
Abstract
Dehalobacter restrictus strain PER-K23 (DSM 9455) is the type strain of the species Dehalobacter restrictus. D. restrictus strain PER-K23 grows by organohalide respiration, coupling the oxidation of H2 to the reductive dechlorination of tetra- or trichloroethene. Growth has not been observed with any other electron donor or acceptor, nor has fermentative growth been shown. Here we introduce the first full genome of a pure culture within the genus Dehalobacter. The 2,943,336 bp long genome contains 2,826 protein coding and 82 RNA genes, including 5 16S rRNA genes. Interestingly, the genome contains 25 predicted reductive dehalogenase genes, the majority of which appear to be full length. The reductive dehalogenase genes are mainly located in two clusters, suggesting a much larger potential for organohalide respiration than previously anticipated.
Similar content being viewed by others


Introduction
Dehalobacter restrictus strain PER-K23 (DSM 9455), is the type strain of the species Dehalobacter restrictus [1]. Currently two pure cultures of D. restrictus have been described, namely D. restrictus strains PER-K23 and TEA [1,2].
We publish here the first full genome of a pure culture within the genus Dehalobacter and a preliminary comparison with a previously obtained metagenome from a co-culture containing Dehalobacter sp. strain E1 and Sedimentibacter sp [3].
Organohalide respiration (OHR) is considered as a key process in bioremediation of sites contaminated with organohalides such as tetrachloroethene (PCE) and trichloroethene (TCE), leading to a great interest in understanding the physiology and metabolism of organohalide respiring bacteria (OHRB). Most OHRBs are facultative organohalide respirers, capable of dehalogenating a limited number of halogenated compounds, as part of a versatile metabolism. This group consists of several genera, including Desulfitobacterium, Geobacter and Sulfurospirillum. Other isolates are obligate OHRB, among which isolates and enrichments of different Dehalococcoides mccartyi strains are the best studied. They have been shown to degrade a large variety of halogenated compounds solely using H2 as the electron donor. Until recently, the genus Dehalobacter had been thought to encompass exclusively obligate OHRB, however, at least some members of this genus have been described as able to ferment dichloromethane [4,5]. D. restrictus strain PER-K23 is an obligate OHRB, and like Dehalococcoides mccartyi, uses H2 as a sole electron donor. These similarities in physiology and ecology are noteworthy since Dehalobacter spp. are phylogenetically closely related to the metabolically versatile Desulfitobacterium spp.
D. restrictus strain PER-K23 was isolated from a packed bed column containing sediment from the river Rhine collected near Wageningen, the Netherlands, and granular sludge from a sugar refinery. This column had been fed with PCE for a prolonged period, prior to isolation of D. restrictus strain PER-K23 [6].
D. restrictus strain PER-K23 was chosen for genome sequencing because it is the type strain of the Dehalobacter restrictus species. Studying the genome gives an improved insight into the physiology and evolution of the genus Dehalobacter and may ultimately lead to unlocking its full potential for bioremediation.
Classification and features
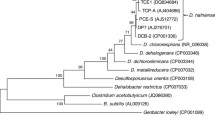
Dehalobacter restrictus is a member of the phylum Firmicutes, class Clostridia, order Clostridiales, and family Peptococcaceae [1],(Table 1). D. restrictus is closely related to the newly sequenced Dehalobacter sp. strain E1 [3], but grows in pure culture. Both Dehalobacter spp. and Desulfitobacterium spp. belong to the family Peptococcaceae (Figure 1). All members of this family are anaerobes, constituting a diverse group with respect to their metabolism and morphology [23]. D. restrictus strain PER-K23 is a rod-shaped bacterium with a single lateral flagellum and has not been reported to form spores. It stains Gram-negative, even though it phylogenetically belongs to the Gram-positive Firmicutes, and does not have an outer membrane, indicating that it should be considered a Gram-positive [1]. D. restrictus strain PER-K23 grows by coupling the oxidation of H2 to the reduction of PCE or TCE, growth has not been observed with any other electron donor or acceptor, nor has fermentative growth been shown [1,6]. D. restrictus strain PER-K23 requires iron as a trace element, the vitamins thiamine and cyanocobalamin, and the amino acids arginine, histidine and threonine for growth [1].

