
Abstract
Brachybacterium faecium Collins et al. 1988 is the type species of the genus, and is of phylogenetic interest because of its location in the Dermabacteraceae, a rather isolated family within the actinobacterial suborder Micrococcineae. B. faecium is known for its rod-coccus growth cycle and the ability to degrade uric acid. It grows aerobically or weakly anaerobically. The strain described in this report is a free-living, nonmotile, Gram-positive bacterium, originally isolated from poultry deep litter. Here we describe the features of this organism, together with the complete genome sequence, and annotation. This is the first complete genome sequence of a member of the actinobacterial family Dermabacteraceae, and the 3,614,992 bp long single replicon genome with its 3129 protein-coding and 69 RNA genes is part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others


Introduction
Strain Schefferle 6–10T (DSM 4810 = ATCC 43885 = JCM 11609 = NCIMB 9860) is the type strain of Brachybacterium faecium, which is the type species of the genus Brachybacterium [1] (Figure 1). B. faecium was described by Collins et al. in 1988 [1] as Gram-positive and nonmotile. The organism is of significant interest for its position in the tree of life where the rapidly growing genus Brachybacterium (12 species) is located in the actinobacterial family Dermabacteraceae [2].
Only two accompanying strains (Schefferle 3–8 = NCIMB 9859, and Schefferle 7–11 = NCIMB 9861) were initially reported from the poultry deep litter from which the type strain Schefferle 6–10T was isolated [7]. Both strains were later reclassified as members of other Brachybacterium species: B. conglomeratum (NCIMB 9859) and B. paraconglomeratum (NCIMB 9861). Some closely related isolates with more than 98.5% 16S rRNA gene sequence identity were reported from marine sediments (JH107; FJ572028), deep-sea sediments (PB10; DQ643203), and living room air (Gauze_W_12_19; FJ267545). Only four sequences from uncultured bacteria are accessible via EMBL, showing at least 98% sequence identity to B. faecium. These sequences were derived from the analyses of urban aerosols (DQ129569), human vaginal epithelium (AY959187), floor dust (FM872846), and water 20m downstream of manure (AY212613). No phylotypes from environmental screening or genomic surveys could be linked to B. faecium (as of February 2009). Here we present a summary classification and a set of features for B. faecium Schefferle 6–10T (Table 1), together with the description of the complete genomic sequencing and annotation.

Phylogenetic tree of B. faecium Schefferle 6–10T and all type strains of the genus Brachybacterium, inferred from 1408 aligned characters [2] of the 16S rRNA sequence under the maximum likelihood criterion [5,6]. The tree was rooted with Dermabacter hominis, another member of the family Dermabacteraceae. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 1000 bootstrap replicates, if larger than 60%. Strains with a genome-sequencing project registered in GOLD [7] are printed in blue; published genomes in bold.
Classification and features
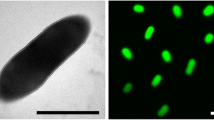
B. faecium Schefferle 6–10T cells vary in shape and exhibit a rod-coccus growth cycle, which is not atypical of this evolutionary group. Cells in the stationary phase are predominantly coccoid (Figure 2), whereas cells in fresh cultures are irregular, slender rods [1]. Cells are frequently arranged at an angle to give V-formations [1] (Figure 2). B. faecium cells are non-acid fast and do not form endospores [1]. B. faecium is essentially aerobic, but is also capable of very weak growth under anaerobic conditions [1].

