
Abstract
Halomonas zhanjiangensis Chen et al. 2009 is a member of the genus Halomonas, family Halomonadaceae, class Gammaproteobacteria. Representatives of the genus Halomonas are a group of halophilic bacteria often isolated from salty environments. The type strain H. zhanjiangensis JSM 078169T was isolated from a sea urchin (Hemicentrotus pulcherrimus) collected from the South China Sea. The genome of strain JSM 078169T is the fourteenth sequenced genome in the genus Halomonas and the fifteenth in the family Halomonadaceae. The other thirteen genomes from the genus Halomonas are H. halocynthiae, H. venusta, H. alkaliphila, H. lutea, H. anticariensis, H. jeotgali, H. titanicae, H. desiderata, H. smyrnensis, H. salifodinae, H. boliviensis, H. elongata and H stevensii. Here, we describe the features of strain JSM 078169T, together with the complete genome sequence and annotation from a culture of DSM 21076T. The 4,060,520 bp long draft genome consists of 17 scaffolds with the 3,659 protein-coding and 80 RNA genes and is a part of Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG) project.
Similar content being viewed by others

Introduction
Strain JSM 078169T (= DSM 21076 = KCTC 22279 = CCTCC AB 208031) is the type strain of the species Halomonas zhanjiangensis [1], one out of 84 species with a validly published name in the genus Halomonas [2], family Halomonadaceae [3]. The family Halomonadaceae currently comprises thirteen genera (Aidingimonas, Carnimonas, Chromohalobacter, Cobetia, Halomonas, Halotalea, Halovibrio, Kushneria, Marinospirillum, Modicisalibacter, Candidtus Portiera, Salinicola and Zymobacter) with Halomonas being the largest genus in this family [3–6]. Members of the genus Halomonas have been isolated from various saline environments and showed halophilic characteristics [7–11]. Strain JSM 078169T was originally isolated from a sea urchin (Hemicentrotus pulcherrimus) that was collected from the South China Sea. The genus name was derived from the Greek words ‘halos’ meaning ‘salt’ and ‘monas’ meaning ‘monad’, yielding the Neo-Latin word ‘halomonas’ [2]; the species epithet was derived from Latin word ‘zhanjiangensis’, of Zhanjiang, a city in China near where the sample was collected [1]. Strain JSM 078169T was found to assimilate several mono- and disaccharides and to produce numerous acid and alkaline phosphatases, leucine arylamidase, naphthol-ASBI-phosphohydrolase and valine arylamidase [1]. There are no PubMed records that document the use of these strain for any biotechnological studies; only comparative analyses performed for the description of later members of the genus Halomonas are recorded. However, the NamesforLife [12] database reports at least 70 patents in which Halomonas ssp. are referenced. Here we present a summary classification and a set of feature for H. zhanjiangensis JSM 078169T, together with the description of the genomic sequencing and annotation of DSM 21076.
Classification and features
16S rRNA analysis
The original assembly of the genome did not contain longer stretches of 16S rRNA copies. Therefore, a 1,413 bp long fragment of the 16S rRNA gene was later patched into the genome sequence assembly. This almost full length version of the 16S rRNA sequence was compared using NCBI BLAST [13,14] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent version of the Greengenes database [15] and the relative frequencies of taxa and unidentified clones (or strains) were calculated by BLAST scores. The most frequently occurring genus was Halomonas (74.8%), and the unidentified clones or isolates represented 25.5% for the total BLAST results. Except for sequences of representatives of the genus Halomonas, no sequences from other genera were observed in the BLAST search. The highest degree of sequence similarity was reported with H. alkantarctica str. CRSS.