Phylogenetic tree highlighting the position of Dehalobacter restrictus relative to phylogenetically closely related organisms. 16S rRNA sequences were retrieved from Genbank (NCBI), and accession numbers are given in parentheses. Strains from which a full genome sequence are available are indicated with an asterisk. Phylogenetic analysis was done using the MEGA5 software package [20]. Sequences were aligned using the MUSCLE algorithm before a neighbor joining tree was constructed and validated with 1,000 bootstraps [21,22]. The reference bar indicates 2% sequence divergence.
Genome project history
Table 2 presents the project information in compliance to MIGS version 2.0 [24].
Growth conditions and DNA isolation
Dehalobacter restrictus strain PER-K23, DSM9455, was cultivated anaerobically as previously described [1]. DNA was extracted from bacterial pellets using the protocol recommended by the JGI. In brief, cell walls were digested with lysozyme before DNA was purified with hexadecyltrimethylammonium bromide, phenol and chloroform, and precipitated with isopropanol. Quality and quantity of the obtained DNA were checked by running aliquots on agarose gels using lambda phage DNA as mass standard and HindIII digested lambda phage DNA as a size marker.
Genome sequencing and assembly
The draft genome of Dehalobacter restrictus PER-K23 was generated at the DOE Joint genome Institute (JGI) using a combination of Illumina [25], and 454 technologies [26]. For this, genome we constructed and sequenced an Illumina GAii shotgun library which generated 77,929,756 reads totaling 5,922.7 Mb, and 1 paired end 454 library with an average insert size of 10 kb which generated 318,117 reads totaling 59.3 Mb of 454 data. All general aspects of library construction and sequencing performed at the JGI can be found at the JGI website [27]. The initial draft assembly contained 90 contigs in 1 scaffold. The 454 paired end data were assembled together with Newbler, version 2.3-PreRelease-6/30/2009. The Newbler consensus sequences were computationally shredded into 2 kb overlapping fake reads (shreds). Illumina sequencing data was assembled with VELVET, version 1.0.13 [28], and the consensus sequence were computationally shredded into 1.5 kb overlapping fake reads (shreds). We integrated the 454 Newbler consensus shreds, the Illumina VELVET consensus shreds and the read pairs in the 454 paired end library using parallel phrap, version SPS - 4.24 (High Performance Software, LLC). The software Consed [29–31] was used in the following finishing process. Illumina data was used to correct potential base errors and increase consensus quality using the software Polisher developed at JGI (Alla Lapidus, unpublished). Possible mis-assemblies were corrected using gapResolution (Cliff Han, unpublished), Dupfinisher [32], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR (J-F Cheng, unpublished) primer walks. A total of 134 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. The total size of the genome is 2,943,336 bp and the final assembly is based on 24.6 Mb of 454 draft data which provides an average 8.5× coverage of the genome and 348 Mb of Illumina draft data which provides an average 120× coverage of the genome.
Genome annotation
Genes of D. restrictus strain PER-K23 were identified using Prodigal [33] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [34]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Non-coding DNA and miscellaneous features were predicted using tRNAscan-SE [35], RNAMMer [36], Rfam [37], TMHMM [38], and signalP [39].
Genome properties
The genome consists of a single chromosome with a total size of 2,943,336 bp with 45% G+C content. A total of 2,908 genes were predicted, 2,826 of which are protein-coding genes. Genes with putative function corresponded to 76.7% (2,168), of all protein coding sequences with the remaining annotated as hypothetical proteins. In addition, 1,174 protein coding genes belong to 356 paralogous families in this genome. The properties and the statistics of the genome are summarized in Tables 3,4 and 5.
