
Abstract
Mesorhizobium opportunistum strain WSM2075T was isolated in Western Australia in 2000 from root nodules of the pasture legume Biserrula pelecinus that had been inoculated with M. ciceri bv. biserrulae WSM1271. WSM2075T is an aerobic, motile, Gram negative, non-spore-forming rod that has gained the ability to nodulate B. pelecinus but is completely ineffective in N2 fixation with this host. This report reveals that the genome of M. opportunistum strain WSM2075T contains a chromosome of size 6,884,444 bp, encoding 6,685 protein-coding genes and 62 RNA-only encoding genes. The genome contains no plasmids, but does harbor a 455.7 kb genomic island from Mesorhizobium ciceri bv. biserrulae WSM1271 that has been integrated into a phenylalanine-tRNA gene.
Similar content being viewed by others


Introduction
Biserrula pelecinus L. is an autogamous annual legume species that is common, though never dominant, on coarse textured and acidic Mediterranean soils [1] and can often be found with other annual legumes including subterranean clover (Trifolium subterraneum) and serradella (Ornithopus) [2]. This reseeding legume was introduced to Western Australia in 1993 in a pasture legume breeding and selection program that sought to develop new pasture legume options for the sandy surfaced duplex, acidic soils in Western Australia, to improve soil fertility and farming system flexibility [1]. At the time of introduction, the Australian resident rhizobial populations were not capable of nodulating B. pelecinus [1,3] and a Mediterranean strain Mesorhizobium ciceri bv. biserrulae WSM1271 had to be used as an inoculant to establish an effective nitrogen fixing symbiosis. After 6 years of cultivation of B. pelecinus under field conditions, an isolate (designated WSM2075) was recovered from root nodules of plants grown near Northam, Western Australia that displayed an ineffective symbiotic phenotype [4]. Accumulated evidence revealed that WSM2075 had gained the ability to nodulate (but not fix with) B. pelecinus by acquiring symbiotic genes from the original inoculant strain Mesorhizobium ciceri bv. biserrulae WSM1271 following a lateral gene transfer event [5]. Strain WSM2075 has now been designated as strain WSM2075T (= LMG 24607 = HAMBI 3007) and is the type strain for a new species described as Mesorhizobium opportunistum [6]. The species name op.por.tu.nis’tum. L. neut. adj. opportunistum reflects the opportunistic behavior of the organism to nodulate a range of legume hosts by acquiring symbiotic genes [4,5]. M. opportunistum WSM2075T is competitive for nodulation of B. pelecinus but cannot fix nitrogen [4] and the finding of such strains that have rapidly evolved in the soil presents a threat to the successful establishment of this valuable pasture species in Australia [5].
Here we present a summary classification and a set of general features for M. opportunistum strain WSM2075T together with the description of the complete genome sequence and annotation. Here we reveal that a 455.7 kb genomic island from the inoculant Mesorhizobium ciceri bv. biserrulae WSM1271 has been horizontally transferred into M. opportunistum strain WSM2075T and integrated into the phenylalanine-tRNA gene.
Classification and general features
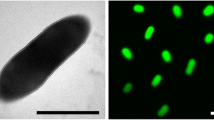
M. opportunistum strain WSM2075T is a motile, Gram-negative, non-spore-forming rod (Figure 1A and Figure 1B in the order Rhizobiales of the class Alphaproteobacteria. They are moderately fast growing, forming 2–4 mm diameter colonies within 3–4 days and have a mean generation time of 4–6 h when grown in half Lupin Agar (½LA) broth [7] at 28°C. Colonies on ½LA are white-opaque, slightly domed, moderately mucoid with smooth margins (Figure 1C).

Image of Mesorhizobium opportunistum strain WSM2075T using scanning electron microscopy
Strains of this organism are able to tolerate a pH range between 5.5 and 9.0. Carbon source utilization and fatty acid profiles have been described previously [6]. Minimum Information about the Genome Sequence (MIGS) is provided in Table 1.

