
Abstract
Halanaerobium praevalens Zeikus et al. 1984 is the type species of the genus Halanaerobium, which in turn is the type genus of the family Halanaerobiaceae. The species is of interest because it is able to reduce a variety of nitro-substituted aromatic compounds at a high rate, and because of its ability to degrade organic pollutants. The strain is also of interest because it functions as a hydrolytic bacterium, fermenting complex organic matter and producing intermediary metabolites for other trophic groups such as sulfate-reducing and methanogenic bacteria. It is further reported as being involved in carbon removal in the Great Salt Lake, its source of isolation. This is the first completed genome sequence of a representative of the genus Halanaerobium and the second genome sequence from a type strain of the family Halanaerobiaceae. The 2,309,262 bp long genome with its 2,110 protein-coding and 70 RNA genes is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others


Introduction
Strain GSLT (= DSM 2228 =ATCC 33744) is the type strain of the species Halanaerobium praevalens, which is the type species of its genus Halanaerobium [1]. Originally described as Haloanaerobium [2], the name was later changed to Halanaerobium to conform with rule 61 of the Bacteriological Code [3]. The genus currently consists of nine validly named species [4]. The genus name is derived from the Latinized Greek word hals; halos meaning salt, the Latinized Greek word an-meaning not, the Latinized Greek word aer meaning air and the Latinized Greek word bios meaning life, yielding the Neo-Latin word ‘Halanaerobium’ meaning ‘salt organism which grows in the absence of air’ [4]. The species epithet is derived from the Latin word ‘praevalens’ (very powerful, very strong, here prevalent) [4]. Strain GSLT was isolated from the hypersaline surface sediments of Great Salt Lake, Utah, USA [2]. Further strains of H. praevalens have been isolated from canned salted Swedish fermented herrings referred to as Surströmming [5] and probably also from the Red Sea [6]. Other members of the genus have been isolated also from high salt environments distributed worldwide [7–10]. The enzymatic activities of the fatty acid synthetase complex and the D-BAPA (N′-benzoyl-arginine-p-nitroanilide)-ase of H. praevalens have been studied in more detail [11,12]. Here we present a summary classification and a set of features for H. praevalens strain GSLT, together with the description of the complete genome sequencing and annotation.
Classification and features
A representative genomic 16S rRNA sequence of H. praevalens was compared using NCBI BLAST under default values (e.g., considering only the best 250 hits) with the most recent release of the Greengenes database [13] and the relative frequencies, weighted by BLAST scores, of taxa and keywords (reduced to their stem [14]) were determined. The five most frequent genera were Halanaerobium (81.9%), Halothermothrix (7.8%), Halanaerobacter (2.7%), Acetohalobium (2.3%) and Natroniella (1.9%). Regarding hits to sequences from other members of the genus, the average identity within HSPs (high-scoring segment pairs) was 97.8%, whereas the average coverage by HSPs was 96.3%. The species yielding the highest score was Halanaerobium saccharolyticum. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The five most frequent keywords within the labels of environmental samples which yielded hits were ‘microbi’ (9.4%), ‘hypersalin’ (9.1%), ‘mat’ (8.6%), ‘len, miniprim, new, view, world’ (8.5%) and ‘food’ (3.4%). The single most frequent keyword within the labels of environmental samples which yielded hits of a higher score than the highest scoring species was ‘hypersalin, len, mat, microbi, miniprim, new, view, world’ (12.5%). These key words are in line with the ecology and the niche from where strains of H. praevalens have been isolated.
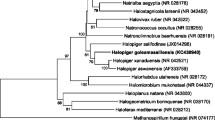
Figure 1 shows the phylogenetic neighborhood of H. praevalens GSLT in a 16S rRNA gene based tree. The sequences of the four 16S rRNA gene copies in the genome differ from each other by up to five nucleotides, and differ by up to five nucleotides from the previously published 16S rRNA gene sequence (AB022034).

Phylogenetic tree highlighting the position of H. praevalens GSLT relative to the other type strains within the family Halanaerobiaceae. The tree was inferred from 1,460 aligned characters [15,16] of the 16S rRNA gene sequence under the maximum likelihood criterion [17] and rooted in accordance with the current taxonomy. The branches are scaled in terms of the expected number of substitutions per site. Numbers to the right of bifurcations are support values from 1,000 bootstrap replicates [18] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [19] are labeled with an asterisk, published genomes with two asterisks [20].