Figure 1 shows the phylogenetic neighborhood of H. zhanjiangensis JSM 078169T in a tree based on 16S rRNA genes. The 1,413 bp long sequence fragment of the 16S rRNA gene differs by three nucleotides from the previously published 16S rRNA sequence (FJ429198). The tree provided a precise insight into the nomenclature and classification of members of the genus Halomonas. The phylogenetic analysis showed that strain H. zhanjiangensis JSM 078169T was most closely related to H. nanhaiensis YIM M 13059T with 98.3% sequence similarity.

Phylogenetic tree highlighting the position of H. zhanjiangensis relative to the closest related type strains of the other species within the family Halomonadaceae. All the 16S rRNA gene sequences of the type strains within the genus Halomonas were included and combined with the representative 16S rRNA gene sequences of the type species in other genera, according to the most recent release of the EzTaxon database. The tree was inferred from 1,381 aligned characters [16] under the neighbor-joining (NJ) [17], and maximum-likelihood (ML) [18] method with 1,000 randomly selected bootstrap replicates using MEGA version 5.2 [19]. The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 1,000 NJ bootstrap (left) and from 1,000 ML bootstrap (right) replicates [20] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [21] are labeled with one asterisk, those also listed as ‘Complete and Published’ with two asterisks [22].
Morphology and physiology
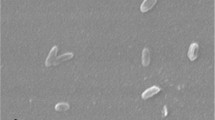
H. zhanjiangensis JSM 078169T is a Gram-negative-staining, non-sporulating, strictly aerobic (Table 1), catalase-positive, oxidase-negative and slightly halophilic bacterium that reduces nitrate [1]. Cells of JSM 078169T are short rods (0.4–0.7 µm × 0.6–1.0 µm) and motile with peritrichous flagella (not visible in Figure 2). Colonies are yellow-pigmented, flat and non-translucent with glistening surfaces and circular/slightly irregular margins, 2–3 mm in diameter after incubation on Marine Agar (MA) at 28 °C for 3–5 days. No diffusible pigments are produced. Growth occurs at 4–40 °C with an optimum growth at 25–30 °C, at pH range of 6.0–10.5 with an optimum pH of 7.5. The salinity range suitable for growth was 1.0–20.0% (w/v) total salts with an optimum between 3.0–5.0% (w/v) total salts. No growth occurs in the absence of NaCl or with NaCl as the sole salt. Strain JSM 078169T grows on Marine Agar and the medium contained the following: 5.0 g peptone, 1.0 g yeast extract, 0.1 g ferric citrate, 19.45 g NaCl, 8.8 g MgCl2, 3.24 g Na2SO4, 1.8 g CaCl2, 0.55 g KCl, 0.16 g NaHCO3, 0.08 g KBr, 0.034 g SrCl2, 0.022 g H3BO3, 0.004 g sodium silicate, 0.0024 g sodium fluoride, 0.0016 g ammonium nitrate, 0008 g disodium phosphate and 15 g agar.

Scanning electron micrograph of H. zhanjiangensis JSM 078169T
Chemotaxonomy
The predominant respiratory quinone is Q-9 which is consistent to the other members of the genus Halomonas [1]. The predominant fatty acids are C18:1 ω 7 c (48.9%), C16:0 (17.0%) and C12:0 3-OH (10.7%). The profile of major fatty acids is also similar to the other representatives of the genus Halomonas [38–41].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [42,43]. Sequencing strain JSM 078169T is part of Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG) project [44], a follow-up of the GEBA project [45], which aims in increasing the sequencing coverage of key reference microbial genomes. The genome project is deposited in the Genomes OnLine Database [21] and the permanent draft genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI) using state of the art sequencing technology [46]. A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
H. zhanjiangensis JSM 078169T, DSM 21076, was grown in DSMZ medium 1510 (modified medium 514 for Halomonas sp.) [47] at 28 °C. DNA was isolated from 0.5–1.0 g of cell paste using MasterPure Gram-positive DNA purification kit (Epicentre MGP04100) following the standard protocol as recommended by the manufacturer with modification st/DL for cell lysis as described by Wu et al. [45]. DNA is available through the DNA Bank Network [48].
