
Abstract
Starkeya novella (Starkey 1934) Kelly et al. 2000 is a member of the family Xanthobacteraceae in the order ‘Rhizobiales’, which is thus far poorly characterized at the genome level. Cultures from this species are most interesting due to their facultatively chemolithoautotrophic lifestyle, which allows them to both consume carbon dioxide and to produce it. This feature makes S. novella an interesting model organism for studying the genomic basis of regulatory networks required for the switch between consumption and production of carbon dioxide, a key component of the global carbon cycle. In addition, S. novella is of interest for its ability to grow on various inorganic sulfur compounds and several C1-compounds such as methanol. Besides Azorhizobium caulinodans, S. novella is only the second species in the family Xanthobacteraceae with a completely sequenced genome of a type strain. The current taxonomic classification of this group is in significant conflict with the 16S rRNA data. The genomic data indicate that the physiological capabilities of the organism might have been underestimated. The 4,765,023 bp long chromosome with its 4,511 protein-coding and 52 RNA genes was sequenced as part of the DOE Joint Genome Institute Community Sequencing Program (CSP) 2008.
Similar content being viewed by others

Introduction
Strain ATCC 8093T (ATCC 8093 = DSM 506 = NBRC 14993) is the type strain of the species Starkeya novella [1] and the type species of the genus Starkeya [1], which currently contains only one other species, S. koreensis [2]. The most prominent feature of S. novella is its ability to grow as a facultative chemolithoautotroph [3], a heterotroph [4], or methylotroph [1,5]. Cultures of strain ATCC 8093T were first isolated from soil samples taken from agricultural land in New Jersey by Robert L. Starkey in the early 1930s [6,7] and deposited in the American Type Culture Collection (ATCC) under the basonym Thiobacillus novellus [3,8]. The bacterium was referred to as the ‘new’ Thiobacillus as it was the first facultatively chemolithoautotrophic sulfur oxidizer to be isolated. Until then, all known dissimilatory sulfur-oxidizing bacteria were also obligate autotrophs. As a result, the metabolism of T. novellus was intensely studied for many years following its discovery, and particularly following the development of more sophisticated biochemical and molecular methods in the 1960s.
During the last fifty years, the strain has been used in numerous molecular studies, both of its oxidative sulfur metabolism and the versatility and regulation of its carbon metabolism. Studies included generation of reducing power in chemosynthesis [9], carbon dioxide fixation and carboxydismutase action [10], catabolite repression in facultative chemoautotrophs [11], regulation of glucose transport and metabolism [12], isolation and characterization of a bacteriophage [13], pathways of thiosulfate oxidation [9,14–17], the formation of sulfite during the oxidation of thiosulfate [18], and the isolation and characterization of a bacterial sulfite dehydrogenase [19–29], a sulfite-oxidizing enzyme.
Based on the 16S rRNA gene sequence in 2000 Kelly et al. [1] proposed the reclassification of T. novellus to S. novella. The genus name Starkeya is in honor of Robert L. Starkey and his important contribution to soil microbiology and sulfur biochemistry [1]; the species epithet was derived from the Latin adjective ‘novella’, new [3]. Here we present a summary classification and a set of features for S. novella ATCC 8093T, together with the description of the genomic sequencing and annotation.
Classification and features
16S rRNA analysis
The single genomic 16S rRNA sequence of strain ATCC 8093T was compared using NCBI BLAST [30,31] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [32] and the relative frequencies of taxa and keywords (reduced to their stem [33]) were determined, weighted by BLAST scores. The most frequently occurring genera were Ancylobacter (30.0%), Starkeya (13.4%), Agrobacterium (13.1%), Xanthobacter (12.4%) and Azorhizobium (11.5%) (98 hits in total). Regarding the three hits to sequences from members of the species, the average identity within HSPs was 99.5%, whereas the average coverage by HSPs was 92.8%. Among all other species, the one yielding the highest score was Ancylobacter rudongensis (AY056830), which corresponded to an identity of 98.1% and an HSP coverage of 98.4%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The highest-scoring environmental sequence was EU835464 (‘structure and quorum sensing reverse osmosis RO membrane biofilm clone 3M02’), which showed an identity of 98.4% and an HSP coverage of 100.0%. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were ‘skin’ (6.0%), ‘microbiom’ (3.0%), ‘human, tempor, topograph’ (2.5%), ‘compost’ (2.1%) and ‘dure’ (2.1%) (152 hits in total) and fit only partially to the known habitat of the species. Environmental samples that yielded hits of a higher score than the highest scoring species were not found.
