
Abstract
Thiothrix nivea (Rabenhorst 1865) Winogradsky 1888 (Approved Lists 1980) emend. Larkin and Shinabarger 1983 is the type species of the genus Thiothrix in the family Thiotrichaceae. The species is of interest not only because of its isolated location in the yet to be genomically characterized region of the tree of life, but also because of its life-style with gliding gonidia, the multilayer sheath, rosettes, and the embedded sulfur granules. Strain JP2T is the neotype strain of the species which was first observed by Rabenhorst in 1865 and later reclassified by Winogradsky in 1888 into the then novel genus Thiothrix. This is the first completed (improved-high-quality-draft) genome sequence to be published of a member of the family Thiotrichaceae. The genome in its current assembly consists of 15 contigs in four scaffolds with a total of 4,691,711 bp bearing 4,542 protein-coding and 52 RNA genes and is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others


Introduction
Strain JP2T (= DSM 5205 = ATCC 35100) is the type strain of Thiothrix nivea [1,2] which is the type species of the genus Thiothrix [1,2]. Cultures of the species were first observed and classified as “Beggiatoa nivea” in 1865 by Rabenhorst [3] and later (1888) placed into the novel genus Thiothrix by Winogradsky [2]. The species was included on the Approved List of Bacterial Names Amended edition in 1980 [4]. Axenic cultures isolated from sulfide-containing well water became available in 1980 through the work of J. M. Larkin [5], with the formal description of strain JP2T as the neotype strain of the species T. nivea in 1983 [1], as well as strain JP1 as a reference strain within the species [1]. The generic name derives from the Neo-Greek words theion, sulfur, and thrix, hair [6]. The species epithet is derived from the Latin word nivea snow-white [6]. The species became well known for its sulfur granules, the gliding motility and the typical rosettes [1], which were first observed by Winogradsky [2]. Here we present a summary classification and a set of features for T. nivea JP2T, together with the description of the complete genomic sequencing and annotation.
Classification and features
A representative genomic 16S rRNA sequence of T. nivea JP2T was compared using NCBI BLAST [7] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [8] and the relative frequencies of taxa and keywords (reduced to their stem [9]) were determined, weighted by BLAST scores. The most frequently occurring genus was Thiothrix (100.0%, 12 hits in total). Regarding the single hit to sequences from members of the species, the average identity within HSPs was 99.5%, whereas the average coverage by HSPs was 99.4%. Regarding the four hits to sequences from other members of the genus, the average identity within HSPs was 94.2%, whereas the average coverage by HSPs was 96.0%. Among all other species, the one yielding the highest score was, Thiothrix fructosivorans (GU269554) which corresponded to an identity of 94.5% and an HSP coverage of 100.0%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The highest-scoring environmental sequence was AM490765 (‘Linking and functional nutrient spiraling mats (USA) microbial mat sulfidic cave spring Lower Kane Cave Big Horn LKC22 clone SS LKC22 UB32’), which showed an identity of 96.7% and an HSP coverage of 100.0%. The most frequently occurring keywords within the labels of environmental samples which yielded hits were ‘sulfid’ (4.2%), ‘microbi’ (4.0%), ‘biofilm’ (3.4%), ‘cave’ (2.8%) and ‘karst’ (2.7%) (238 hits in total). Environmental samples which yielded hits of a higher score than the highest scoring species were not found. These keywords reflect the ecological properties reported for the species and strain JP2T in the original description [1,2].
Figure 1 shows the phylogenetic neighborhood of T. nivea in a 16S rRNA based tree. The sequences of the two identical 16S rRNA gene copies in the genome do not differ from the previously published 16S rRNA sequence (L40993), which contains six ambiguous base calls.

Phylogenetic tree highlighting the position of T. nivea relative to the other type strains within the family Thiotrichaceae. The tree was inferred from 1,332 aligned characters [10,11] of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion [12]. Rooting was done initially using the midpoint method [13] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 200 ML bootstrap replicates [14] (left) and from 1,000 maximum parsimony bootstrap replicates [15] (right) if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [16] are labeled with one asterisk, those also listed as ‘Complete and Published’ with two asterisks.