Scanning electron micrograph of B. faecium Schefferle 6–10T
B. faecium is capable of degrading uric acid, and fermenting cellobiose, glucose, maltose, and mannose, but not cellulose, chitin, or gelatin. The optimal growth temperature is 25–30°C. Nitrate is reduced to nitrite by some B. faecium strains [1] as a candidate for terminal electron acceptor during anaerobic growth.
Figure 1 shows the phylogenetic neighborhood of B. faecium strain Schefferle 6–10T in a 16S rRNA based tree. The sequences of the three 16S rRNA genes in the B. faecium Schefferle 6–10T genome differ by up to two nucleotides (nts) from each other, and by three nts from the reference sequence of strain DSM 4810 (X91032). The slight differences between the genome data and the previously reported 16S rRNA gene sequence is most likely due to sequencing errors in the previously reported sequence data.
Chemotaxonomy
Strain Schefferle 6–10T was originally described as a coryneform bacterium. This descriptive term applies to a diverse range of taxa and indicates that the comparisons made in the original publication need to be reviewed. The murein of B. faecium contains meso-diaminopimelic acid, alanine and glutamic acid. The strain possesses a type A4γ peptidoglycan, type A31.2 according to the German Collection of Microorganisms and Cell Cultures. Galactose and glucose are the cell wall sugars [1]. As in other Brachybacterium strains, the fatty acid pattern of strain Schefferle 6–10T is dominated by branched-chain saturated anteiso- (ai-) fatty acids: ai-C15:0 (40%), ai-C17:0 (37%), and C16:0 and iso-C16:0 7.5%, each, with smaller amounts of iso-C15:0 (3.5%), iso-C17:0 (2.0%) [1]. Straight chain and unsaturated fatty acids are absent [1]. As usual for most members of the Actinomycetales, mycolic acids were not reported [1]. A menaquinone with seven isoprene units (MK-7) predominates (88%) complemented by 11% MK-8 [1]. Phosphatidylglycerol and diphosphatidylglycerol were identified as the dominant polar lipids, together with several glycolipids and an unknown phospholipid [1]. The Rf values of the glycolipids suggest that they contain different numbers of sugars (one, two or possibly three) and may also show differences in the nature and linkage of the sugars. It is not known whether these glycolipids are based on a diglyceride or whether they contain an acylated sugar, directly linked to a monoglyceride. The chemical composition is typical of members of the genus Brachybacterium and similar, but not identical with the members of the only other genus placed in the family Dermabacteraceae, Dermabacter. In addition to cytochrome aa3, B. faecium possesses cytochrome d626, and cytochrome c550 [8].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position, and is part of the Genomic Encyclopedia of Bacteria and Archaea project. The genome project is deposited in the Genomes OnLine Database [7] and the complete genome sequence in GenBank (CP001643). Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
B. faecium Schefferle 6–10T, DSM 4810, was grown in DSMZ medium 92 (with 3% trypticase soy broth, 0.3% yeast extract) at 28°C. DNA was isolated from 1–1.5 g of cell paste using Qiagen Genomic 500 DNA Kit (Qiagen, Hilden, Germany) without modification of the manufacturer’s protocol for cell lysis.
Genome sequencing and assembly
The genome was sequenced using a combination of Sanger, 454 and Illumina sequencing platforms. All general aspects of library construction and sequencing performed at the JGI can be found on the JGI website. 454 Pyrosequencing reads were assembled using the Newbler assembler version 1.1.02.15 (Roche). Large Newbler contigs were broken into 4,074 overlapping fragments of 1,000 bp and entered into the assembly as pseudo-reads. The sequences were assigned quality scores based on Newbler consensus q-scores with modifications to account for overlap redundancy and to adjust inflated q-scores. A hybrid 454/Sanger assembly was made using the PGA assembler. Possible mis-assemblies were corrected and gaps between contigs were closed by custom primer walks from sub-clones or PCR products. 258 Sanger finishing reads were produced. Illumina reads were used to improve the final consensus quality using an in-house developed tool (the Polisher). The error rate of the completed genome sequence is less than 1 in 100,000. Together all sequence types provided 50x coverage of the genome.
Genome annotation
Genes were identified using GeneMark [12] as part of the genome annotation pipeline in the Integrated Microbial Genomes Expert Review (IMG-ER) system [13], followed by a round of manual curation using the JGI GenePRIMP pipeline [14]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. The tRNAScanSE tool [15] was used to find tRNA genes, whereas ribosomal RNAs were found by using the tool RNAmmer [16]. Other non-coding RNAs were identified by searching the genome for the Rfam profiles using INFERNAL (v0.81) [17]. Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes (IMG) platform [18].
Metabolic network analysis
The metabolic Pathway/Genome Database (PGDB) was computationally generated using Pathway Tools software version 12.5 [19] and MetaCyc version 12.5 [20], based on annotated EC numbers and a customized enzyme name mapping file. It has undergone no subsequent manual curation and may contain errors, similar to a Tier 3 BioCyc PGDB [21].
Genome properties
The genome is 3,614,992 bp long and comprises one circular chromosome with a 72.1% GC content (Table 3 and Figure 3). Of the 3,198 genes predicted, 3,129 were protein coding genes, and 69 RNAs. Sixty pseudogenes were also identified. The majority of genes (77.3%) of the genes were assigned with a putative function while the remaining ones are annotated as hypothetical proteins. The properties and the statistics of the genome are summarized in Table 3. The distribution of genes into COGs functional categories is presented in Table 4, and a cellular overview diagram is presented in Figure 4, followed by a summary of metabolic network statistics shown in Table 5.