Image of Mesorhizobium opportunistum strain WSM2075T using transmission electron microscopy

Image of Mesorhizobium opportunistum strain WSM2075T colony morphology on a solid medium.
Figure 2 shows the phylogenetic neighborhood of Mesorhizobium opportunistum strain WSM2075T in a 16S rRNA sequence based tree. This strain clusters in a tight group which included M. amorphae, M. huakuii, M. plurifarium and M. septentrionale and has >99% sequence identity with all four type strains. However, based on a polyphasic taxonomic study we have identified that this strain belongs to a new species [6].

Phylogenetic tree showing the relationships of Mesorhizobium opportunistum WSM2075T with other root nodule bacteria in the order Rhizobiales based on aligned sequences of the 16S rRNA gene (1,290 bp internal region). All positions containing gaps and missing data were eliminated. Phylogenetic analyses were performed using MEGA, version 3.1 [20]. The tree was built using the Maximum-Likelihood method with the General Time Reversible model and bootstrap analysis [21] with 500 replicates to construct a consensus tree. Type strains are indicated with a superscript T. Brackets after the strain name contain a DNA database accession number and/or a GOLD ID (beginning with the prefix G) for a sequencing project registered in GOLD [22]. Published genomes are indicated with an asterisk.
Symbiotaxonomy
M. opportumistum strain WSM2075T forms an ineffective (non-N fixing) symbiosis with its original host of isolation, B. pelecinus L., as well as with Astragalus adsurgens, A. membranaceus, Lotus peregrinus and Macroptilium atropurpureum [4,6]. In all cases the root nodules formed are small, white and seem incapable of fixing nitrogen [6]. Strain WSM2075T has a broader host range for nodulation than Mesorhizobium ciceri bv. biserrulae WSM1271 [6].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its environmental and agricultural relevance to issues in global carbon cycling, alternative energy production, and biogeochemical importance, and is part of the Community Sequencing Program at the U.S. Department of Energy, Joint Genome Institute (JGI) for projects of relevance to agency missions. The genome project is deposited in the Genomes OnLine Database [22] and the complete genome sequence in GenBank. Sequencing, finishing and annotation were performed by the JGI. A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
M. opportunistum strain WSM2075T was grown to mid logarithmic phase in TY rich medium [23] on a gyratory shaker at 28°C. DNA was isolated from 60 mL of cells using a CTAB (Cetyl trimethyl ammonium bromide) bacterial genomic DNA isolation method [24].
Genome sequencing and assembly
The genome of Mesorhizobium opportunistum WSM2075T was sequenced at the Joint Genome Institute (JGI) using a combination of Illumina [25] and 454 technologies [26]. An Illumina GAii shotgun library comprising 370 Mb in reads of 36 bases, a 454 Titanium library with read length of 480–495 bases containing approximately 1.05 million reads, and a paired end 454 library containing 63840 reads with average insert size of 39 Kb were generated for this genome. All general aspects of library construction and sequencing performed at the JGI can be found at [24]. Illumina sequencing data was assembled with VELVET [27], and the consensus sequences were shredded into 1.5 Kb overlapped fake reads and assembled together with the 454 data. Draft assemblies were based on 375 Mb 454 standard data, and all of the 454 paired end data. Newbler parameters used were ‘-consed -a 50 -l 350 -g -mi 96 -ml 96’. The initial Newbler assembly contained 44 contigs in 1 scaffold. We converted the initial 454 assembly into a phrap assembly by making fake reads from the consensus, collecting the read pairs in the 454 paired end library. The Phred/Phrap/Consed software package was used for sequence assembly and quality assessment [28–30] in the subsequent finishing process. Illumina data was used to correct potential base errors and increase consensus quality using software developed at JGI (Polisher, Alla Lapidus, unpublished). After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Gaps were closed in silico using software developed at JGI (gapResolution, unpublished), and mis-assemblies were corrected using Dupfinisher [31], or sequencing cloned bridging PCR fragments. Remaining gaps between contigs were manually closed by editing in Consed, by PCR, and by Bubble PCR primer walks. A total of 464 additional reactions and 3 shatter libraries were necessary to close all gaps and to improve the quality of the finished sequence.