Figure 1 shows the phylogenetic neighborhood of in a 16S rRNA based tree. The sequence of the single 16S rRNA gene copy in the genome differs by nine nucleotides from the previously published 16S rRNA sequence (D32247), which contains one ambiguous base call.

Phylogenetic tree highlighting the position of S. novella relative to the type strains of the other species within the family Xanthobacteraceae (blue font color). The tree was inferred from 1,381 aligned characters [34,35] of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion [36]. Hyphomicrobiaceae (green font color for those species that caused conflict according to the Parafit test, black color for the remaining ones; see below for the difference) were included in the dataset for use as outgroup taxa but then turned out to be intermixed with the target family; hence, the rooting shown was inferred by the midpoint-rooting method [29]. The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 550 ML bootstrap replicates [37] (left) and from 1,000 maximum-parsimony bootstrap replicates [38] (right) if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [39] are labeled with one asterisk, those also listed as ‘Complete and Published’ with two asterisks (see [40] and CP000781 for Xanthobacter autotrophicus, CP002083 for Hyphomicrobium denitrificans and CP002292 for Rhodomicrobium vannielii).
To measure conflict between 16S rRNA data and taxonomic classification in detail, we followed a constraint-based approach as described recently in detail [41], conducting both unconstrained searches and searches constrained for the monophyly of both families and using our own re-implementation of CopyCat [42] in conjunction with AxPcoords and AxParafit [43] was used to determine those leaves (species) whose placement significantly deviated between the constrained and the unconstrained tree.
The best-supported ML tree had a log likelihood of −12,191.55, whereas the best tree found under the constraint had a log likelihood of −12,329.92. The constrained tree was significantly worse than the globally best one in the SH test as implemented in RAxML [37,44] (α = 0.01). The best supported MP trees had a score of 1,926, whereas the best constrained trees found had a score of 1.982 and were also significantly worse in the KH test as implemented in PAUP [8,44] (α < 0.0001). Accordingly, the current classification of the family as used in [45,46], on which the annotation of Figure 1 is based, is in significant conflict with the 16S rRNA data. Figure 1 also shows those species that cause phylogenetic conflict as detected using the ParaFit test (i.e., those with a p value > 0.05 because ParaFit measures the significance of congruence) in green font color. According to our analyses, the Hyphomonadaceae genera (Blastochloris and Prosthecomicrobium) nested within the Xanthobacteraceae display significant conflict. In the constrained tree (data not shown), the Angulomicrobium-Methylorhabdus clade is placed at the base of the Xanthobacteraceae clade (forced to be monophyletic). For this reason, Angulomicrobium and Methylorhabdus were not detected as causing conflict (note that the ParaFit test essentially compares unrooted trees). A taxonomic revision of the group would probably need to start with the reassignment of these genera to different families.
Morphology and physiology
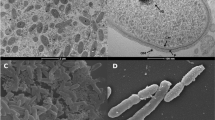
Cells of S. novella ATCC 8093T are non-motile, Gram-negative staining short rods or coccobacilli with a size of 0.4–0.8 µm × 0.8–2.0 µm, occurring singly or in pairs (Figure 2, Table 1) [1]. Colonies grown on thiosulfate agar turn white with sulfur on biotin supplemented growth media [1], while in the presence of small amounts of yeast extract (DSMZ medium 69) the colonies have a pale pink appearance following growth on thiosulfate and no sulfur formation is observed. Cells grow on thiosulfate and tetrathionate under aerobic conditions, but not on sulfur or thiocyanate [1]. Ammonium salts, nitrates, urea and glutamate can serve as nitrogen sources [1]. Several surveys of substrates supporting heterotrophic growth have been published, and include glucose, formate, methanol, oxalate [1,2,4,6]. The growth range spans from 10–37°C, with an optimum at 25–30°C, and a pH range from 5.7–9.0 with an optimum at pH 7.0 [1].