Genome sequencing and assembly
The draft genome sequence was generated using the Illumina technology [49]. An Illumina Standard shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform which generated 15,593,002 reads totaling 2,339.0 Mbp. All general aspects of library construction and sequencing performed at the JGI can be found at [50]. All raw Illumina sequence data was passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artifacts [51]. Following steps were then performed for assembly: (1) filtered Illumina reads were assembled using Velvet [52], (2) 1–3 kbp simulated paired end reads were created from Velvet contigs using wgsim [53], (3) Illumina reads were assembled with simulated read pairs using Allpaths-LG [54]. Parameters for assembly steps were: 1) Velvet (velveth: 63 –shortPaired and velvetg: –very clean yes –export-Filtered yes –min contig lgth 500 –scaffolding no –cov cutoff 10) 2) wgsim (–e 0 –1 100 –2 100 –r 0 –R 0 –X 0) 3) Allpaths-LG (PrepareAllpathsInputs: PHRED 64=1 PLOIDY=1 FRAG COVERAGE=125 JUMP COVERAGE=25 LONG JUMP COV=50, RunAllpathsLG: THREADS=8 RUN=std shredpairs TARGETS=standard VAPI WARN ONLY=True OVERWRITE=True). The final draft assembly contained 18 contigs in 17 scaffolds. The total size of the genome is 4.1 Mbp and the final assembly is based on 501.3 Mbp of Illumina data, which provides an average 123.5 × coverage of the genome.
Genome annotation
Genes were identified using Prodigal [55] as part of the DOE-JGI genome annotation pipeline [56], following by a round of manual curation using the JGI GenePRIMP pipeline [57]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro database. These data sources were combined to assert a product description for each predicted protein. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes-Expert Review (IMG-ER) platform [58].
Genome properties
The assembly of the draft genome sequence consists of 17 scaffolds amounting to 4,060,520 bp, and the G+C content is 54.5% (Table 3 and Figure 3). Of the 3,739 genes predicted, 3,659 were protein-coding genes, and 80 RNAs. The majority of the protein-coding genes (87.1%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4 and Figure 3.

The graphical map of the largest scaffold of the genome. From bottom to the top: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNA green, rRNA red, other RNAs black), GC content, GC skew (purple/olive).
Insights into the genome sequence
One complete genome sequence from a type strain of the family Halomonas - H. elongata [22] is available in GenBank, and four other permanent draft genomes of H. anticariensis, H. lutea, H. jeotgali and H. halocynthiae are available from IMG. The genome size of H. zhanjiangensis is smaller than those of H. elongata, H. lutea and H. anticariensis (4.06–5.02 Mbp), but much larger than those of H. jeotgali and H. halocynthiae (2.85–2.88 Mbp). Using the genome-to-genome distance calculator [59–61] version 2.0 revealed that all digital DNA-DNA hybridization (DDH) values are much lower than 70% using the program NCBI-BLAST, which demonstrated that H. zhanjiangensis is distinct from H. elongata, H. anticariensis, H. lutea, H. jeotgali and H. halocynthiae at the species level. Distance is 0.1845 between the type strain genomes of H. zhanjiangensis and H. elongata, which corresponds to a DDH value of 13.00 ± 2.99%. The distances of H. zhanjiangensis from H. anticariensis, H. lutea, H. jeotgali and H. halocynthiae are 0.1842, 0.1837, 0.1835 and 01849, which correspond to DDH values of 20.30 ±2.41%, 20.30 ±2.41%, 20.40 ±2.41% and 20.20 ±2.41%, respectively.
