour S, Cramer-Borde E (2010) Platelets and viruses: an ambivalent relationship. Cell Mol Life Sci 67(4):545–556
Fleming FE, Graham KL, Takada Y, Coulson BS (2011) Determinants of the specificity of rotavirus interactions with the alpha2beta1 integrin. J Biol Chem 286(8):6165–6174
Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER (1999) Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta(3) integrins. J Virol 73(5):3951–3959
Gavrilovskaya IN, Gorbunova EE, Mackow ER (2010) Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J Virol 84(9):4832–4839
Ghigo E (2010) A dilemma for viruses and giant viruses: which endocytic pathway to use to enter cells? Intervirology 53(5):274–283
Ghosh K, Gangodkar S, Jain P, Shetty S, Ramjee S, Poddar P, Basu A (2008) Imaging the interaction between dengue 2 virus and human blood platelets using atomic force and electron microscopy. J Electron Microsc (Tokyo) 57(3):113–118
Giannini E, Borro P, Botta F, Fumagalli A, Malfatti F, Podesta E, Romagnoli P, Testa E, Chiarbonello B, Polegato S, Mamone M, Testa R (2002) Serum thrombopoietin levels are linked to liver function in untreated patients with hepatitis C virus-related chronic hepatitis. J Hepatol 37(5):572–577
Gibellini D, Vitone F, Buzzi M, Schiavone P, De CE, Cicola R, Conte R, Ponti C, Re MC (2007) HIV-1 negatively affects the survival/maturation of cord blood CD34(+) hematopoietic progenitor cells differentiated towards megakaryocytic lineage by HIV-1 gp120/CD4 membrane interaction. J Cell Physiol 210(2):315–324
Gibellini D, Clo A, Morini S, Miserocchi A, Ponti C, Re MC (2013) Effects of human immunodeficiency virus on the erythrocyte and megakaryocyte lineages. World J Virol 2(2):91–101
Goeijenbier M, van Wissen M, van de Weg C, Jong E, Gerdes VE, Meijers JC, Brandjes DP, van Gorp EC (2012) Review: Viral infections and mechanisms of thrombosis and bleeding. J Med Virol 84(10):1680–1696
Greenberg J, Packham MA, Cazenave JP, Reimers HJ, Mustard JF (1975) Effects on platelet function of removal of platelet sialic acid by neuraminidase. Lab Invest 32(4):476–484
Gresele P, Falcinelli E, Sebastiano M, Baldelli F (2012) Endothelial and platelet function alterations in HIV-infected patients. Thromb Res 129(3):301–308
Grozovsky R, Hoffmeister KM, Falet H (2010) Novel clearance mechanisms of platelets. Curr Opin Hematol 17(6):585–589
Gupalo E, Buriachkovskaia L, Othman M (2011) Human platelets express CAR with localization at the sites of intercellular interaction. Virol J 8:456
Gupalo E, Kuk C, Qadura M, Buriachkovskaia L, Othman M (2013) Platelet-adenovirus vs. inert particles interaction: effect on aggregation and the role of platelet membrane receptors. Platelets 24(5):383–391
Guzman MG, Alvarez M, Halstead SB (2013) Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: an historical perspective and role of antibody-dependent enhancement of infection. Arch Virol 158(7):1445–1459
Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA (1998) CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391(6667):591–594
Holm PW, Slart RH, Zeebregts CJ, Hillebrands JL, Tio RA (2009) Atherosclerotic plaque development and instability: a dual role for VEGF. Ann Med 41(4):257–264
Hottz E, Tolley ND, Zimmerman GA, Weyrich AS, Bozza FA (2011) Platelets in dengue infection. Drug Discov Today 8:e33–e38
Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza FA, Bozza PT (2013a) Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 122(20):3405–3414
Hottz ED, Oliveira MF, Nunes PC, Nogueira RM, Valls-de-Souza R, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza PT, Bozza FA (2013b) Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost 11(5):951–962
Hunt CL, Kolokoltsov AA, Davey RA, Maury W (2011) The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J Virol 85(1):334–347