Insights from genome sequencing
Reductive dehalogenase paralogs
The genome of D. restrictus contains 25 loci predicted to code for proteins with sequence homology to reductive dehalogenases (RDHs). Among these 25 genes, one is a partial sequence and four are truncated due to possible frame-shift mutations (Table 5). This high number is in contrast to those found to date for metabolically versatile organohalide respirers. These possess a limited number of RDHs typically in the range of 1 to7 [43,44]. The number of RDHs in D. restrictus lies in the same range as seen in specialized organohalide respirers, such as Dehalococcoides mccartyi strains and Dehalogenimonas lykanthroporepellens, which have been predicted to possess between 10 and up to 36 RDHs [45,46].
For D. restrictus however, this finding is intriguing since, PCE and TCE, currently, are the only electron acceptors known to be utilized by strain PER-K23 [1]. The identification of a total of 25 rdhA genes suggests that D. restrictus possesses a much larger potential for OHR metabolism, than previously anticipated.
The majority of the rdhA genes are located in two clusters, one on each chromosome arm, with all but two RDHs being encoded on the leading strand. Cluster A is approximately 54 kb long, located on the right chromosome arm and contains 10 reductive dehalogenase genes including two truncated ones. Cluster B is approximately 61 kb long, located on the left chromosome arm and contains 11 reductive dehalogenase genes, of which two appear truncated (Table 5 & Figure 2).

Circular map of the chromosome of D. restrictus strain PER-K23. Labeling from the outside circle towards the inside circle. Numbers outside the map indicate nucleotide positions; Circles 1 and 2: predicted coding sequences, including pseudogenes, on the forward and reverse strand, respectively (colored by COG categories); Circle 3: RNA genes (tRNAs green, rRNAs red, other RNAs black); Circle 4: Position of reductive dehalogenase genes, in red, both functional and truncated, A and B indicate two rdh clusters; Circle 5: Position of transposases including inactive derivatives, in green; Circle 6: Position of genes related to corrinoid synthesis and uptake, in blue; Circle 7: GC content (peaks out/inside the circle indicate above or below average GC content, respectively: Circle 8: GC skew, calculated as (G-C)/(G+C), purple or olive indicates values lower or higher than 1, respectively.
The remaining three complete RDH genes and one partial RDH encoding gene are scattered throughout the genome (Table 5 & Figure 2). A similar pattern has previously been observed in the genomes of Dehalococcoides mccartyi strains, where the majority of the RDHs are located on each side of, and close to the origin of replication [47]. These regions were described as high plasticity regions, where frequent events of rearrangement and horizontal gene transfer are thought to occur. It was suggested that these regions enable fast adaptation to dehalogenation of new organohalides, while at the same time protecting key metabolic functions from being disrupted by horizontal gene transfer events [47].
We identified transcriptional regulators of the CRP/FNR type being encoded by genes in the vicinity of most of the RDH encoding genes, with PceA (encoded by Dehre_2398) as a notable exception [48]. A regulator of this type has been demonstrated to regulate the expression of the genes that code for chlorophenol reductive dehalogenase (cpr operon in Desulfitobacterium dehalogenans and Desulfitobacterium hafniense strain DCB-2 [49]. The presence of transcriptional regulator genes close to almost all rdhA genes suggest that their transcription is regulated. This was confirmed by a recent study looking at transcription of rdh genes and the proteome of Dehalobacter restrictus strain PER-K23 growing in the presence of H2 and PCE. In this study we found that PceA (encoded by Dehre_2398) was highly present at both RNA and proteomic level, whereas the remaining RDHs and the corresponding transcripts were either not detected at all or at very low levels, suggesting that the RDH encoding genes are tightly regulated, and probably only expressed in the presence of their specific substrate [48].