A major feature of the previously sequenced genomes from this family is the presence of large numbers of proteins for the TRAP-type C4-dicarboxylate transport systems. A total of 267 genes in the genome of H. zhanjiangensis encode proteins for carbohydrate transport and metabolism, 68 genes are related to TRAP-type C4-dicarboxylate transport systems and encoded 22 large permease proteins, 24 periplasmic proteins and 22 small permease proteins. Genomic analysis of H. elongata, H. anticariensis, H. lutea, H. jeotgali and H. halocynthiae showed that they encode 58, 65, 61, 7 and 32 proteins related to TRAP-type C4-dicarboxylate transport system respectively. Proteins for TRAP-type C4-dicarboxylate transport systems constitute 1.86% as the total protein-coding sequences of the H. zhanjiangensis genome. In the genomes of H. elongata, H. anticariensis, H. lutea, H. jeotgali and H. halocynthiae, TRAP-type C4-dicarboxylate transport system related proteins are accounted for 1.67%, 1.37%, 1.42%, 0.27% and 1.18% of the total protein-coding genes respectively. Therefore, H. zhanjiangensis has the highest percentage of TRAP-type C4-dicarboxylate transport system related encoding proteins in this group of bacteria to date.
Of the signal transduction mechanisms, Methyl-accepting Chemotaxis Proteins (MCPs) are transmembrane sensor proteins of bacteria. The MCPs allow bacteria to detect concentrations of molecules in the extracellular matrix so that they may smoothly swim or tumble accordingly [62,63]. Various environmental conditions give rise to diversity in bacterial signaling receptors, and consequently there are many genes encoding MCPs [64]. A number of MCPs (23) are present in H. zhanjiangensis, while H. elongata, H. anticariensis, H. lutea, and H. jeotgali have only 4, 21, 16, and 17 MCPs, respectively. MCPs are not found in the genome of H. halocynthiae. H. zhanjiangensis has the largest numbers of MCPs in this family. The analysis of bacterial genomes reveals that the family Halomonadaceae differs enormously in the number of MCPs from E. coli, and the number of MCPs in Halomonadaceae is about two times than that of E. coli strains.
References
Chen YG, Zhang YQ, Huang HY, Klenk HP, Tang SK, Huang K, Chen QH, Cui XL, Li WJ. Halomonas zhanjiangensis sp. nov., a halophilic bacterium isolated from a sea urchin. Int J Syst Evol Microbiol 2009; 59:2888–2893. PubMed http://dx.doi.org/10.1099/ijs.0.010173-0
Vreeland R, Litchfield CD, Martin EL, Elliot E. Halomonas elongata, a new genus and species of extremely salt-tolerant bacteria. Int J Syst Evol Microbiol 1980; 30:485–495.
Franzmann PD, Wehmeyer U, Stackebrandt E. Halomonadaceae fam. nov., a new family of the class Proteobacteria to accommodate the genera Halomonas and Deleya. Syst Appl Microbiol 1988; 11:16–19. http://dx.doi.org/10.1016/S0723-2020(88)80043-2
Anzai Y, Kim H, Park JY, Wakabayashi H, Oyaizu H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int J Syst Evol Microbiol 2000; 50:1563–1589. PubMed http://dx.doi.org/10.1099/00207713-50-4-1563
Arahal DR, Ludwig W, Schleifer KH, Ventosa A. Phylogeny of the family Halomonadaceae based on 23S and 16S rDNA sequence analyses. Int J Syst Evol Microbiol 2002; 52:241–249. PubMed
Mellado E, Moore ERB, Nieto JJ, Ventosa A. Phylogenetic inferences and taxonomic consequences of 16S ribosomal DNA sequence comparison of Chromohalobacter marismortui, Volcaniella eurihalina, and Deleya salina and reclassification of V. eurihalina as Halomonas eurihalina comb. nov. Int J Syst Evol Microbiol 1995; 45:712–716. PubMed