Hutt-Fletcher LM (2007) Epstein-Barr virus entry. J Virol 81(15):7825–7832
Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, Chisari FV, Ruggeri ZM, Guidotti LG (2005) Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med 11(11):1167–1169
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang CY, Penninger JM (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436(7047):112–116
Jedlitschky G, Greinacher A, Kroemer HK (2012) Transporters in human platelets: physiologic function and impact for pharmacotherapy. Blood 119(15):3394–3402
Jenne CN, Kubes P (2015) Platelets in inflammation and infection. Platelets 26(4):286–292
Jin C, Liang MF, Ning JY, Gu W, Jiang H, Wu W, Zhang FS, Li C, Zhang QF, Zhu H, Chen T, Han Y, Zhang WL, Zhang S, Wang Q, Sun L, Liu QZ, Li JD, Wang T, Wei Q, Wang SW, Deng Y, Qin C, Li D (2012) Pathogenesis of emerging severe fever with thrombocytopenia syndrome virus in C57/BL6 mouse model. Proc Natl Acad Sci U S A 109(25):10053–10058
Kasper B, Petersen F (2011) Molecular pathways of platelet factor 4/CXCL4 signaling. Eur J Cell Biol 90(6–7):521–526
Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, Coughlin SR, Carmeliet P, Lina B, Rimmelzwaan GF, Planz O, Ludwig S, Riteau B (2013) PAR1 contributes to influenza A virus pathogenicity in mice. J Clin Invest 123:206–214
Kota S, Sabbah A, Chang TH, Harnack R, Xiang Y, Meng X, Bose S (2008) Role of human beta-defensin-2 during tumor necrosis factor-alpha/NF-kappaB-mediated innate antiviral response against human respiratory syncytial virus. J Biol Chem 283(33):22417–22429
Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE (2014) Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 124(5):791–802
Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, Kahr WH, Lindemann S, Seizer P, Yost CC, Zimmerman GA, Weyrich AS (2011) Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog 7(11), e1002355
Kunz S, Rojek JM, Perez M, Spiropoulou CF, Oldstone MB (2005) Characterization of the interaction of lassa fever virus with its cellular receptor alpha-dystroglycan. J Virol 79(10):5979–5987
Langer HF, Daub K, Braun G, Schonberger T, May AE, Schaller M, Stein GM, Stellos K, Bueltmann A, Siegel-Axel D, Wendel HP, Aebert H, Roecken M, Seizer P, Santoso S, Wesselborg S, Brossart P, Gawaz M (2007) Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol 27(6):1463–1470
Le VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S, Antunes L, Lina B, Bordet JC, Jandrot-Perrus M, Ludwig S, Riteau B (2015) Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med 191(7):804–819
Leikina E, Delanoe-Ayari H, Melikov K, Cho MS, Chen A, Waring AJ, Wang W, Xie Y, Loo JA, Lehrer RI, Chernomordik LV (2005) Carbohydrate-binding molecules inhibit viral fusion and entry by crosslinking membrane glycoproteins. Nat Immunol 6(10):995–1001
Li X, Jeffers LJ, Garon C, Fischer ER, Scheffel J, Moore B, Reddy KR, Demedina M, Schiff ER (1999) Persistence of hepatitis C virus in a human megakaryoblastic leukaemia cell line. J Viral Hepat 6(2):107–114
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426(6965):450–454
Li Z, Nardi MA, Karpatkin S (2005) Role of molecular mimicry to HIV-1 peptides in HIV-1-related immunologic thrombocytopenia. Blood 106(2):572–576
Liebman HA (2008) Viral-associated immune thrombocytopenic purpura. Hematol Am Soc Hematol Educ Program 2008:212–218
Lin CF, Lei HY, Liu CC, Liu HS, Yeh TM, Wang ST, Yang TI, Sheu FC, Kuo CF, Lin YS (2001) Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol 63(2):143–149
Long KM, Whitmore AC, Ferris MT, Sempowski GD, Mcgee C, Trollinger B, Gunn B, Heise MT (2013) Dendritic cell immunoreceptor regulates chikungunya virus pathogenesis in mice. J Virol 87(10):5697–5706
Madoff MA, Ebbe S, Baldini M (1964) Sialic acid of human blood platelets. J Clin Invest 43:870–877