Recently the draft genome of Dehalobacter sp. strain E1 was published [3]. This genome contains nine potentially functional rdhA genes, and one pseudogene. Six of these are conserved between D. restrictus strain PER-K23 and strain E1 (Table 5). Two of the conserved rdhA genes are located at the edge of cluster A and one at the edge of cluster B. Interestingly all four rdhA genes present outside cluster A or B are conserved between the two strains, which may indicate that both cluster A and B represent high plasticity regions unique to D. restrictus (Table 5). Currently, pceA (encoded by Dehre_2398) is the only RDH-encoding gene from Dehalobacter restrictus to be characterized in detail. The corresponding gene product PceA has been shown to catalyze the reduction of PCE to TCE and TCE to cis-DCE, the only two electron acceptors demonstrated to support growth of D. restrictus [1,42]. The pceA gene belongs to a gene cluster, pceABCT (Dehre_2395-2398), which is highly similar to a gene cluster identified in a composite transposon structure identified in several Desulfitobacterium strains [50–52]. The transposon structure is not conserved in D. restrictus although the gene cluster is flanked by sequences resembling transposase genes in a late state of decay (Dehre_2394 and 2399). This combined with the fact that the pceABCT gene cluster including the cryptic transposases and the surrounding genomic context are conserved between D. restrictus and D. strain E1 (data not shown) suggest that the presence of pceABCT is the result of an ancient horizontal gene transfer event.
Corrinoid synthesis and uptake
Corrinoid is the key cofactor in characterized RD catalytic subunits. Dehalobacter restrictus strain PER-K23 requires vitamin B12 in the medium for growth [1].
Therefore it is noteworthy to report the presence of a full set of corrinoid biosynthesis genes in the genome of D. restrictus, although cbiH (Dehre_2856) encoding precorrin-3B C17-methyltransferase displays a frame-shift mutation, and consequently is annotated as a pseudogene. The vitamin B12 synthesis pathway is encoded by two distinct gene clusters in D. restrictus strain PER-K23, where Dehre_2848-2865 encode enzymes of the upper pathway, and Dehre_1606-1615 the lower pathway. One additional gene (Dehre_1488) belonging to the lower pathway is located elsewhere in the genome (Figure 2) [48]. The genome encodes several gene clusters associated with corrinoid uptake and salvaging pathways. Preliminary studies of the proteome from cultures grown at standard conditions or with partial vitamin B12 depletion showed that gene products encoded by one of the salvaging pathways (Dehre_0281-0291) were much more abundant in the vitamin B12 starved cells than in the cells grown under standard concentrations (J. Maillard and T. Kruse unpublished data). These findings suggest that the de novo corrinoid synthesis pathway is not functional and that Dehalobacter restrictus strain PER-K23 is dependent on salvaging corrinoids from the environment.
Hydrogenases
Another interesting feature is the presence of genes predicted to code for eight different hydrogenases. These include three periplasmic membrane-bound Ni/Fe uptake hydrogenases, consisting of three subunits: a catalytic unit, an Fe/S cluster protein and a membrane-bound b-type cytochrome (Dehre_551-553, 1061–1063 and 2405–2007), two six-subunits membrane-bound energy-conserving Ni/Fe hydrogenases (Dehre_1568-1573 and 1645–1650), and three Fe-only hydrogenases (Dehre_1739-1741, 2317–2320 and 2372–2374). The Fe-only hydrogenases consist of the catalytic subunit and two to three putative electron transferring subunits.
The presence of multiple uptake hydrogenases has also been observed in Desulfitobacterium spp., whereas Dehalococcoides mccartyi strains only have one uptake hydrogenase [43,44,53]. The two six-subunits Ni/Fe resemble the Hyc and Ech complexes found in Dehalococcoides mccartyi strain 195 [54], as well as the Hyc complex found in Desulfitobacterium spp [43,44,55].
Disrupting either one uptake hydrogenase or the six-subunits energy-conserving hydrogenase in Desulfitobacterium dehalogenans led to loss of the ability to grow using lactate or formate as electron donor and 3-chloro-4-hydroxyphenylacetate as electron acceptor, indicating that hydrogenases may play an important role in the electron transport chain to RD catalytic subunits, even when hydrogen is not used as the initial electron donor [55].