Transmission electron micrograph of S. novella ATCC 8093T. Scale bar: 500 nm
Chemotaxonomy
The lipopolysaccharide of strain ATCC 8093T lacks heptoses and has only 2,3-diamino-2,3-dideoxyglucose as the backbone sugar [1]; other data on the cell wall structure of strain ATCC 8093T are not available. The major isoprenoid quinone is ubiquinone Q-10 [1], and the major cellular fatty acids are octadecenoid acid (C18:1) and C19 cyclopropane acid; no hydroxyl acids are present [1]. Cells contain putrescine and homospermidine.
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of the DOE Joint Genome Institute Community Sequencing Program (CSP) 2008. The genome project is deposited in the Genomes On Line Database [39] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
Strain ATCC 8093T was grown from a culture of DSMZ 506 in DSMZ medium 69 at 28°Cg DNA was purified using the Genomic-tip 100 System (Qiagen) following the directions provided by the supplier. The purity, quality and size of the bulk gDNA preparation were assessed by JGI according to DOE-JGI guidelines.
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [57]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 13 contigs in one scaffold was converted into a phrap [58] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (211.3 Mb) were assembled with Velvet [59] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 259.9 Mb 454 draft data and all of the 454 paired-end data. Newbler parameters were -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [58] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [58], Dupfinisher [60], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 43 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [61]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 97.8 × coverage of the genome. The final assembly contained 865,253 pyrosequence and 6,036,863 Illumina reads.
Genome annotation
Genes were identified using Prodigal [62] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [63]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Non-coding genes and miscellaneous features were predicted using tRNAscan-SE [64, RNAMMer [65], Rfam [66], TMHMM [67], and SignalP [68].
Genome properties
The genome consists of a circular 4,765,023 bp chromosome a 67.9% G+C content (Table 3 and Figure 3). Of the 4,563 genes predicted, 4,511 were protein-coding genes, and 52 RNAs; 80 pseudogenes were also identified. The majority of the protein-coding genes (74.8%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4. A total of 388 genes are predicted to encode proteins involved in signal transduction, including 284 one-component systems, 41 histidine kinases, 47 response regulators, seven chemotaxis proteins and two additional unclassified proteins.

Graphical map of the chromosome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content (black), GC skew (purple/olive).
Insights into the genome
As indicated in the introduction, because S. novella was the first facultative sulfur chemolithotrophic bacterium to be isolated, many studies of its metabolic capabilities were carried out following its discovery. Several groups worked on the carbon metabolism of S. novella, which led to the discovery of an operational pentose phosphate pathway in this bacterium [69], which is also the only reported pathway of glucose metabolism in the description of S. novella [1]. However, analysis of the genome sequence revealed that in addition to a pentose phosphate pathway, S. novella also contains enzymes required for the Entner-Doudoroff pathway (Snov_2999 & Snov_3400, 2-dehydro-3-deoxyphosphogluconate aldolase; 6-phosphogluconate dehydratase; biocyc database) and the enzymes required for the Embden-Meyerhoff pathway, although this pathway appears to lack a phosphofructokinase (EC 2.7.1.11), indicating that it may only be able to be used for gluconeogenesis.
The respiratory chain of S. novella has also been studied and an aa3 type terminal oxidase was identified and characterized in some detail [70–73]. It was also discovered that the cytochrome c that interacts with this cytochrome oxidase (most likely this cytochrome is encoded by Snov_1033) has properties that are reminiscent of the mitochondrial respiratory chain cytochrome c [70–75], including a high pI and an ability to transfer electrons to the bovine cytochrome oxidase [76]. The analysis of the genome revealed a much greater diversity of respiratory chain complexes than previously recognized, including two NADH oxidases (gene regions Snov_1853 & Snov_2407), one succinate dehydrogenase (Snov_3317 gene region) and a cytochrome bc1 complex (Snov_2477 gene region). In addition to these components, the genome encodes two aa3 type cytochrome oxidases (gene regions Snov_0584 & 4240), two cytochrome bd type quinol oxidases (pfam02322, gene regions Snov_0620 & 3535), a cbb3 type cytochrome oxidase (gene region Snov_4464), and a cyoB type quinol oxidase (COG0843, cd01662, gene region Snov_1015) indicating a significant versatility of respiration in S. novella as well as the potential to grow at low oxygen tensions as both the cbb3 and bd type oxidases are known to have high affinities for oxygen, enabling growth under microaerophilic conditions. Experiments in our laboratory have shown that final OD600 values reached by cultures grown on thiosulfate (5g/l) and hydrogen carbonate (20 mM) supplemented DSMZ medium 69 were the same regardless of whether 25, 50, 100 or 200 ml of medium were used in a 250 ml flask. This clearly confirms that, as indicated by the genome data, S. novella is capable of growth under microaerophilic as well as aerobic conditions.