Cabrera A, Aguilera M, Fuentes S, Incerti C, Russell NJ, Ramos-Cormenzana A, Monteoliva-Sanchez M. Halomonas indalinina sp. nov., a moderately halophilic bacterium isolated from a solar saltern in Cabo de Gata, Almería, southern Spain. Int J Syst Evol Microbiol 2007; 57:376–380. PubMed http://dx.doi.org/10.1099/ijs.0.64702-0
Kim KK, Jin L, Yang HC, Lee ST. Halomonas gomseomensis sp. nov., Halomonas janggokensis sp. nov., Halomonas salaria sp. nov. and Halomonas denitrificans sp. nov., moderately halophilic bacteria isolated from saline water. Int J Syst Evol Microbiol 2007; 57:675–681. PubMed http://dx.doi.org/10.1099/ijs.0.64767-0
Soto-Ramírez N, Sánchez-Porro C, Rosas S, González W, Quiñones M, Ventosa A, Montalvo-Rodríguez R. Halomonas avicenniae sp. nov., isolated from the salty leaves of the black mangrove Avicennia germinans in Puerto Rico. Int J Syst Evol Microbiol 2007; 57:900–905. PubMed http://dx.doi.org/10.1099/ijs.0.64818-0
Ventosa JJ. Nieto, Oren A. Biology of moderately halophilic aerobic bacteria. Microbiol Mol Biol Rev 1998; 62:504–544. PubMed
Wang YN, Cai H, Chi CQ, Lu AH, Lin XG, Jiang ZF, Wu XL. Halomonas shengliensis sp. nov., a moderately halophilic, denitrifying, crude-oilutilizing bacterium. Int J Syst Evol Microbiol 2007; 57:1222–1226. PubMed http://dx.doi.org/10.1099/ijs.0.64973-0
Garrity GM. Names for Life Browser Tool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403–410. PubMed http://dx.doi.org/10.1016/S0022-2836(05)80360-2
Korf I, Yandell M, Bedell J. BLAST, O’Reilly, Sebastopol, 2003.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed http://dx.doi.org/10.1128/AEM.03006-05
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed http://dx.doi.org/10.1093/bioinformatics/18.3.452
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987; 4:406–425. PubMed
Strimmer K, von Haeseler A. Quartet puzzling: a quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol 1996; 13:964–969. http://dx.doi.org/10.1093/oxfordjournals.molbev.a025664
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731–2739. PubMed http://dx.doi.org/10.1093/molbev/msr121
Pattengale ND, Alipour M, Bininda-Emonds OR, Moret BM, Stamatakis A. How many bootstrap replicates are necessary? [doi:10.1089/cmb.2009.0179]. [pmid:20377449]. J Comput Biol 2010; 17:337–354.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40:D571–D579. PubMed http://dx.doi.org/10.1093/nar/gkr1100
Schwibbert K, Marin-Sanguino A, Bagyan I, Heidrich G, Lentzen G, Seitz H, Rampp M, Schuster SC, Klenk HP, Pfeiffer F, et al. A blueprint of ectoine metabolism from the genome of the industrial producer Halomonas elongata DSM 2581T. Environ Microbiol 2011; 13:1973–1994. PubMed http://dx.doi.org/10.1111/j.1462-2920.2010.02336.x
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, Gilbert J, Glöckner FO, Hirschman L, Karsch-Mzrachi I, et al. PLoS Biol 2011; 9:e1001088. PubMed http://dx.doi.org/10.1371/journal.pbio.1001088
Euzéby JP. List of bacterial names with standing in nomenclature: A folder available on the Internet. Int J Syst Bacteriol 1997; 47:590–592. PubMed http://dx.doi.org/10.1099/00207713-47-2-590
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Garrity GM, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Springer, New York, 2005.
Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class. nov. In: Brenner DJ, Krieg NR, Staley JT and Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1.