The role of the six-subunit hydrogenase complexes are still poorly understood. It has been speculated that they play a role in generating low potential electrons for OHR by reverse electron flow. However, this was considered as unlikely in one study where Dehalococcoides mccartyi strain 195 was cultivated in the presence of varying concentrations of hydrogen [56]. The exact role of the different hydrogenases in Dehalobacter restrictus strain PER-K23 still needs further studies.
The genome also encodes an intact Wood-Ljungdahl pathway (Dehre_0130-0155 and 2348–2351). The presence of a whole or partial Wood-Ljungdahl pathway has been observed in other OHRB. The closely related Desulfitobacterium hafniense strains Y51 and DCB-2 both contain genes predicted to encode a full Wood-Ljungdahl pathway, and strain DCB-2 has been shown to fix CO2 [43,44]. The more distantly related Dehalococcoides mccartyi strains have been shown to contain partial Wood-Ljungdahl pathways, but its exact role in the metabolism of these organisms remains unclear [57,58].
The genome of D. restrictus contains 72 genes annotated as encoding transposases or inactive derivatives thereof, whereas it only contains few phage-associated genes despite the lack of a CRISPR phage immunity system.
Cells of Dehalobacter restrictus strain PER-K23 are motile [1]. The genome contains genes for synthesis of flagella and several genes predicted to be involved in chemotaxis. The role of chemotaxis in OHRB is currently understudied. Chemotactic behavior towards metals has been described for Geobacter, some members of this genus have been shown to be OHRB. Chemotactic behavior towards organohalides has, however, not been described for Geobacter spp [59–61].
Conclusion
The presence of an unexpectedly large number of putative RDH encoding genes suggests a far larger potential for use in bioremediation than previously anticipated, especially if Dehalobacter restrictus strain PER-K23 is attracted by organohalides in a chemotactic manner. The complete genome sequence of Dehalobacter restrictus strain PER-K23, the type strain of the genus Dehalobacter, represents a significant leap towards understanding the physiology, ecology and evolution of this specialized organohalide respiring group of bacteria. Current work focuses on obtaining a deeper understanding of the expression and regulation of the RDH genes, and thereby expanding the known organohalide substrate range of this organism. Shot-gun proteome analysis will aid in deciphering the metabolism of D. restrictus strain PER-K23 and allow generation of refined genome scale metabolic models of these dedicated degraders.
Abbreviations
- OHR:
-
organohalide respiration
- OHRB:
-
organohalide respiring bacteria
- RDH:
-
reductive dehalogenase homologue
- PCE:
-
tetrachloroethene
References
Holliger C, Hahn D, Harmsen H, Ludwig W, Schumacher W, Tindall B, Vazquez F, Weiss N, Zehnder AJB. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch Microbiol 1998; 169:313–321. PubMed http://dx.doi.org/10.1007/s002030050577
Wild A, Hermann R, Leisinger T. Isolation of an anaerobic bacterium which reductively dechlorinates tetrachloroethene and trichloroethene. Biodegradation 1996; 7:507–511. PubMed http://dx.doi.org/10.1007/BF00115297
Maphosa F, Van Passel MWJ, De Vos WM, Smidt H. Metagenome analysis reveals yet unexplored reductive dechlorinating potential of Dehalobacter sp. E1 growing in co-culture with Sedimentibacter sp. Environmental Microbiology Reports 2012; 4:604–616. PubMed
Lee M, Low A, Zemb O, Koenig J, Michaelsen A, Manefield M. Complete chloroform dechlorination by organochlorine respiration and fermentation. Environ Microbiol 2012; 14:883–894. PubMed http://dx.doi.org/10.1111/j.14622920.2011.02656.x
Justicia-Leon SD, Ritalahti KM, Mack EE, Löffler FE. Dichloromethane fermentation by a Dehalobacter sp. in an enrichment culture derived from pristine river sediment. Appl Environ Microbiol 2012; 78:1288–1291. PubMed http://dx.doi.org/10.1128/AEM.07325-11
Holliger C, Schraa G, Stams AJ, Zehnder AJ. A highly purified enrichment culture couples the reductive dechlorination of tetrachloroethene to growth. Appl Environ Microbiol 1993; 59:2991–2997. PubMed
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J Syst Bacteriol 1978; 28:1–6. http://dx.doi.org/10.1099/00207713-28-1-1
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119–169.