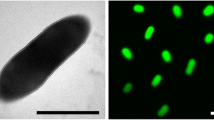
Cells of strain JP2T are rod shaped with various lengths (Figure 2). Cultures of T. nivea contain gliding gonidia, filaments and rosettes (= aggregations of gonidial cells, not visible in Figure 2) [1]. The presence of a sheath was first reported in the 19th century [2] and later confirmed for the neotype strain [1]. The sheath contains several separate layers [1] of so far unknown structure. Motility was observed, but no flagella [1]. Numerous genes allocated to the functional role category motility were identified in the genome (see below). Many of these genes might be involved in the formation of the polar located fimbriae [32]. The typical rosettes generated by T. nivea are known from sulfide-containing waters [1,2]. Sulfur granules are invaginated by the cells, as reported in detail by Larkin and Shinabarger [1]. Strain JP2T stains Gram-negative, and grows only aerobically, best within a temperature range of 20–30°C [1]. Both the neotype strain and reference strain JP1 produce oxidase, but not catalase. The strains also produce poly-β-hydroxybutyrate [1]. Strain JP2T uses only four carbon sources; acetate, malate, pyruvate and oxalacetate [1]. Ammonia and nitrate (but not nitrite) are used as sole nitrogen sources [1]. The sole sulfur sources are sulfide and thiosulfate. What remains unresolved, based on the literature is whether or not T. nivea is autotrophic, obtaining carbon from CO2 and energy via oxidation of sulfide as reported by Winogradsky [13] or not, as reported by Larkin and Shinabarger [1]. In the case in which strain JP2T could use CO2 as a carbon source as well as acetate, malate, pyruvate and oxalacetate, while oxidizing the reduced sulfur compounds, it could be considered to be a mixotroph [1].

Scanning electron micrograph of T. nivea JP2T
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [33], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [34]. The genome project is deposited in the Genome On Line Database [16] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
T. nivea JP2T, DSM 5205, was grown in DSMZ medium 1300 (Thiothrix Medium) [35] at 25°C. DNA was isolated from 0.5–1 g of cell paste using Jetflex Genomic DNA Purification Kit (GENOMED 600100) following the standard protocol as recommended by the manufacturer, but adding 10µl proteinase K for one hour extended lysis at 58°C. DNA is available through the DNA Bank Network [36].
Chemotaxonomy
There are no chemotaxonomic data on cell wall structure, cellular lipids, quinones or polar lipids of strain JP2T.
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [37]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 269 contigs in four scaffolds was converted into a phrap assembly by [38] making fake reads from the consensus to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (518.8 Mb) was assembled with Velvet [39], and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 162.5 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [38] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [37], Dupfinisher, or sequencing cloned bridging PCR fragments with subcloning or transposon bombing (Epicentre Biotechnologies, Madison, WI). Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 632 additional reactions were necessary to close gaps and to raise the quality of the final sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [40]. This genome is not finished. The improved high quality draft consists of 15 contigs in four scaffolds. Some mis-assemblies are possible in the final assembly. Together, the combination of the Illumina and 454 sequencing platforms provided 140.4 × coverage of the genome. The final assembly contained 444,417 pyrosequence and 14,381,947 Illumina reads.
Genome annotation
Genes were identified using Prodigal [41] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [42]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [43].
Genome properties
The genome consists in the current assembly of 15 contigs in four scaffolds with a length of 5,599 bp, 7,015 bp, 40,927 bp, and 4,638,170 bp, respectively, and a G+C content of 54.9% (Table 3). Of the 4,594 genes predicted, 4,542 were protein-coding genes, and 52 RNAs; 213 pseudogenes were also identified. The majority of the protein-coding genes (98.8%) were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Insight into the genome sequence
The genomic basis for gliding motility is not yet completely resolved, but the requirement of the genes gldA, gldF and gldG was described [44]. Genes gldA, gldF and gldG exhibit a high degree of sequence similarity to components of ABC transporters [44]. A closer examination of the JP2T genome revealed a region of three genes (Thini_0004.00016790, Thini_0004.00016780, Thini_0004.00016770, currently annotated as ABC-type uncharacterized transport system), for which the derived protein sequences show high similarity to GldA, GldF and GldG of Bdellovibrio bacteriovorus HD100 (Bd1023, Bd1024 and Bd1025) [45]. The requirement of gldA for gliding motility was experimentally shown for Flavobacterium johnsoniae. A non-motile mutant lacking the intact gldA gene was complemented by a vector carrying an intact gldA gene. The motility of the mutant was restored [46].
While we were able to locate a phosphoenolpyruvate carboxylase gene in the genome, (Thini_0004.00035050), we could not identify a gene for malate dehydrogenase. Unless T. nivea encodes a malate dehydrogenase that is not homologous to other malate dehydrogenases, we can not confirm for the neotype strain the genomic basis for the speculation that the original T. nivea culture can fix CO2 as reported by Winogradsky [13] and is a mixotroph [1].
References
Larkin JM, Shinabarger DL. Characterization of Thiothrix nivea. Int J Syst Bacteriol 1983; 33:841–846. doi:10.1099/00207713-33-4-841
Winogradsky S. Beiträge zur Morphologie und Physiologie der Bakterien. In: Zur Morphologie und Physiologie der Schwefelbakterien. Felix A (ed) Leipzig 1888; 1:1–120.