We also re-evaluated the range of substrates that support growth of S. novella. In the description of the genus Starkeya [1] only glucose, formate, methanol and oxalate were listed as growth-supporting substrates in addition to thiosulfate and tetrathionate. An early paper reporting a test of the heterotrophic potential of S. novella was published in 1969 by Taylor and Hoare [4] in which they identified 16 potential growth substrates (Table no. 7 in [4]) including all of the above except oxalate, which was identified subsequently by [5] who were seeking to evaluate the C1 compound metabolism of S. novella and also identified formamide as a potential substrate. It is unclear why the description of the genus Starkeya did not list all of the 16 growth substrates identified by Taylor and Hoare. To confirm the earlier data, we carried out a growth substrate screen using the Biolog system (GN2 assay plates) as well as an api20NE test for bacterial identification. Some substrates that are not part of this Biolog GN2 plate (e.g. oxalate, fructose, succinate etc.) were independently tested in the laboratory for their ability to support growth. In the API20NE test, in addition to a positive oxidase response, S. novella tested positive for ESC/Fecit and p-nitrophenyl hydrolysis, glucose, mannitol and gluconate utilization. The Biolog assay clearly showed that the heterotrophic potential of this bacterium is greater than previously identified, with a total of 28 growth-supporting substrates being identified in the screen (Table 5). The metabolic profile could not be identified as such, and was most closely related to that of Ancylobacter aquaticus (SIM: 0.45, Dist: 8.96), which supports the phylogenetic placement of S. novella in the Ancylobacter subgroup of the Xanthobacteriaceae. When combining all the data from the various studies, there are now 39 substrates that have been identified as supporting heterotrophic growth of S. novella. In addition to sugars such as glucose, fructose and arabinose, several sugar alcohols and amino acids as well as some organic acids can be used as growth substrates (Table 5). This reasonably large range of growth substrates is reflected in the size and the diversity of metabolic pathways present in the S. novella genome which, with a size of 4.6 Mb, is comparable to the genomes of e.g., Escherichia coli and Rhodopseudomonas palustris.
Although the analyses presented above are limited, they clearly illustrate that while the genome data confirm many of the results from early studies of the physiology of this bacterium, the metabolic capabilities of S. novella as indicated by the genome data clearly exceed those previously published in the literature and suggest that the versatility and adaptability to changing environments likely is a significant factor for its survival.
References
Kelly DP, McDonald IR, Wood AP. Proposal for the reclassification of Thiobacillus novellus as Starkeya novella gen. nov., comb. nov., in the alpha-subclass of the Proteobacteria. Int J Syst Evol Microbiol 2000; 50:1797–1802. PubMed
Im WT, Aslam Z, Lee M, Ten LN, Yang DC, Lee ST. Starkeya koreensis sp. nov. isolated from rice straw. Int J Syst Evol Microbiol 2006; 56:2409–2414. PubMed http://dx.doi.org/10.1099/ijs.0.64093-0
Santer M, Boyer J, Santer U. Thiobacillus novellus: I. Growth on organic and inorganic media. J Bacteriol 1959; 78:197–202. PubMed
Taylor BF, Hoare DS. New facultative Thiobacillus and a reevaluation of the heterotrophic potential of Thiobacillus novellus. J Bacteriol 1969; 100:487–497. PubMed
Chandra TS, Shethna YI. Oxalate, formate, formamide, and methanol metabolism in Thiobacillus novellus. J Bacteriol 1977; 131:389–398. PubMed
Starkey RL. Isolation of some bacteria which oxidize thiosulfate. Soil Sci 1935; 39:197–220. http://dx.doi.org/10.1097/00010694-193503000-00004
Starkey RL. Cultivation of organisms concerned in the oxidation of thiosulfate. J Bacteriol 1934; 28:365–386. PubMed
Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225–420. http://dx.doi.org/10.1099/00207713-30-1-225
Aleem MIH. Thiosulfate oxidation and electron transport in Thiobacillus novellus. J Bacteriol 1965; 90:95–101. PubMed