Murray RGE. The Higher Taxa, or, a Place for Everything…? In: Holt JG (ed), Bergey’s Manual of Systematic Bacteriology, First Edition, Volume 1, The Williams and Wilkins Co., Baltimore, 1984, p. 31–34.
List of new names and new combinations previously effectively, but not validly, published. List no. 132. Int J Syst Evol Microbiol 2010; 60:469–472. http://dx.doi.org/10.1099/ijs.0.022855-0
Rainey FA. Class II. Clostridia class nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 3, Springer-Verlag, New York, 2009, p. 736.
Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225–420. http://dx.doi.org/10.1099/00207713-30-1-225
Prévot AR. In: Hauderoy P, Ehringer G, Guillot G, Magrou. J., Prévot AR, Rosset D, Urbain A (eds), Dictionnaire des Bactéries Pathogènes, Second Edition, Masson et Cie, Paris, 1953, p. 1–692.
Rogosa M. Peptococcaceae, a new family to include the Gram-positive, anaerobic cocci of the genera Peptococcus, Peptostreptococcus and Ruminococcus. Int J Syst Bacteriol 1971; 21:234–237. http://dx.doi.org/10.1099/00207713-21-3-234
Validation of publication of new names and new combinations previously effectively published outside the IJSB. List No. 66. Int J Syst Bacteriol 1998; 48:631–632. http://dx.doi.org/10.1099/00207713-48-3-631
Holliger C, Hahn D, Harmsen H, Ludwig W, Schumacher W, Tindall B, Vazquez F, Weiss N, Zehnder AJ. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch Microbiol 1998; 169:313–321. PubMed http://dx.doi.org/10.1007/s002030050577
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol 2011. PubMed http://dx.doi.org/10.1093/molbev/msr121
Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792–1797. PubMed http://dx.doi.org/10.1093/nar/gkh340
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987; 4:406–425. PubMed
Takayuki EH. C. The Firmicutes. In: Vos PG, G.; Jones, D.; Krieg, N.R.; Ludwig, W.; Rainey, F.A.; Schleifer, K.-H.; Whitman, W.B., editor. Bergey’s Manual of Systematic Bacteriology. Volume 32009. p 969–975.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Bennett S. Solexa Ltd. Pharmacogenomics 2004; 5:433–438. PubMed http://dx.doi.org/10.1517/14622416.5.4.433
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:376–380. PubMed
The DOE Joint Genome Institute. http://www.jgi.doe.gov
Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed http://dx.doi.org/10.1101/gr.074492.107
Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998; 8:175–185. PubMed http://dx.doi.org/10.1101/gr.8.3.175
Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998; 8:186–194. PubMed http://dx.doi.org/10.1101/gr.8.3.175
Gordon D, Abajian C, Green P. Consed: A graphical tool for sequence finishing. Genome Res 1998; 8:195–202. PubMed http://dx.doi.org/10.1101/gr.8.3.195
Han C, Chain P. 2006 Finishing Repetitive Regions Automatically with Dupfinisher. In Proceedings of the 2006 International Conference on Bioinformatics Computational Biology, BIOCOMP’06.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; PubMed
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457
Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955–964. PubMed
Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007; 35:3100–3108. PubMed http://dx.doi.org/10.1093/nar/gkm160
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: An RNA family database. Nucleic Acids Res 2003; 31:439–441. PubMed http://dx.doi.org/10.1093/nar/gkg006
Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 2001; 305:567–580. PubMed http://dx.doi.org/10.1006/jmbi.2000.4315
Bendtsen JD, Nielsen H, Von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004; 340:783–795. PubMed http://dx.doi.org/10.1016/j.jmb.2004.05.028
Hug LA, Maphosa F, Leys D, Loffler FE, Smidt H, Edwards EA, Lorenz A. Overview of organohalide respiration and introduction of a simple classification system for reductive dehalogenases. Philos Trans R Soc Lond B Biol Sci 2012; ⋯:368.
