
Abstract
In this paper, we propose a novel image processing method, DeepHCS, to transform bright-field microscopy images into synthetic fluorescence images of cell nuclei biomarkers commonly used in high-content drug screening. The main motivation of the proposed work is to automatically generate virtual biomarker images from conventional bright-field images, which can greatly reduce time-consuming and laborious tissue preparation efforts and improve the throughput of the screening process. DeepHCS uses bright-field images and their corresponding cell nuclei staining (DAPI) fluorescence images as a set of image pairs to train a series of end-to-end deep convolutional neural networks. By leveraging a state-of-the-art deep learning method, the proposed method can produce synthetic fluorescence images comparable to real DAPI images with high accuracy. We demonstrate the efficacy of this method using a real glioblastoma drug screening dataset with various quality metrics, including PSNR, SSIM, cell viability correlation (CVC), the area under the curve (AUC), and the IC50.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
- High Content Screening (HCS)
- Bright Field Image
- Convolution Neural Network
- Cell Nuclei Staining
- DAPI Fluorescence Image
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
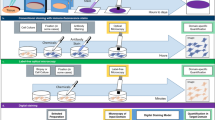
A glioblastoma (GBM) is a brain tumor that is commonly found in the cerebral hemisphere of the brain. GBM is considered an obstinate brain tumor because even after medical advances in the past few decades, no effective treatment has been discovered that greatly improves life expectancy in patients. When patients are diagnosed with a GBM, in most cases, the best treatment option is surgery to eliminate as many tumor cells as possible. In addition to surgical treatments, patient-specific chemotherapy by analyzing patient-driven GBM tumor cells to find the most effective drug for the target patient, called precision medicine, has become popular. High-throughput screening (HTS) and high-content screening (HCS) have demonstrated their effectiveness in precision medicine in recent studies [2, 8]. Both approaches for precision medicine involve readouts of various drug responses to patient-derived cell cultures. Among them, HCS uses high-throughput imaging and automatic image analysis to evaluate changes in the phenotype of the whole cells, such as counting the number of living cells versus dead cells, measuring the size of the cells, comparing the shape of the cells, etc. In HCS, multiple imaging modalities are commonly used together to image various aspects of the cell phenotypes (Fig. 1). Such imaging modalities include bright-field and fluorescence microscopy, in which the former can capture the overall morphology of the cells, while the latter can image various fluorescent biomarkers. One advantage of using bright-field images in HCS is its ability to acquire a photographic record of cells without any extra preparation while fluorescence images require time-consuming cell fixation and staining procedures. Another advantage of the bright-field image method is its ability to capture the dynamics of cells because cell fixation and cell staining are not required (Fig. 1 lower row). However, fluorescence imaging can capture only a snapshot of the cells at any given point in time because cells die during fixation and staining (Fig. 1 upper row).

DeepHCS eliminates the cell fixation and staining progress in the original HCS workflow and generates corresponding fluorescence image based bright-field image by Operetta. DeepHCS can keep the cells alive during the entire progress.
There have been many research efforts to develop image processing techniques for bright-field imaging to extract cell phenotypes without fluorescence imaging. Selinummi et al. [9] used multi-focal bright-field images to extract the shape of cells without whole cell fluorescence images. Their method calculates the intensity variation along the z-stack of multi-focal bright-field images to robustly detect cell boundaries. Ali et al. [1] proposed detection and segmentation of adherent HT1080 and HeLa cells from bright-field images. This method extracts local phase and local orientation from multi-focal bright-field images using the monogenic single framework to guide the evolution of the active contour. Tikkanen et al. [10] employed a machine learning approach using the histogram of oriented gradient (HOG) [3] feature for detecting cells in 25 focal images. The extracted features and their neighboring intensity histograms are combined for classification using a support vector machine (SVM). Liimatainen et al. [6] employed a logistic regression with a \(\ell _{1}\) penalty to classify the location of cells and non-cells using the intensity values from 25 focal images as features. However, most previous work focused only on cell segmentation and detection directly from bright-field images, and no state-of-the-art deep learning methods are leveraged. In addition, in spite of ongoing research efforts in bright-field image analysis, the standard HCS workflow still relies on detecting and analyzing biomarkers presented in fluorescence images.
Based on these observations, we propose DeepHCS, a novel data-driven image conversion technique for high-content screening. Unlike most existing methods that directly analyze bright-field images, DeepHCS converts bright-field images to fluorescence images as accurately as possible using end-to-end convolutional neural networks. By doing this, DeepHCS effectively avoids the time-consuming and laborious cell preparation process for generating biomarkers while providing accurate image analysis results by using the well-established conventional HCS workflow (Fig. 1 bottom row). We evaluate the accuracy of DeepHCS using widely used image quality metrics (e.g., PSNR and SSIM). In addition, we compare cell viability [4], the area under curve (AUC) and the IC50 of the results and real DAPI images to demonstrate that DeepHCS can replace the tissue preparation and fluorescence imaging process in the conventional HCS workflow with the software-based image conversion process.