Graphical circular map of the genome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.

Schematic cellular overview of all pathways of the B. faecium strain Schefferle 6–10T metabolism. Nodes represent metabolites, with shape indicating class of metabolite. Lines represent reactions.
References
Collins M, Brown J, Jones D. Brachybacterium faecium gen. nov., sp. nov., a coryneform bacterium from poultry deep litter. Int J Syst Bacteriol 1988; 38:45–48 doi:10.1099/00207713-38-1-45
Stackebrandt E, Rainey F, Ward-Rainey N. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol 1997; 47:479–491 doi:10.1099/00207713-47-2-479
Schefferle H. Coryneform bacteria in poultry deep litter. J Appl Bacteriol 1966; 29:147–160 doi:10.1111/j.1365-2672.1966.tb03462.x
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464 P-MID:11934745 doi:10.1093/bioinformatics/18.3.452
Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 1981; 17:368–376 PMID:7288891 doi:10.1007/BF01734359
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 2008; 57:758–771 PMID:18853362 doi:10.1080/10635150802429642
Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2008; 36:D475–479 PMID:17981842 doi:10.1093/nar/gkm884
Schleifer K, Lang K. Close relationship among strains of Micrococcus conglomeratus and Arthrobacter sp. FEMS Microbiol 1980; 9:223–226 doi:10.1111/j.1574-6968.1980.tb05641.x
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547 PMID:18464787 doi:10.1038/nbt1360
Anonymous. Biological Agents: Technical rules for biological agents. <www.baua.de>.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29 PMID:10802651 doi:10.1038/75556
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 2001; 29:2607–2618 PMID:11410670 doi:10.1093/nar/29.12.2607
Markowitz V, Mavromatis K, Ivanova N, Chen I-M, Chu K, Palaniappan K, Szeto E, Anderson I, Lykidis A, Kyrpides N. Expert Review of Functional Annotations for Microbial Genomes. Bioinformatics 2009; in press.
Pati A. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes (In preparation 2009).
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955–964 PMID:9023104 doi:10.1093/nar/25.5.955
Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007; 35:3100–3108 PMID:17452365 doi:10.1093/nar/gkm160
Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005; 33:D121–D124 PMID:15608160 doi:10.1093/nar/gki081
Markowitz VM, Szeto E, Palaniappan K, Grechkin Y, Chu K, Chen IM, Dubchak I, Anderson I, Lykidis A, Mavromatis K, et al. The integrated microbial genomes (IMG) system in 2007: data content and analysis tool extensions. Nucleic Acids Res 2008; 36:D528–D533 PMID:17933782 doi:10.1093/nar/gkm846
Karp P, Paley S, Romero P. The pathway tools software. Bioinformatics 2000; 18:S225–232
Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 2008; 36:D623–631 PMID:17965431 doi:10.1093/nar/gkm900
Karp PD, Ouzounis CA, Moore-Kochlacs C, Goldovsky L, Kaipa P, Ahren D, Tsoka S, Darzentas N, Kunin V, Lopez-Bigas N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res 2005; 33:6083–6089 PMID:16246909 doi:10.1093/nar/gki892
Acknowledgements
We gratefully acknowledge the help of Gabriele Gehrich-Schröter for growing B. faecium cultures and Susanne Schneider for DNA extraction and quality analysis (both at DSMZ). This work was performed under the auspices of the US Department of Energy’s Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, as well as German Research Foundation (DFG) INST 599/1-1.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Lapidus, A., Pukall, R., LaButtii, K. et al. Complete genome sequence of Brachybacterium faecium type strain (Schefferle 6–10T). Stand in Genomic Sci 1, 3–11 (2009). https://doi.org/10.4056/sigs.492
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.492