Maugeri N, Franchini S, Campana L, Baldini M, Ramirez GA, Sabbadini MG, Rovere-Querini P, Manfredi AA (2012) Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity 45(8):584–587
Maurice A, Marchand-Arvier M, Edert D, Le FA, Gondrexon G, Vigneron C (2002) The virucidal effect of platelet concentrates: preliminary study and first conclusions. Platelets 13(4):219–222
Mayne E, Funderburg NT, Sieg SF, Asaad R, Kalinowska M, Rodriguez B, Schmaier AH, Stevens W, Lederman MM (2012) Increased platelet and microparticle activation in HIV infection: upregulation of P-selectin and tissue factor expression. J Acquir Immune Defic Syndr 59(4):340–346
Mercer J, Schelhaas M, Helenius A (2010) Virus entry by endocytosis. Annu Rev Biochem 79:803–833
Messaoudi I, Basler CF (2015) Immunological features underlying viral hemorrhagic fevers. Curr Opin Immunol 36:38–46
Morrell CN, Aggrey AA, Chapman LM, Modjeski KL (2014) Emerging roles for platelets as immune and inflammatory cells. Blood 123(18):2759–2767
Nardi M, Tomlinson S, Greco MA, Karpatkin S (2001) Complement-independent, peroxide-induced antibody lysis of platelets in HIV-1-related immune thrombocytopenia. Cell 106(5):551–561
Noisakran S, Gibbons RV, Songprakhon P, Jairungsri A, Ajariyakhajorn C, Nisalak A, Jarman RG, Malasit P, Chokephaibulkit K, Perng GC (2009) Detection of dengue virus in platelets isolated from dengue patients. Southeast Asian J Trop Med Public Health 40(2):253–262
Onlamoon N, Noisakran S, Hsiao HM, Duncan A, Villinger F, Ansari AA, Perng GC (2010) Dengue virus-induced hemorrhage in a nonhuman primate model. Blood 115(9):1823–1834
Othman M, Labelle A, Mazzetti I, Elbatarny HS, Lillicrap D (2007) Adenovirus-induced thrombocytopenia: the role of von Willebrand factor and P-selectin in mediating accelerated platelet clearance. Blood 109(7):2832–2839
Packham MA, Rand ML (2011) Historical perspective on ADP-induced platelet activation. Purinergic Signal 7(3):283–292
Pryzdial EL, Sutherland MR, Ruf W (2014) The procoagulant envelope virus surface: contribution to enhanced infection. Thromb Res 133(Suppl 1):S15–S17
Pugliese A, Gennero L, Cutufia M, Enrietto M, Morra E, Pescarmona P, Ponzetto A (2004) HCV infective virions can be carried by human platelets. Cell Biochem Funct 22(6):353–358
Quinones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, Marotta ML, Mirza M, Jiang B, Kiser P, Medvik K, Sieg SF, Weinberg A (2003) Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. Auto Immune Defic Syndr 17(16):F39–F48
Rajan SK, Espina BM, Liebman HA (2005) Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 129(6):818–824
Rand ML, Wright JF (1998) Virus-associated idiopathic thrombocytopenic purpura. Transfusion Science 19(3):253–259
Rivadeneyra L, Pozner RG, Meiss R, Fondevila C, Gomez RM, Schattner M (2015) Poly (I:C) downregulates platelet production and function through type I interferon. Thromb Haemost 114(5):982–993
Rothmeier A, Ruf W (2012) Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol 34:133–149
Saito M, Oishi K, Inoue S, Dimaano EM, Alera MT, Robles AM, Estrella BD Jr, Kumatori A, Moji K, Alonzo MT, Buerano CC, Matias RR, Morita K, Natividad FF, Nagatake T (2004) Association of increased platelet-associated immunoglobulins with thrombocytopenia and the severity of disease in secondary dengue virus infections. Clin Exp Immunol 138(2):299–303
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, Akira S (2012) Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12(1):109–116
Satchell CS, Cotter AG, O’Connor EF, Peace AJ, Tedesco AF, Clare A, Lambert JS, Sheehan GJ, Kenny D, Mallon PWG (2010) Platelet function and HIV: a case-control study. Auto Immune Defic Syndr 24(5):649–657
Sato T, Sekine H, Kakuda H, Miura N, Sunohara M, Fuse A (2000) HIV infection of megakaryocytic cell lines. Leuk Lymphoma 36(3–4):397–404