Overview of data acquisition and preprocessing: (a) layout of a 384-well plate, (b) nine overlapped images for a single well, (c) before and after stitching images (yellow line on the left is the border between adjacent images)
2 Method
2.1 Data
We acquired the image data from patients who had been diagnosed with a GBM brain tumor. The GBM tumor cells were divided evenly into a 384-well plate organized into a \(24\times 16\) grid (Fig. 2a) and stained with Hoechst 33342 solution. For drug screening, biologists added various FDA-approved drugs into the wells. Each drug was administered to a \(1\times 8\) column, starting with a 20 \(\upmu \)/mol dosage and tripling the dosage in each subsequent well (green box of Fig. 2a). The last wells in the \(1\times 8\) column contained no drugs and were used as a control (blue box of Fig. 2a). Each well was imaged with the Operetta CLS™ high-content analysis system equipped with an high resolution 14bit CCD camera for cell imaging and the Harmony 3.5 software. Nine-field image montage per well (Fig. 2b) is generated by using an image stitching algorithm (Fig. 2c). The resolution of each image is \(1360\times 1024\) pixels. We took images from various locations with different drug dosages and evenly distributed cells, and made pairs for training set in which each pair consists of a bright-field image and its fluorescence nuclei image.

DeepHCS consists of two sub-networks: a Transformation Network (green box); and a Refinement Network (pink box). Convolution layers (blue layer) include ReLU as a non-linear function. Residual blocks (purple layer) consist of three identical convolution layers. All filter sizes used in this system are \(3\times 3\).
2.2 Proposed Method: DeepHCS
DeepHCS is built upon two deep neural networks, Transformation Network (TN) and Refinement Network (RN) (see Fig. 3).
Transformation Network is the first part of DeepHCS, consisting of two sets of FusionNet variant networks [7]. The first network in the TN is used to gradually transform the input bright-field image into the intermediate feature image, and the second network in the TN is used to actually perform the translation into the DAPI fluorescence image. The first network in the TN can effectively expand the depth of the network when the size of the input is relatively small, and adequately performs drop-out in which 16 feature maps are merged into one feature map at the end. The second network in the TN has more neuron weights by using residual blocks and actually translates the input image into DAPI fluorescence images. The number of filters in the entire network is expressed under each layer in Fig. 3.

Refinement Network improves the cell shapes and restores missing cells. A false positive (top row) and a false negative (bottom row) from the TN are corrected by the RN.
Refinement Network is the second part of DeepHCS and is designed to improve the image quality of the translated result from the TN in terms of the noise and the cell shape. In contrast to the TN, the RN takes a concatenation of the translated TN result and the input bright-field image of the TN, which provides a clue to rectify errors in the translated image generated by the TN. For example, as shown in Fig. 4, the RN can improve the cell shapes and restore falsely removed cells. Another benefit of using the concatenated input image is to help reducing the gradient-vanishing problem caused by the black background in the translated result by the TN.
Loss Function. For the TN, the mean-square error (MSE) is used to define the loss function to measure the pixel-wise error between the ground truth and the output image of the TN, as follows:
where x is the input bright-field image, \(y_{i}\) is the real fluorescence image, and \(\hat{y}_{i}\) is the output of the TN. For the RN, the mean-absolute error (MAE) and the SSIM are used as the loss function to deal with the shape of cells and the pixel intensity at the same time. The MAE is define as follows:
where \(\hat{x}\) is the concatenation of the translated result of the TN and the input bright-field image, and \(r_{i}\) is the output of the RN. In contrast to the TN, we employed the MAE to handle the translated result of the TN because the MSE penalizes larger errors and is more tolerant to smaller errors. The SSIM is defined as follows:
where \(\mu _x\) and \(\sigma _x\) represent the mean and the variance of image x, respectively; \(\sigma _{xy}\) represents the covariance of image x and y, and \(c_1\) and \(c_2\) are two constant variables for division stability. Based on Eq. 3, we can measure the degree of structural change in the image and additionally recognize the difference between the two images based on luminance and contrast. The SSIM values range between 0 and 1; therefore, we defined the loss function using the SSIM as follows:
By combining the two error measures, we can define the loss function for the RN as follows (\(\alpha \) is empirically set to 0.8):
3 Results
We used the training set consisting of 2,860 pairs of bright-field images and their corresponding fluorescence images, each measuring \(256\times 256\) pixels (we cropped the center of each image to reduce boundary effects). To validate DeepHCS, we used eight cases (C1 to C8), including either 1,386 or 2,646 images.

(a) Ground truth fluorescence image, (b) the result of our method. Zoom-in shows the similarity of the cell shapes between the ground truth and ours.
To assess the quality of the images generated by DeepHCS, we used two image error metrics (PSNR and SSIM) and cell viability correlation (CVC) that measures the similarity between the actual and generated DAPI fluorescence images using \(R^2\) correlation, as shown in Table 1. In the experiment, we achieved an average of 0.9092 and a maximum of 0.9702 correlation with the ground truth. In addition, the shape of the cells and the status of the cells (living or dead) are clearly distinguished as shown in Fig. 5.
To further demonstrate the feasibility of DeepHCS for replacing biomarker generation in the conventional HCS workflow, we used seven other cases for the validation test. Figure 6 shows the correlation of real DAPI images and our synthetic fluorescence images in terms of AUC and IC50, respectively. In addition, the responses of two anti-cancer drugs (AMG232 and RG7112) measured by AUC and IC50 are also shown using heatmaps; clear separation of two groups in drug responses are identically shown in DAPI images and ours. These results confirm that the images generated by DeepHCS can be used to compute AUC and IC50 for the estimation of drug responses, which shows potential to replace the conventional fluorescence imaging process in the HSC workflow.