The cells of strain GSLT are straight and rod-shaped (1 × 2.4 µm) (Figure 2) when grown to the mid-log phase at 37ºC on CS medium containing 12.5% NaCl and 0.5% glucose [2]. When grown at higher NaCl concentrations (> 20%) the cells appear granulated and shorter in length [2]. Single colonies were reported as white to translucent in color, 0.5–2.0 mm in diameter, and glistened, when grown on agar plates containing CS medium, 12.5% NaCl, and 0.5% glucose [2]. H. praevalens cells stain Gram-negative [2] and electron microscopy in thin section revealed architectural features typical of Gram-negative bacteria [2]. However, the positive D-BAPA-ase reaction [11] confirms its phylogenetic affiliation to the endospore-forming firmicutes (Table 1). In this respect, H. praevalens is able to hydrolyze only the D- but not the L-isomer of N′-benzoyl-arginine-p-nitroanilide (BAPA) [11]. The activity of D-BAPA-ase was highest at low NaCl concentration (100 mM) and completely inhibited at NaCl concentration equal or higher than 1.0 M (∼12%) [11]. Strain GSLT is described to be non-motile, although many flagellar genes have been identified in the genome (see below). Other isolates of H. praevalens were described as motile [5,6], as were other members of the genus [7,9,10], suggesting strain GSLT is atypical with regard to motility. The organism is a strictly anaerobic chemoorganotroph [2]. It grows at NaCl concentrations between 2% and 30%, with optimal growth at approximately 13% [2]. The doubling time is 4 h at 12.5% NaCl and 7 h at 25% NaCl in complex CS medium [2]. The temperature range for growth ranges from 5ºC to 60ºC, with an optimum at 37ºC [2]. The pH range for growth is between 6.0 and 9.0, with an optimum at pH between 7.0 and 7.4 [2]. Strain GSLT is able to utilize carbohydrates (including pectin and N-acetylglucosamine), amino acids, yeast extract, and trypticase; the two latter serving as carbon and energy sources on complex medium [2]. The fermentation of glucose yielded butyrate, acetate, propionate, H2, and CO2 as major products [2]. Also, fructose, D-mannose and maltose are utilized and methionine is transformed to methylmercaptan [2]. Penicillin, tetracycline, cycloserine, chloramphenicol (each at 100 µg/ml culture) or sodium azide (500 µg/ml) completely inhibit the growth of H. praevalens [2]. Strain GSLT was also able to degrade nitro-substituted aromatic compounds such as nitrobenzene, o-nitrophenol, m-nitrophenol, p-nitrophenol, 2,4-dinitrophenol, and 2,4-dinitroaniline [34]. The fatty acid synthetase of H. praevalens is only slightly inhibited at 17.5% and was the first reported to be active in the presence of high salt concentrations [12]. H. praevalens was reported to be involved in carbon sequestration in the Great Salt Lake [35], since it is present in the sediments of this lake in high numbers (≥ 108 cells/ml) [2,36]. H. praevalens regulates its internal osmotic pressure by the accumulation of salts (Na+, K+, Cl-) rather than by compatible solutes [36]. High concentrations of these salts were measured inside the cells, in sufficient concentration to be isotonic or hypertonic with the medium [37]. Thiosulfate reduction and rhodanese-like enzyme (thiosulfate:cyanide sulfur-transferase) activities also tested positive in strain GSLT [8]. Early in 1987, Matheson et al. [38] established the primary structure of the ribosomal A-protein of the strain GSLT, which is the equivalent to the ribosomal protein L12 from Escherichia coli.

Scanning electron micrograph of H. praevalens GSLT
Chemotaxonomy
When grown on CS medium, at 5% NaCl, more lipids are produced than at 25% NaCl (3.74% and 2.54% of the dry weight of the organism, respectively) [2]. At 5% NaCl, the fractions of glycolipids and phospholipids are 46.9% and 44.5% of the total lipids, respectively. At 25%, the proportion changes in favor of phospholipids (49.1%), whereas glycolipids decrease (43.0%) [2]. The glycolipids consist of a single component diacylglycerol derivative, while the phospholipids consist mainly of cardiolipin (CL), phosphatidyl glycerol (PG), and three minor unidentified constituents [2]. When grown on CS medium, at 5%, the major fatty acids are C14:0 (49.3%), C16:1 (31.3%) and C16:0 (11.4%). At 25%, these fractions change to 36.8%, 39%, and 22.7%, respectively. Similar though more detailed results on the fatty acid composition have been reported recently [6].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [39], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [40]. The genome project is deposited in the Genome On Line Database [19] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
H. praevalens GSLT, DSM 2228, was grown anaerobically in DSMZ medium 210 (‘Haloanaerobium’ medium) [41] at 30–37°C. DNA was isolated from 0.5–1 g of cell paste using MasterPure Gram-positive DNA purification kit (Epicentre MGP04100) following the standard protocol as recommended by the manufacturer, with modification st/DL for cell lysis as described in Wu et al. [40]. DNA is available through the DNA Bank Network [42].