Genome annotation
Genes were identified using Prodigal [32] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePrimp pipeline [33]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Non-coding genes and miscellaneous features were predicted using tRNAscan-SE [34], RNAMMer [35], Rfam [36], TMHMM [37], and SignalP [38]. Additional gene prediction analyses and functional annotation were performed within the Integrated Microbial Genomes (IMG-ER) platform [39].
Genome properties
The genome is 6,884,444 nucleotides with 62.87% GC content (Table 3) and comprised of a single chromosome and no plasmids. From a total of 6,747 genes, 6,685 were protein encoding and 62 RNA only encoding genes. Within the genome, 177 pseudogenes were also identified. The majority of genes (71.11%) were assigned a putative function while the remaining genes were annotated as hypothetical. The distribution of genes into COGs functional categories is presented in Table 4 and Figure 3.

Graphical circular map of the chromosome of Mesorhizobium opportunistum WSM2075T. From outside to the center: Genes on forward strand (color by COG categories as denoted by the IMG platform), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, sRNAs red, other RNAs black), GC content, GC skew.
References
Howieson JG, Loi A, Carr SJ. Biserrula pelecinus L. — a legume pasture species with potential for acid, duplex soils which is nodulated by unique root-nodule bacteria. Aust J Agric Res 1995; 46:997–1009. http://dx.doi.org/10.1071/AR9950997
Hackney B, Dear B, Crocker G. Biserrula. In: Industries DoP, editor New South Wales 2007. p 1–5.
Nandasena KG, O’Hara GW, Tiwari RP, Yates RJ, Kishinevsky BD, Howieson JG. Symbiotic relationships and root nodule ultrastructure of the pasture legume Biserrula pelecinus L. - a new legume in agriculture. Soil Biol Biochem 2004; 36:1309–1317. http://dx.doi.org/10.1016/j.soilbio.2004.04.012
Nandasena KG, O’Hara GW, Tiwari RP, Sezmis E, Howieson JG. In situ lateral transfer of symbiosis islands results in rapid evolution of diverse competitive strains of mesorhizobia suboptimal in symbiotic nitrogen fixation on the pasture legume Biserrula pelecinus L. Environ Microbiol 2007; 9:2496–2511. PubMed http://dx.doi.org/10.1111/j.1462-2920.2007.01368.x
Nandasena KG, O’Hara GW, Tiwari RP, Howieson JG. Rapid in situ evolution of nodulating strains for Biserrula pelecinus L. through lateral transfer of a symbiosis island from the original mesorhizobial inoculant. Appl Environ Microbiol 2006; 72:7365–7367. PubMed http://dx.doi.org/10.1128/AEM.00889-06
Nandasena KG, O’Hara GW, Tiwari RP, Willems A, Howieson JG. Mesorhizobium australicum sp. nov. and Mesorhizobium opportunistum sp. nov., isolated from Biserrula pelecinus L. in Australia. Int J Syst Evol Microbiol 2009; 59:2140–2147. PubMed http://dx.doi.org/10.1099/ijs.0.005728-0
Howieson JG, Ewing MA, D’antuono MF. Selection for acid tolerance in Rhizobium meliloti. Plant Soil 1988; 105:179–188. http://dx.doi.org/10.1007/BF02376781
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen M, Angiuoli SV, et al. Towards a richer description of our complete collection of genomes and metagenomes “Minimum Information about a Genome Sequence” (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1.
Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. In: Garrity GM, Brenner DJ, Kreig NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second ed: New York: Springer-Verlag; 2005.
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6. PubMed http://dx.doi.org/10.1099/ijs.0.64188-0
Kuykendall LD. Order VI. Rhizobiales ord. nov. In: Garrity GM, Brenner DJ, Kreig NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second ed: New York: Springer-Verlag; 2005. p 324.