Aleem MI, Huang E. Carbon dioxide fixation and carboxydismutase in Thiobacillus novellus. Biochem Biophys Res Commun 1965; 20:515–520. PubMed http://dx.doi.org/10.1016/0006-291X(65)90610-8
Léjohn HB, van Caeseele L, Lees H. Catabolite repression in the facultative chemoautotroph Thiobacillus novellus. J Bacteriol 1967; 94:1484–1491. PubMed
Matin A, Schleiss M, Perez RC. Regulation of glucose transport and metabolism in Thiobacillus novellus. J Bacteriol 1980; 142:639–644. PubMed
Johnson K, Chow CT, Lyric RM, van Caeseele L. Isolation and characterization of bacteriophage for Thiobacillus novellus. J Virol 1973; 12:1160–1163. PubMed
Kappler U, Friedrich CG, Trüper HG, Dahl C. Evidence for two pathways of thiosulfate oxidation in Starkeya novella (formerly Thiobacillus novellus). Arch Microbiol 2001; 175:102–111. PubMed http://dx.doi.org/10.1007/s002030000241
Charles AM, Suzuki I. Mechanism of thiosulfate oxidation by Thiobacillus novellus. Biochim Biophys Acta 1966; 128:510–521. http://dx.doi.org/10.1016/0926-6593(66)90012-9
Oh JK, Suzuki I. Isolation and characterization of a membrane-associated thiosulphate-oxidising system of Thiobacillus novellus. J Gen Microbiol 1977; 99:397–412. http://dx.doi.org/10.1099/00221287-99-2-397
Oh JK, Suzuki I. Resolution of a membrane-associated thiosulphate-oxidising complex of Thiobacillus novellus. J Gen Microbiol 1977; 99:413–423. http://dx.doi.org/10.1099/00221287-99-2-413
De Ley J, van Poucke M. The formation of sulphite during the oxidation of thiosulphate by Thiobacillus novellus. Biochim Biophys Acta 1961; 50:371–373. PubMed http://dx.doi.org/10.1016/0006-3002(61)90342-0
Aguey-Zinsou KF, Bernhardt PV, Kappler U, McEwan AG. Direct electrochemistry of a bacterial sulfate dehydrogenase. J Am Chem Soc 2003; 125:530–535. PubMed http://dx.doi.org/10.1021/ja028293e
Charles AM, Suzuki I. Purification and properties of sulfite:cytochrome c oxidoreductase from Thiobacillus novellus. Biochim Biophys Acta 1966; 128:522–534. http://dx.doi.org/10.1016/0926-6593(66)90013-0
Yamanaka T, Yoshioka T, Kimura K. Purification of sulphite cytochrome c reductase of Thiobacillus novellus and reconstitution of its sulphite oxidase system with the purified constituents. Plant Cell Physiol 1981; 22:613–622.
Southerland WM, Toghrol F. Sulfite oxidase activity in Thiobacillus novellus. J Bacteriol 1983; 156:941–944. PubMed
Toghrol F, Southerland WM. Purification of Thiobacillus novellus sulfite oxidase. Evidence for the presence of heme and molybdenum. J Biol Chem 1983; 258:6762–6766. PubMed
Kappler U, Bennett B, Rethmeier J, Schwarz G, Deutzmann R, McEwan AG, Dahl C. Sulfite: cytochrome c oxidoreductase from Thiobacillus novellus — purification, characterization and molecular biology of a heterodimeric member of the sulfite oxidase family. J Biol Chem 2000; 275:13202–13212. PubMed http://dx.doi.org/10.1074/jbc.275.18.13202
Kappler U, Bailey S. Molecular basis of intramolecular electron transfer in sulfite-oxidizing enzymes is revealed by high resolution structure of a heterodimeric complex of the catalytic molybdopterin subunit and a c-type cytochrome subunit. J Biol Chem 2005; 280:24999–25007. PubMed http://dx.doi.org/10.1074/jbc.M503237200
Kappler U, Bailey S, Feng CJ, Honeychurch MJ, Hanson GR, Bernhardt PV, Tollin G, Enemark JH. Kinetic and structural evidence for the importance of Tyr236 for the integrity of the Mo active site in a bacterial sulfite dehydrogenase. Biochemistry 2006; 45:9696–9705. PubMed http://dx.doi.org/10.1021/bi060058b
Bailey S, Rapson T, Winters-Johnson K, Astashkin AV, Enemark JH, Kappler U. Molecular basis for enzymatic sulfite oxidation — how three conserved active site residues shape enzyme activity. J Biol Chem 2009; 284:2053–2063. PubMed http://dx.doi.org/10.1074/jbc.M807718200
Rapson TD, Kappler U, Hanson GR, Bernhardt PV. Short circuiting a sulfite oxidising enzyme with direct electrochemistry: Active site substitutions and their effect on catalysis and electron transfer. Biochim Biophys Acta (BBA) — Bioenergetics 2011; 1807:108–118. http://dx.doi.org/10.1016/j.bbabio.2010.09.005
Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669–674. http://dx.doi.org/10.1111/j.1095-8312.2007.00864.x
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403–410. PubMed
Korf I, Yandell M, Bedell J. BLAST, O’Reilly, Sebastopol, 2003.