Schenk BI, Petersen F, Flad HD, Brandt E (2002) Platelet-derived chemokines CXC chemokine ligand (CXCL)7, connective tissue-activating peptide III, and CXCL4 differentially affect and cross-regulate neutrophil adhesion and transendothelial migration. J Immunol 169(5):2602–2610
Schubert S, Weyrich AS, Rowley JW (2014) A tour through the transcriptional landscape of platelets. Blood 124(4):493–502
Semple JW, Italiano JE Jr, Freedman J (2011) Platelets and the immune continuum. Nat Rev Immunol 11(4):264–274
Shimojima M, Ikeda Y, Kawaoka Y (2007) The mechanism of Axl-mediated Ebola virus infection. J Infect Dis 196(Suppl 2):S259–S263
Shimojima M, Stroher U, Ebihara H, Feldmann H, Kawaoka Y (2012) Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J Virol 86(4):2067–2078
Shimony N, Elkin G, Kolodkin-Gal D, Krasny L, Urieli-Shoval S, Haviv YS (2009) Analysis of adenoviral attachment to human platelets. Virol J 6:25
Simon AY, Sutherland MR, Pryzdial ELG (2015) Dengue virus binding and replication by platelets. Blood 126:378–85
Singh MV, Davidson DC, Jackson JW, Singh VB, Silva J, Ramirez SH, Maggirwar SB (2014) Characterization of platelet-monocyte complexes in HIV-1-infected individuals: possible role in HIV-associated neuroinflammation. J Immunol 192(10):4674–4684
Sloutskin A, Yee MB, Kinchington PR, Goldstein RS (2014) Varicella-Zoster virus and herpes simplex virus 1 can infect and replicate in the same neurons whether co-or superinfected. J Virol 88(9):5079–5086
Smith SA, Morrissey JH (2014) Polyphosphate: a new player in the field of hemostasis. Curr Opin Hematol 21(5):388–394
Solomon TT, Gnirss K, Rahe-Meyer N, Kiene M, Kramer-Kuhl A, Behrens G, Munch J, Pohlmann S (2013) Platelet activation suppresses HIV-1 infection of T cells. Retrovirology 10:48
Sorensen AL, Rumjantseva V, Nayeb-Hashemi S, Clausen H, Hartwig JH, Wandall HH, Hoffmeister KM (2009) Role of sialic acid for platelet life span: exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood 114(8):1645–1654
Spear PG (2004) Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6(5):401–410
Sridharan A, Chen Q, Tang KF, Ooi EE, Hibberd ML, Chen J (2013) Inhibition of megakaryocyte development in the bone marrow underlies dengue virus-induced thrombocytopenia in humanized mice. J Virol 87(21):11648–11658
Stevenson SC, Rollence M, MarshallNeff J, McClelland A (1997) Selective targeting of human cells by a chimeric adenovirus vector containing a modified fiber protein. J Virol 71(6):4782–4790
Stone D, Liu Y, Shayakhmetov D, Li ZY, Ni SH, Lieber A (2007) Adenovirus-platelet interaction in blood causes virus sequestration to the reticuloendothelial system of the liver. J Virol 81(9):4866–4871
Sutherland MR, Ruf W, Pryzdial ELG (2012) Tissue factor and glycoprotein C on herpes simplex virus type 1 are protease-activated receptor 2 cofactors that enhance infection. Blood 119:3638–3645
Sutherland MR, Simon AY, Serrano K, Schubert P, Acker JP, Pyzdial ELG (2016) Dengue virus persists and replicates during storage of platelet and red blood cell units. Transfusion 56(5):1129–37
Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, Eller MA, Pattanapanyasat K, Sarasombath S, Birx DL, Steinman RM, Schlesinger S, Marovich MA (2003) DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med 197(7):823–829
Tohidnezhad M, Varoga D, Podschun R, Wruck CJ, Seekamp A, Brandenburg LO, Pufe T, Lippross S (2011) Thrombocytes are effectors of the innate immune system releasing human beta defensin-3. Injury 42(7):682–686
Tohidnezhad M, Varoga D, Wruck CJ, Podschun R, Sachweh BH, Bornemann J, Bovi M, Sonmez TT, Slowik A, Houben A, Seekamp A, Brandenburg LO, Pufe T, Lippross S (2012) Platelets display potent antimicrobial activity and release human beta-defensin 2. Platelets 23(3):217–223