Mergaert J, Swings J. Family IV. Phyllobacteriaceae In: Garrity GM, Brenner DJ, Kreig NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second ed: New York: Springer-Verlag; 2005. p 393.
Jarvis BDW, Van Berkum P, Chen WX, Nour SM, Fernandez MP, Cleyet-Marel JC, Gillis M. Transfer of Rhizobium loti, Rhizobium huakuii, Rhizobium cicer, Rhizobium mediterraneum, Rhizobium tianshanense to Mesorhizobium gen.nov. Int J Syst Evol Microbiol 1997; 47:895–898. http://dx.doi.org/10.1099/00207713-47-3-895
Chen WX, Wang ET, Kuykendall LD. The Proteobacteria. New York: Springer-Verlag; 2005.
Agents B. Technical rules for biological agents. TRBA (http://www.baua.de):466.
Nandasena KG, O’Hara GW, Tiwari RP, Willlems A, Howieson JG. Mesorhizobium cicer biovar biserrulae, a novel biovar nodulating the pasture legume Biserrula pelecinus L. Int J Syst Evol Microbiol 2007; 57:1041–1045. PubMed http://dx.doi.org/10.1099/ijs.0.64891-0
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Kumar S, Tamura K, Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 2004; 5:150–163. PubMed http://dx.doi.org/10.1093/bib/5.2.150
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 1985; 39:783–791. http://dx.doi.org/10.2307/2408678
Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2008; 36:D475–D479. PubMed http://dx.doi.org/10.1093/nar/gkm884
Reeve WG, Tiwari RP, Worsley PS, Dilworth MJ, Glenn AR, Howieson JG. Constructs for insertional mutagenesis, transcriptional signal localization and gene regulation studies in root nodule and other bacteria. Microbiology 1999; 145:1307–1316. PubMed http://dx.doi.org/10.1099/13500872-145-6-1307
DOE Joint Genome Institute. http://my.jgi.doe.gov/general/index.html
Bennett S. Solexa Ltd. Pharmacogenomics 2004; 5:433–438. PubMed http://dx.doi.org/10.1517/14622416.5.4.433
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:376–380; 10.1038/nature03959. PubMed
Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Current Protocols in Bioinformatics 2010; Chapter 11:Unit 11 5.
Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998; 8:175–185. PubMed http://dx.doi.org/10.1101/gr.8.3.175
Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998; 8:175–185. PubMed http://dx.doi.org/10.1101/gr.8.3.175
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998; 8:195–202. PubMed http://dx.doi.org/10.1101/gr.8.3.195
Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Valafar HRAH, editor. Proceeding of the 2006 international conference on bioinformatics & computational biology: CSREA Press; 2006. p 141–146.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed http://dx.doi.org/10.1186/1471-2105-11-119
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955–964. PubMed
Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007; 35:3100–3108. PubMed http://dx.doi.org/10.1093/nar/gkm160
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439–441. PubMed http://dx.doi.org/10.1093/nar/gkg006
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305:567–580. PubMed http://dx.doi.org/10.1006/jmbi.2000.4315
Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004; 340:783–795. PubMed http://dx.doi.org/10.1016/j.jmb.2004.05.028
Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed http://dx.doi.org/10.1093/bioinformatics/btp393
Acknowledgements
This work was performed under the auspices of the US Department of Energy’s Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396. We gratefully acknowledge the funding received from Australian Research Council Discovery grant (DP0880896), Murdoch University Strategic Research Fund through the Crop and Plant Research Institute (CaPRI) and the Centre for Rhizobium Studies (CRS) at Murdoch University. The authors would like to thank the Australia-China Joint Research Centre for Wheat Improvement (ACCWI) and SuperSeed Technologies (SST) for financially supporting Mohamed Ninawi’s PhD project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Reeve, W., Nandasena, K., Yates, R. et al. Complete genome sequence of Mesorhizobium opportunistum type strain WSM2075T. Stand in Genomic Sci 9, 294–303 (2013). https://doi.org/10.4056/sigs.4538264
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.4538264