Rabenhorst L. Flora Europa algarum aquae dulcis et submarinae, sect II. E. Kummer, Leipzig 1865.
Skerman VBD, McGowan V, Sneath PHA, eds. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225–420. doi:10.1099/00207713-30-1-225
Larkin JM. Isolation of Thiothrix in pure culture and observation of a filamentous epiphyte on Thiothrix. Curr Microbiol 1980; 4:155–158. doi:10.1007/BF02602820
Euzéby JP. List of Bacterial Names with Standing in Nomenclature: a folder available on the Internet. Int J Syst Bacteriol 1997; 47:590–592. PubMed doi:10.1099/00207713-47-2-590
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Bascic local alignment search tool. J Mol Biol 1990; 215:403–410. PubMed
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed doi:10.1128/AEM.03006-05
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130–137.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669–674. doi:10.1111/j.1095-8312.2007.00864.x
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-7_13
Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002.
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed doi:10.1093/nar/gkp848
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1.
Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, second edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria), Springer, New York, 2005, p. 1.
Validation list No. 106. Int J Syst Evol Microbiol 2005; 55:2235–2238. doi:10.1099/ijs.0.64108-0
Garrity GM, Bell JA, Lilburn T. Order V. Thiotrichales ord. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, second edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria), Springer, New York, 2005, p. 131.
Garrity GM, Bell JA, Lilburn T. Family I. Thiotrichaceae fam. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, second edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria), Springer, New York, 2005, p. 131.
Brock TD. Genus II. Thiothrix Winogradsky 1888, 39. In: Buchanan RE, Gibbons NE (eds), Bergey’s Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 119–120.
Howarth R, Unz RF, Seviour EM, Seviour RJ, Blackall LL, Pickup RW, Jones JG, Yaguchi J, Head IM. Phylogenetic relationships of filamentous sulfur bacteria (Thiothrix spp. and Eikelboom type 021N bacteria) isolated from wastewater-treatment plants and description of Thiothrix ei-kelboomii sp. nov., Thiothrix unzii sp. nov., Thiothrix fructosivorans sp. nov. and Thiothrix defluvii sp. nov. Int J Syst Bacteriol 1999; 49:1817–1827. PubMed doi:10.1099/00207713-49-4-1817
Aruga S, Kamagata Y, Kohno T, Hanada S, Nakamura K, Kanagawa T. Characterization of filamentous Eikelboom type 021N bacteria and description of Thiothrix disciformis sp. nov. and Thiothrix flexilis sp. nov. Int J Syst Evol Microbiol 2002; 52:1309–1316. PubMed doi:10.1099/ijs.0.02177-0
Bland JA, Staley JT. Observations on the biology of Thiothrix. Arch Microbiol 1978; 117:79–87. doi:10.1007/BF00689355
Lackey JB. ELackey EW, Morgan GB. Taxonomy and ecology of the sulfur bacteria. Eng Prog Univ Fla Bull Ser 1965; 199:3–23.
BAuA. 2010, Classification of Bacteria and Archaea in risk groups. http://www.baua.de TRBA 466, p. 241.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Larkin JM, Nelson R. Mechanism of attachment of swarm cells ot Thiothrix nivea. J Bacteriol 1987; 169:5877–5879. PubMed
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven Genomic Encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
List of growth media used at DSMZ: http://www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011; 9:51–55. doi:10.1089/bio.2010.0029
The DOE Joint Genome Institute. http://www.jgi.doe.gov
Phrap and Phred for Windows. MacOS, Linux, and Unix. http://www.phrap.com
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed doi:10.1101/gr.074492.107
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
McBride MJ. Bacterial gliding motility: Multiple mechanisms for cell movement over surfaces. Annu Rev Microbiol 2001; 55:49–75. PubMed doi:10.1146/annurev.micro.55.1.49
Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C, Keller H, Lambert C, Evans KJ, Goesmann A, et al. A Predator Unmasked: Life Cycle of Bdellovibrio bacteriovorus from a Genomic Perspective. Science 2004; 303:689–692. PubMed doi:10.1126/science.1093027
Agarwal S, Hunnicutt DW, McBride MJ. Cloning and characterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) gliding motility gene, gldA. Proc Natl Acad Sci USA 1997; 94:12139–12144. PubMed doi:10.1073/pnas.94.22.12139
Acknowledgements
We would like to gratefully acknowledge the help of Anja Frühling (DSMZ) for growing T. nivea cultures. This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-1.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Lapidus, A., Nolan, M., Lucas, S. et al. Genome sequence of the filamentous, gliding Thiothrix nivea neotype strain (JP2T). Stand in Genomic Sci 5, 398–406 (2011). https://doi.org/10.4056/sigs.2344929
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.2344929