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130–137
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed http://dx.doi.org/10.1128/AEM.03006-05
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed http://dx.doi.org/10.1093/bioinformatics/18.3.452
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed http://dx.doi.org/10.1093/oxfordjournals.molbev.a 026334
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 2008; 57:758–771. PubMed http://dx.doi.org/10.1080/10635150802429642
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. http://dx.doi.org/10.1007/978-3-642-02008-7_13
Swofford DL. PAUP: Phylogenetic Analysis Using Parsimony (and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40:D571–D579. PubMed http://dx.doi.org/10.1093/nar/gkr1100
Lee KB, De Backer P, Aono T, Liu CT, Suzuki S, Suzuki T, Kaneko T, Yamada M, Tabata S, Kupfer DM, et al. The genome of the versatile nitrogen fixer Azorhizobium caulinodans ORS571. BMC Genomics 2008; 9:271. PubMed http://dx.doi.org/10.1186/1471-2164-9-271
Abt B, Han C, Scheuner C, Lu M, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, et al. Complete genome sequence of the termite hindgut bacterium Spirochaeta coccoides type strain (SPN1T), reclassification in the genus Sphaerochaeta as Sphaerochaeta coccoides comb. nov. and emendations of the family Spirochaetaceae and the genus Sphaerochaeta. Stand Genomic Sci 2012; 6:194–209. PubMed http://dx.doi.org/10.4056/sigs.2796069
Meier-Kolthoff JP, Auch AF, Huson DH, Göker M. COPYCAT: Co-phylogenetic Analysis tool. Bioinformatics 2007; 23:898–900. PubMed http://dx.doi.org/10.1093/bioinformatics/btm027
Stamatakis A, Auch AF, Meier-Kolthoff J, Göker M. AxPcoords & parallel AxParafit: statistical co-phylogenetic analyses on thousands of taxa. BMC Bioinformatics 2007; 8:405. PubMed http://dx.doi.org/10.1186/1471-2105-8-405
Felsenstein J. Inferring phylogenies. Sinauer Associates Inc., Sunderland, Massachusetts 2004.
Euzéby JP. List of bacterial names with standing in nomenclature: A folder available on the Internet. Int J Syst Bacteriol 1997; 47:590–592. PubMed http://dx.doi.org/10.1099/00207713-47-2-590
Yarza P, Ludwig W, Euzéby J, Amann R, Schleifer KH, Glöckner FO, Rosselló-Móra R. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Syst Appl Microbiol 2010; 33:291–299. PubMed http://dx.doi.org/10.1016/j.syapm.2010.08.001
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, second edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria), Springer, New York, 2005, p. 1.
Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1.
Validation List no. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6. PubMed http://dx.doi.org/10.1099/ijs.0.64188-0
Kuykendall LD. Order VI. Rhizobiales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 324.