Validation of publication of new names and new combinations previously effectively published outside the IJSEM. List no. 106. Int J Syst Evol Microbiol 2005; 55:2235–2238. http://dx.doi.org/10.1099/ijs.0.64108-0
Williams KP, Kelly DP. Proposal for a new class within the phylum Proteobacteria, Acidithiobacillia classis nov., with the type order Acidithiobacillales, and emended description of the class Gammaproteobacteria. Int J Syst Evol Microbiol 2013; 63:2901–2906. PubMed http://dx.doi.org/10.1099/ijs.0.049270-0
Garrity GM, Bell JA, Lilburn T. Order VIII. Oceanospirillales ord. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, second edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria), Springer, New York, 2005, p. 270.
Validation List no. 29. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1989; 39:205–206. http://dx.doi.org/10.1099/00207713-39-2-205
Dobson SJ, Franzmann PD. Unification of the genera Deleya (Baumann et al. 1983), Halomonas (Vreeland et al. 1980), and Halovibrio (Fendrich 1988) and the species Paracoccus halodenitrificans (Robinson and Gibbons 1952) into a single genus, Halomonas, and placement of the genus Zymobacter in the family Halomonadaceae. Int J Syst Bacteriol 1996; 46:550–558. http://dx.doi.org/10.1099/00207713-46-2-550
Ntougias S, Zervakis GI, Fasseas C. Halotalea alkalilenta gen. nov., sp. nov., a novel osmotolerant and alkalitolerant bacterium from alkaline olive mill wastes, and emended description of the family Halomonadaceae Franzmann et al. 1989, emend. Dobson and Franzmann 1996. Int J Syst Evol Microbiol 2007; 57:1975–1983. PubMed http://dx.doi.org/10.1099/ijs.0.65078-0
Ben Ali Gam Z, Abdelkafi S, Casalot L, Tholozan JL, Oueslati R, Labat M. Modicisalibacter tunisiensis gen. nov., sp. nov., an aerobic, moderately halophilic bacterium isolated from an oilfield-water injection sample, and emended description of the family Halomonadaceae Franzmann et al. 1989 emend Dobson and Franzmann 1996 emend. Ntougias et al. 2007. Int J Syst Evol Microbiol 2007; 57:2307–2313. PubMed http://dx.doi.org/10.1099/ijs.0.65088-0
Dobson SJ, Franzmann PD. Unification of the genera Deleya (Baumann et al. 1983), Halomonas (Vreeland et al. 1980), and Halovibrio (Fendrich 1988) and the species Paracoccus halodenitrificans (Robinson and Gibbons 1952) into a single genus, Halomonas, and placement of the genus Zymobacter in the family Halomonadaceae. Int J Syst Bacteriol 1996; 46:550–558. http://dx.doi.org/10.1099/00207713-46-2-550
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Berendes F, Gottschalk G, Heine-Dobbernack E, Moore ERB, Tindall BJ. Halomonas desiderata sp. nov, a new alkaliphilic, halotolerant and denitrifying bacterium isolated from a municipal sewage works. Syst Appl Microbiol 1996; 19:158–167. http://dx.doi.org/10.1016/S0723-2020(96)80041-5
Romano I, Gottschalk G, Heine-Dobbernack E, Moore ERB, Tindall BJ. Characterization of a haloalkalophilic strictly aerobic bacterium, isolated from Pantelleria island. Syst Appl Microbiol 1996; 19:326–333. http://dx.doi.org/10.1016/S0723-2020(96)80059-2
Heyrman J, Balcaen A, De Vos P, Swings J. Halomonas muralis sp. nov., isolated from microbial biofilms colonizing the walls and murals of the Saint-Catherine chapel (Castle Herberstein, Austria). Int J Syst Evol Microbiol 2002; 52:2049–2054. PubMed http://dx.doi.org/10.1099/ijs.0.02166-0
Jeon CO, Lim JM, Lee JR, Lee GS, Park DJ, Lee JC, Oh HW, Kim CJ. Halomonas kribbensis sp. nov., a novel moderately halophilic bacterium isolated from a solar saltern in Korea. Int J Syst Evol Microbiol 2007; 57:2194–2198. PubMed http://dx.doi.org/10.1099/ijs.0.65285-0
Klenk HP, Göker M. En route to a genome — based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed http://dx.doi.org/10.1016/j.syapm.2010.03.003