Campanella JJ, Bitincka L, Smalley J. MatGAT: An application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinformatics 2003; 4:29. PubMed http://dx.doi.org/10.1186/1471-2105-4-29
Maillard J, Schumacher W, Vazquez F, Regeard C, Hagen WR, Holliger C. Characterization of the corrinoid iron-sulfur protein tetrachloroethene reductive dehalogenase of Dehalobacter restrictus. Appl Environ Microbiol 2003; 69:4628–4638. PubMed http://dx.doi.org/10.1128/AEM.69.8.4628-4638.2003
Nonaka H, Keresztes G, Shinoda Y, Ikenaga Y, Abe M, Naito K, Inatomi K, Furukawa K, Inui M, Yukawa H. Complete genome sequence of the dehalorespiring bacterium Desulfitobacterium hafniense Y51 and comparison with Dehalococcoides ethenogenes 195. J Bacteriol 2006; 188:2262–2274. PubMed http://dx.doi.org/10.1128/JB.188.6.2262-2274.2006
Kim SH, Harzman C, Davis J, Hutcheson R, Broderick J, Marsh T, Tiedje J. Genome sequence of Desulfitobacterium hafniense DCB-2, a Grampositive anaerobe capable of dehalogenation and metal reduction. BMC Microbiol 2012; 12:21. PubMed http://dx.doi.org/10.1186/1471-2180-12-21
Siddaramappa S, Challacombe JF, Delano SF, Green LD, Daligault H, Bruce D, Detter C, Tapia R, Han S, Goodwin L, et al. Complete genome sequence of Dehalogenimonas lykanthroporepellens type strain (BL-DC-9 T) and comparison to “Dehalococcoides” strains. Stand Genomic Sci 2012; 6:251–264. PubMed http://dx.doi.org/10.4056/sigs.2806097
Taş N, Van Eekert MHA, De Vos WM, Smidt H. The little bacteria that can — Diversity, genomics and ecophysiology of “Dehalococcoides” spp. in contaminated environments. Microb Biotechnol 2010; 3:389–402. PubMed http://dx.doi.org/10.1111/j.1751-7915.2009.00147.x
McMurdie PJ, Behrens SF, Muller JA, Goke J, Ritalahti KM, Wagner R, Goltsman E, Lapidus A, Holmes S, Loffler FE, et al. Localized Plasticity in the Streamlined Genomes of Vinyl Chloride Respiring Dehalococcoides. PLoS Genet 2009. PubMed http://dx.doi.org/10.1371/journal.pgen.1000714
Rupakula A, Kruse T, Boeren S, Holliger C, Smidt H, Maillard J. Evaluation of the restricted metabolism of the obligate organohalide respiring bacterium Dehalobacter restrictus — lessons from a tiered functional genomics approach. Philos Trans R Soc Lond B Biol Sci 2013; ⋯:368.