Lee KB, Liu CT, Anzai Y, Kim H, Aono T, Oyaizu H. The hierarchical system of the ‘Alphaproteobacteria’: description of Hyphomonadaceae fam. nov., Xanthobacteraceae fam. nov. and Erythrobacteraceae fam. nov. Int J Syst Evol Microbiol 2005; 55:1907–1919. PubMed http://dx.doi.org/10.1099/ijs.0.63663-0
BAuA. 2010, Classification of Bacteria and Archaea in risk groups. http://www.baua.de TRBA 466, p. 209.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
The DOE Joint Genome Institute. www.jgi.doe.gov
Phrap and Phred for Windows. MacOS, Linux, and Unix. www.phrap.com
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed http://dx.doi.org/10.1101/gr.074492.107
Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Proceeding of the 2006 international conference on bioinformatics & computational biology. Arabnia HR, Valafar H (eds), CSREA Press. June 26–29, 2006:141–146.
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. BMC Bioinformatics 2010; 11:119. PubMed http://dx.doi.org/10.1186/1471-2105-11-119
Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes. Nat Methods 2010; 7:455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955–964. PubMed
Lagesen K, Hallin PF, Rødland E, Stærfeldt HH, Rognes T, Ussery DW. RNammer: consistent annotation of rRNA genes in genomic sequences. Nucleic Acids Res 2007; 35:3100–3108. PubMed http://dx.doi.org/10.1093/nar/gkm160
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439–441. PubMed http://dx.doi.org/10.1093/nar/gkg006
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 2001; 305:567–580. PubMed http://dx.doi.org/10.1006/jmbi.2000.4315
Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004; 340:783–795. PubMed http://dx.doi.org/10.1016/j.jmb.2004.05.028
Greenley DE, Smith DW. Novel pathway of glucose catabolism in Thiobacillus novellus. Arch Microbiol 1979; 122:257–261. http://dx.doi.org/10.1007/BF00411288
Yamanaka T, Fujii K, Kamita Y. Subunits of cytochrome a-type terminal oxidases derived from Thiobacillus novellus and Nitrobacter agilis. J Biochem 1979; 86:821–824. PubMed
Yamanaka T, Fujii K. Cytochrome a-type terminal oxidase derived from Thiobacillus novellus — molecular and enzymatic properties. Biochim Biophys Acta 1980; 591:53–62. PubMed http://dx.doi.org/10.1016/0005-2728(80)90219-4
Yamanaka T, Fukumori Y, Yamazaki T, Kato H, Nakayama K. A comparative survey of several bacterial aa3-type cytochrome c oxidases. J Inorg Biochem 1985; 23:273–277. PubMed http://dx.doi.org/10.1016/0162-0134(85)85035-2
Shoji K, Yamazaki T, Nagano T, Fukumori Y, Yamanaka T. Thiobacillus novellus cytochrome c oxidase contains one heme alpha molecule and one copper atom per catalytic unit. J Biochem 1992; 111:46–53. PubMed
Yamanaka T, Fukumori Y. Thiobacillus novellus cytochrome oxidase can separate some eucaryotic cytochromes c. FEBS Lett 1977; 77:155–158. PubMed http://dx.doi.org/10.1016/0014-5793(77)80224-X
Shoji K, Tanigawa M, Hori K, Tomozawa Y, Yamanaka T. The effects of several nucleotides on the molecular state and catalytic activity of Thiobacillus novellus cytochrome c oxidase — atp affects the oxidase uniquely. Eur J Biochem 1999; 264:960–964. PubMed http://dx.doi.org/10.1046/j.1432-1327.1999.00703.x
Yamanaka T, Nagano T, Shoji K, Fukumori Y. Cytochromes c of Nitrobacter winogradskyi and Thiobacillus novellus: structure, function and evolution. Biochim Biophys Acta 1991; 1058:48–51. PubMed http://dx.doi.org/10.1016/S0005-2728(05)80267-1
Acknowledgements
The work conducted by the U.S. Department of Energy Joint Genome Institute was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, and a Fellowship and grant to UK (DP 0878525). We would like to thank Dr. Richard Webb from the Center for Microscopy and Microanalysis at the University of Queensland for preparing the electron micrograph of S. novella.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Kappler, U., Davenport, K., Beatson, S. et al. Complete genome sequence of the facultatively chemolithoautotrophic and methylotrophic alpha Proteobacterium Starkeya novella type strain (ATCC 8093T). Stand in Genomic Sci 7, 44–58 (2012). https://doi.org/10.4056/sigs.3006378
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.3006378