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [43]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly, consisting of 85 contigs in 31 scaffolds, was converted into a phrap [44] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina sequencing data (360 Mb) was assembled with Velvet [45] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 401.6 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [44] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [43], Dupfinisher [46], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 417 additional reactions and ten shatter libraries were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [47]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 375.4 × coverage of the genome. The final assembly contained 838,597 pyrosequence and 12,903,210 Illumina reads.
Genome annotation
Genes were identified using Prodigal [48] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [49]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [50].
Genome properties
The genome consists of a 2,309,262 bp long chromosome with a G+C content of 30.3% (Table 3 and Figure 3). Of the 2,180 genes predicted, 2,110 were protein-coding genes, and 70 RNAs; 42 pseudogenes were also identified. The majority of the protein-coding genes (77.7%) were assigned with a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the chromosome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
References
Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List No. 16. Int J Syst Bacteriol 1984; 34:503–504. doi:10.1099/00207713-34-4-503
Zeikus JG, Hegge PW, Thompson TE, Phelps TJ, Langworthy TA. Isolation and description of Haloanaerobium praevalens gen. nov. and sp. nov., an obligately anaerobic halophile common to Great Salt Lake sediments. Curr Microbiol 1983; 9:225–233. doi:10.1007/BF01567586
Oren A. Change of the names Haloanaerobiales, Haloanaerobiaceae and Haloanaerobium to Halanaerobiales, Halanaerobiaceae and Halanaerobium, respectively, and further nomenclatural changes within the order Halanaerobiales. Int J Syst Evol Microbiol 2000; 50:2229–2230. PubMed doi:10.1099/00207713-50-6-2229
Euzéby JP. List of bacterial names with standing in nomenclature: a folder available on the Internet. Int J Syst Bacteriol 1997; 47:590–592. PubMed doi:10.1099/00207713-47-2-590
Kobayashi T, Kimura B, Fujii T. Strictly anaerobic halophiles isolated from canned Swedish fermented herrings (Surströmming). Int J Food Microbiol 2000; 54:81–89. PubMed doi:10.1016/S0168-1605(99)00172-5
Eder W, Jahnke LL, Schmidt M, Huber R. Microbial diversity of the brine-seawater interface of the Kebrit Deep, Red Sea, studied via 16S rRNA gene sequences and cultivation methods. Appl Environ Microbiol 2001; 67:3077–3085. PubMed doi:10.1128/AEM.67.7.3077-3085.2001
Tsai CR, Garcia JL, Patel BKC, Cayol JL, Baresi L, Mah RA. Haloanaerobium alcaliphilum sp. nov., an anaerobic moderate halophile from the sediments of Great Salt Lake, Utah. Int J Syst Bacteriol 1995; 45:301–307. PubMed doi:10.1099/00207713-45-2-301
Ravot G, Casalot L, Ollivier B, Loison G, Magot M. rdlA, a new gene encoding a rhodanese-like protein in Halanaerobium congolense and other thiosulfate-reducing anaerobes. Res Microbiol 2005; 156:1031–1038. PubMed doi:10.1016/j.resmic.2005.05.009
Cayol JL, Ollivier B, Patel BKC, Ageron E, Grimont PAD, Prensier G, Garcia JL. Haloanaerobium lacusroseus sp. nov., an extremely halophilic fermentative bacterium from the sediments of a hypersaline Lake. Int J Syst Bacteriol 1995; 45:790–797. PubMed doi:10.1099/00207713-45-4-790
Bhupathiraju VK, Mclnerney MJ, Woese CR, Tanner RS. Haloanaerobium kushneri sp. nov., an obligately halophilic, anaerobic bacterium from an oil brine. Int J Syst Bacteriol 1999; 49:953–960. PubMed doi:10.1099/00207713-49-3-953
Oren A, Gofshtein-Gandman LV, Keynan A. Hydrolysis of N′-benzoyl-D-arginine-p-nitroanilide by members of the Haloanaerobiaceae: additional evidence that Haloanaerobium praevalens is related to endospore-forming bacteria. FEMS Microbiol Lett 1989; 58:5–10.