Comparison of AUC and IC50 values from the real DAPI images (ground truth) and our results from the seven patients’ data. The heat maps show the drug response (green is low, and red is high).
Finally, we compared DeepHCS with the latest GAN-based image translation method used in the Pix2Pix network [5]. As shown in Fig. 7, DeepHCS reproduces cell images close to real DAPI images while Pix2Pix fails to accurately generate cell shapes in some cases. The GAN attempts to approximate the data distribution of the training set as much as possible; therefore, the reconstructed images look like cell images. However, this does not imply that the reconstructed image satisfies the accuracy up to the HCS analysis. Even though Pix2Pix learns the morphological characteristics in the cell image by minimizing the \(\ell _{1}\) distance from the ground truth, it is not enough to satisfy this problem.

(a) Ground truth fluorescence image, (b) the results of the proposed method, (c) the results of the Pix2Pix network [5]. The results are generated after 300 training epochs. Our method can generate most of cell structures close to the ground truth.
4 Conclusion
In this paper, we introduced DeepHCS, a novel deep end-to-end convolution neural network for generating DAPI fluorescence images directly from bright-field images. We showed that the DeepHCS can generate results similar to real DAPI images and outperforms state-of-the-art image translation methods. The proposed method demonstrates the potential to reduce the laborious biomarker preparation process and to improve the throughput of the large-scale image-based drug screening process using deep learning. In the future, we plan to apply the proposed method to time-lapse bright-field images, and assess the efficacy of generating other biomarker images.
References
Ali, R., Gooding, M., Szilágyi, T., Vojnovic, B., Christlieb, M., Brady, M.: Automatic segmentation of adherent biological cell boundaries and nuclei from brightfield microscopy images. Mach. Vis. Appl. 23(4), 607–621 (2012)
Boutros, M., Heigwer, F., Laufer, C.: Microscopy-based high-content screening. Cell 163(6), 1314–1325 (2015)
Dalal, N., Triggs, B.: Histograms of oriented gradients for human detection. In: 2005 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR 2005), vol. 1, pp. 886–893. IEEE (2005)
Darzynkiewicz, Z., Li, X., Gong, J.: Assays of cell viability: discrimination of cells dying by apoptosis. Methods Cell Biol. 41, 15–38 (1994)
Isola, P., Zhu, J.Y., Zhou, T., Efros, A.A.: Image-to-image translation with conditional adversarial networks. arXiv preprint arXiv:1611.07004 (2016)
Liimatainen, K., Ruusuvuori, P., Latonen, L., Huttunen, H.: Supervised method for cell counting from bright field focus stacks. In: 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI), pp. 391–394. IEEE (2016)
Quan, T.M., Hilderbrand, D.G., Jeong, W.K.: FusionNet: a deep fully residual convolutional neural network for image segmentation in connectomics. arXiv preprint arXiv:1612.05360 (2016)
Quartararo, C.E., Reznik, E., DeCarvalho, A.C., Mikkelsen, T., Stockwell, B.R.: High-throughput screening of patient-derived cultures reveals potential for precision medicine in glioblastoma. ACS Med. Chem. Lett. 6(8), 948–952 (2015)
Selinummi, J., et al.: Bright field microscopy as an alternative to whole cell fluorescence in automated analysis of macrophage images. PloS One 4(10), e7497 (2009)
Tikkanen, T., Ruusuvuori, P., Latonen, L., Huttunen, H.: Training based cell detection from bright-field microscope images. In: 2015 9th International Symposium on Image and Signal Processing and Analysis (ISPA), pp. 160–164. IEEE (2015)
Acknowledgements
This work was partially supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1D1A1A09000841) and Bio & Medical Technology Development Program of the National Research Foundation of Korea (NRF) funded by MSIT (NRF-2015M3A9A7029725).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this paper

Cite this paper
Lee, G., Oh, JW., Kang, MS., Her, NG., Kim, MH., Jeong, WK. (2018). DeepHCS: Bright-Field to Fluorescence Microscopy Image Conversion Using Deep Learning for Label-Free High-Content Screening. In: Frangi, A., Schnabel, J., Davatzikos, C., Alberola-López, C., Fichtinger, G. (eds) Medical Image Computing and Computer Assisted Intervention – MICCAI 2018. MICCAI 2018. Lecture Notes in Computer Science(), vol 11071. Springer, Cham. https://doi.org/10.1007/978-3-030-00934-2_38
Download citation
DOI: https://doi.org/10.1007/978-3-030-00934-2_38
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-00933-5
Online ISBN: 978-3-030-00934-2
eBook Packages: Computer ScienceComputer Science (R0)