Göker M, Klenk HP. Phylogeny-driven target selection for genome-sequencing (and other) projects. Stand Genomic Sci 2013; 8:360–374. PubMed http://dx.doi.org/10.4056/sigs.3446951
Kyrpides NC, Woyke T, Eisen JA, Garrity G, Lilburn TG, Beck BJ, Whitman WB, Hugenholz P, Klenk HP. Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG-I) project. Stand Genomic Sci 2013; 9:628–634. http://dx.doi.org/10.4056/sigs.5068949
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed http://dx.doi.org/10.1038/nature08656
Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS ONE 2012; 7:e48837. PubMed http://dx.doi.org/10.1371/journal.pone.0048837
List of growth media used at DSMZ. http://www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011; 9:51–55. http://dx.doi.org/10.1089/bio.2010.0029
Bennett S. Solexa Ltd. Pharmacogenomics 2004; 5:433–438. PubMed http://dx.doi.org/10.1517/14622416.5.4.433
The DOE Joint Genome Institute. http://www.jgi.doe.gov
Mingkun L, Copeland A, Han J. DUK, unpublished, 2011.
Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed http://dx.doi.org/10.1101/gr.074492.107
Gnerre S, MacCallum I. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA 2011; 108:1513–1518. PubMed http://dx.doi.org/10.1073/pnas.1017351108
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiati on site identification. BMC Bioinformatics 2010; 11:119. PubMed http://dx.doi.org/10.1186/1471-2105-11-119
Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009; 1:63–67. PubMed http://dx.doi.org/10.4056/sigs.632
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A. Kyrp ides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed http://dx.doi.org/10.1093/bioinformatics/btp393
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013; 14:60. PubMed http://dx.doi.org/10.1186/1471-2105-1460
Auch AF, von Jan M, Klenk HP, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2010; 2:117–134. PubMed http://dx.doi.org/10.4056/sigs.531120
Auch AF, Klenk HP, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2010; 2:142–148. PubMed http://dx.doi.org/10.4056/sigs.541628
Derr P, Boder E, Goulian M. Changing the specificity of a bacterial chemoreceptor. J Mol Biol 2006; 355:923–932. PubMed http://dx.doi.org/10.1016/j.jmb.2005.11.025
Alexander RP, Zhulin IB. Evolutionary genomics reveals conserved structural determinants of signaling and adaptation in microbial chemoreceptors. Proc Natl Acad Sci USA 2007; 104:2885–2890. PubMed http://dx.doi.org/10.1073/pnas.0609359104
Harrison DM, Skidmore J, Armitage JP, Maddock JR. Localization and environmental regulation of MCP-like proteins in Rhodobacter sphaeroides. Mol Microbiol 1999; 31:885–892. PubMed http://dx.doi.org/10.1046/j.1365-2958.1999.01226.x
Acknowledgements
The authors gratefully acknowledge the assistance of Susanne Schneider for growing H. zhanjiangensis cultures and Evelyne-Marie Brambilla for DNA extraction and quality control (both at the DSMZ). This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344 Genome analysis was supported by the National Basic Research Program of China (No. 2010CB833801). W-J Li was also supported by ‘Hundred Talents Program’ of the Chinese Academy of Sciences. A.L. was supported in part by Russian Ministry of Science Mega-grant no.11.G34.31.0068 (PI. Dr Stephen J O’Brien)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhou, Y., Li, R., Gao, XY. et al. High quality draft genome sequence of the slightly halophilic bacterium Halomonas zhanjiangensis type strain JSM 078169T (DSM 21076T) from a sea urchin in southern China. Stand in Genomic Sci 9, 1020–1030 (2014). https://doi.org/10.4056/sigs.5449586
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.5449586