Oren A, Gurevich P. The fatty acid synthetase complex of Haloanaerobium praevalens is not inhibited by salt. FEMS Microbiol Lett 1993; 108:287–290. doi:10.1111/j.1574-6968.1993.tb06117.x
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie E, Keller K, Huber T, Dalevi D, Hu P, Andersen G. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed doi:10.1128/AEM.03006-05
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems. 1980; 14:130–137.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-7_13
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed doi:10.1093/nar/gkp848
Mavromatis K, Ivanova N, Anderson I, Lykidis A, Hooper SD, Sun H, Kunin V, Lapidus A, Hugenholtz P, Patel B, Kyrpides NC. Genome analysis of the anaerobic thermohalophilic bacterium Halothermothrix orenii. PLoS ONE 2009; 4:e4192. PubMed doi:10.1371/journal.pone.0004192
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1. Springer, New York 2001:119–169.
Gibbons NE, Murray RGE. Proposals concerning the higher taxa of Bacteria. Int J Syst Bacteriol 1978; 28:1–6. doi:10.1099/00207713-28-1-1
Validation list 132. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2010; 60:469–472. doi:10.1099/ijs.0.022855-0
Rainey FA. Class II. Clostridia class nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 3, Springer-Verlag, New York, 2009, p. 736.
Validation List No. 55. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1995; 45:879–880. doi:10.1099/00207713-45-4-879
Judicial Commission of the International Committee on Systematics of Prokaryotes. The nomenclatural types of the orders Acholeplasmatales, Halanaerobiales, Halobacteriales, Methanobacteriales, Methanococcales, Methanomicrobiales, Planctomycetales, Prochlorales, Sulfolobales, Thermococcales, Thermoproteales and Verrucomicrobiales are the genera Acholeplasma, Halanaerobium, Halobacterium, Methanobacterium, Methanococcus, Methanomicrobium, Planctomyces, Prochloron, Sulfolobus, Thermococcus, Thermoproteus and Verrucomicrobium, respectively. Opinion 79. Int J Syst Evol Microbiol 2005; 55:517–518. PubMed doi:10.1099/ijs.0.63548-0
Rainey FA, Zhilina TN, Boulygina ES, Stackebrandt E, Tourova TP, Zavarzin GA. The taxonomic status of the fermentative halophilic anaerobic bacteria: description of Haloanaerobiales ord. nov., Halobacteroidaceae fam. nov., Orenia gen. nov. and further taxonomic rearrangements at the genus and species level. Anaerobe 1995; 1:185–199. PubMed doi:10.1006/anae.1995.1018
Oren A, Paster BJ, Woese CR. Haloanaerobiaceae: a new family of moderately halophilic, obligately anaerobic bacteria. Syst Appl Microbiol 1984; 5:71–80.
BAuA. Classification of bacteria and archaea in risk groups. TRBA 2005; 466:84.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Oren A, Gurevich P, Henis Y. Reduction of nitrosubstituted aromatic compounds by the halophilic anaerobic eubacteria Haloanaerobium praevalens and Sporohalobacter marismortui. Appl Environ Microbiol 1991; 57:3367–3370. PubMed
Oren A, Weisburg WG, Kessel M, Woese CR. Halobacteroides halobius gen. nov., sp. nov., a moderately halophilic anaerobic bacterium from the bottom sediments of the dead sea. Syst Appl Microbiol 1984; 5:58–70.
Oren A. The ecology and taxonomy of anaerobic halophilic eubacteria. FEMS Microbiol Rev 1986; 39:23–29. doi:10.1111/j.1574-6968.1986.tb01838.x
Oren A. 2006. Life at high salt concentrations. In: M Dworkin, S Falkow, E Rosenberg, KH Schleifer E Stackebrandt (eds), The Prokaryotes, 3. ed, vol. 2. Springer, New York, p. 263–282.
Matheson AT, Louie KA, Tak BD, Zuker M. The primary structure of the ribosomal A-protein (L12) from the halophilic eubacterium Haloanaerobium praevalens. Biochimie 1987; 69:1013–1020. PubMed doi:10.1016/0300-9084(87)90001-0
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreservation and Biobanking 2011; 9:51–55. doi:10.1089/bio.2010.0029
JGI website. http://www.jgi.doe.gov
The Phred/Phrap/Consed software package. http://www.phrap.com
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed doi:10.1101/gr.074492.107
Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Proceeding of the 2006 international conference on bioinformatics & computational biology. Arabnia HR, Valafar H (eds), CSREA Press. June 26–29, 2006: 141–146.
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Acknowledgements
We would like to gratefully acknowledge the help of Olivier D. Ngatchou-Djao (HZI) for drafting the manuscript, and Helga Pomrenke (DSMZ) for growing H. praevalens cultures. This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-2.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ivanova, N., Sikorski, J., Chertkov, O. et al. Complete genome sequence of the extremely halophilic Halanaerobium praevalens type strain (GSLT). Stand in Genomic Sci 4, 312–321 (2011). https://doi.org/10.4056/sigs.1824509
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.1824509