Gabor K, Veríssimo CS, Cyran BC, Ter Horst P, Meijer NP, Smidt H, De Vos WM, Van Der Oost J. Characterization of CprK1, a CRP/FNR-type transcriptional regulator of halorespiration from Desulfitobacterium hafniense. J Bacteriol 2006; 188:2604–2613. PubMed http://dx.doi.org/10.1128/JB.188.7.2604-2613.2006
Duret A, Holliger C, Maillard J. The opportunistic physiology of Desulfitobacterium hafniense strain TCE1 towards organohalide respiration with tetrachloroethene. Appl Environ Microbiol 2012; 78:6121–6127. PubMed http://dx.doi.org/10.1128/AEM.01221-12
Maillard J, Regeard C, Holliger C. Isolation and characterization of Tn-Dha1, a transposon containing the tetrachloroethene reductive dehalogenase of Desulfitobacterium hafniense strain TCE1. Environ Microbiol 2005; 7:107–117. PubMed http://dx.doi.org/10.1111/j.1462-2920.2004.00671.x
Futagami T, Tsuboi Y, Suyama A, Goto M, Furukawa K. Emergence of two types of nondechlorinating variants in the tetrachloroethene-halorespiring Desulfitobacterium sp strain Y51. Appl Microbiol Biotechnol 2006; 70:720–728. PubMed http://dx.doi.org/10.1007/s00253-005-0112-9
Maphosa F, de Vos WM, Smidt H. Exploiting the ecogenomics toolbox for environmental diagnostics of organohalide-respiring bacteria. Trends Biotechnol 2010; 28:308–316. PubMed http://dx.doi.org/10.1016/j.tibtech.2010.03.005
Seshadri R, Adrian L, Fouts DE, Eisen JA, Phillippy AM, Methe BA, Ward NL, Nelson WC, Deboy RT, Khouri HM, et al. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes. Science 2005; 307:105–108. PubMed http://dx.doi.org/10.1126/science.1102226
Smidt H, Song DL, van der Oost J, de Vos WM. Random transposition by Tn916 in Desulfitobacterium dehalogenans allows for isolation and characterization of halorespiration-deficient mutants. J Bacteriol 1999; 181:6882–6888. PubMed
Morris RM, Sowell S, Barofsky D, Zinder S, Richardson R. Transcription and mass-spectroscopic proteomic studies of electron transport oxidoreductases in Dehalococcoides ethenogenes. Environ Microbiol 2006; 8:1499–1509. PubMed http://dx.doi.org/10.1111/j.1462-2920.2006.01090.x
Ahsanul Islam M, Edwards EA, Mahadevan R. Characterizing the metabolism of Dehalococcoides with a constraint-based model. PLOS Comput Biol 2010; 6:e1000887. PubMed http://dx.doi.org/10.1371/journal.pcbi.1000887
Tang YJ, Yi S, Zhuang WQ, Zinder SH, Keasling JD, Alvarez-Cohen L. Investigation of carbon metabolism in “Dehalococcoides ethenogenes” strain 195 by use of isotopomer and transcriptomic analyses. J Bacteriol 2009; 191:5224–5231. PubMed http://dx.doi.org/10.1128/JB.00085-09
Childers SE, Ciufo S, Lovley DR. Geobacter metallireducens accesses insoluble Fe(III) oxide by chemotaxis. Nature 2002; 416:767–769. PubMed http://dx.doi.org/10.1038/416767a
Lovley DR, Ueki T, Zhang T, Malvankar NS, Shrestha PM, Flanagan KA, Aklujkar M, Butler JE, Giloteaux L, Rotaru AE, et al. Geobacter. The Microbe Electric’s Physiology, Ecology, and Practical Applications. Adv Microb Physiol 2011; 59:1–100. PubMed http://dx.doi.org/10.1016/B978-0-12-387661-4.00004-5
Tran HT, Krushkal J, Antommattei FM, Lovley DR, Weis RM. Comparative genomics of Geobacter chemotaxis genes reveals diverse signaling function. BMC Genomics 2008; 9:471. PubMed http://dx.doi.org/10.1186/1471-2164-9-471
Acknowledgements
The work of Thomas Kruse and Hauke Smidt were supported by the Netherlands Genomics Initiative as well as the European Community’s Seventh Framework Programme (FP7/ 2007–2013) through the Ecogenomics program and the BACSIN and METAEXPLORE projects (grant agreements No. 211684 and 222625), respectively. The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Kruse, T., Maillard, J., Goodwin, L. et al. Complete genome sequence of Dehalobacter restrictus PER-K23T. Stand in Genomic Sci 8, 375–388 (2013). https://doi.org/10.4056/sigs.3787426
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.3787426