Triantafilou K, Triantafilou M (2001) A biochemical approach reveals cell-surface molecules utilised by Picornaviridae: Human Parechovirus 1 and Echovirus 1. J Cell Biochem 80(3):373–381
Triantafilou K, Triantafilou M, Takada Y, Fernandez N (2000) Human parechovirus 1 utilizes integrins alpha v beta 3 and alpha v beta 1 as receptors. J Virol 74(13):5856–5862
Vanderstocken G, Bondue B, Horckmans M, Di Pietrantonio L, Robaye B, Boeynaems JM, Communi D (2010) P2Y(2) receptor regulates VCAM-1 membrane and soluble forms and eosinophil accumulation during lung inflammation. J Immunol 185(6):3702–3707
Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, Zaki SR, Swanepoel R, Ansari AA, Peters CJ (1999) Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J Infect Dis 179(Suppl 1):S188–S191
Wang J, Zhang W, Nardi MA, Li Z (2011) HIV-1 Tat-induced platelet activation and release of CD154 contribute to HIV-1-associated autoimmune thrombocytopenia. J Thromb Haemost 9(3):562–573
Wat JM, Foley JH, Krisinger MJ, Ocariza LM, Lei V, Wasney GA, Lameignere E, Strynadka NC, Smith SA, Morrissey JH, Conway EM (2014) Polyphosphate suppresses complement via the terminal pathway. Blood 123(5):768–776
Weksler BB (2007) Review article: the pathophysiology of thrombocytopenia in hepatitis C virus infection and chronic liver disease. Aliment Pharmacol Ther 26(Suppl 1):13–19
Wenzel F, Gruber W, Giers G (2010) Alteration of serum thrombopoietin levels in patients with chronic hepatitis C under interferon therapy. Clin Hemorheol Microcirc 44(2):137–144
Wickham TJ, Mathias P, Cheresh DA, Nemerow GR (1993) Integrin-alpha-v-beta-3 and integrin-alpha-V-beta-5 promote adenovirus internalization but not virus attachment. Cell 73(2):309–319
Wolins N, Lozier J, Eggerman TL, Jones E, Aguilar-Cordova E, Vostal JG (2003) Intravenous administration of replication-incompetent adenovirus to rhesus monkeys induces thrombocytopenia by increasing in vivo platelet clearance. Br J Haematol 123(5):903–905
Yang KD, Lee CS, Hwang KP, Chu ML, Shaio MF (1995) A model to study cytokine profiles in primary and heterologously secondary dengue-2 virus-infections. Acta Virol 39(1):19–21
Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM (2002) Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 99(11):4021–4029
Zahn A, Allain JP (2005) Hepatitis C virus and hepatitis B virus bind to heparin: purification of largely IgG-free virions from infected plasma by heparin chromatography. J Gen Virol 86:677–685
Zahn A, Jennings N, Ouwehand WH, Allain JP (2006) Hepatitis C virus interacts with human platelet glycoprotein VI. J Gen Virol 87(Pt 8):2243–2251
Zapata JC, Cox D, Salvato MS (2014) The role of platelets in the pathogenesis of viral hemorrhagic fevers. PLoS Negl Trop Dis 8(6), e2858
Zarbock A, Polanowska-Grabowska RK, Ley K (2007) Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev 21(2):99–111
Zhang Y, Bergelson JM (2005) Adenovirus receptors. J Virol 79(19):12125–12131
Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S (2009) Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 113(17):4086–4093
Zhu ZL, Gershon MD, Ambron R, Gabel C, Gershon AA (1995) Infection of cells by varicella-zoster virus—inhibition of viral entry by mannose 6-phosphate and heparin. Proc Natl Acad Sci U S A 92(8):3546–3550
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Pryzdial, E.L.G., Lin, B.H., Sutherland, M.R. (2017). Virus–Platelet Associations. In: Gresele, P., Kleiman, N., Lopez, J., Page, C. (eds) Platelets in Thrombotic and Non-Thrombotic Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-47462-5_72
Download citation
DOI: https://doi.org/10.1007/978-3-319-47462-5_72
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47460-1
Online ISBN: 978-3-319-47462-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)