
Abstract
Isosphaera pallida (ex Woronichin 1927) Giovannoni et al. 1995 is the type species of the genus Isosphaera. The species is of interest because it was the first heterotrophic bacterium known to be phototactic, and it occupies an isolated phylogenetic position within the Planctomycetaceae. Here we describe the features of this organism, together with the complete genome sequence and annotation. This is the first complete genome sequence of a member of the genus Isosphaera and the third of a member of the family Planctomycetaceae. The 5,472,964 bp long chromosome and the 56,340 bp long plasmid with a total of 3,763 protein-coding and 60 RNA genes are part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others


Introduction
Strain IS1BT (= ATCC 43644) is the type strain of Isosphaera pallida which in turn is the type and sole species of the genus Isosphaera [1–2]. The genus Isosphaera is one out of nine genera in the family Planctomycetaceae [3]. The genus name is derived from the Greek adjective isos, equal and sphaera, a ball, globe, yielding Isosphaera, sphere of equal size [4]. The species epithet pallida is derived from the Latin adjective pallida, pale [1]. Strain IS1BT was isolated from a hot spring in Kah-nee-tah, Oregon, USA [1]. Other closely related strains belonging to the species were isolated from several warm springs in North America [1]. The cells resemble Isocystis pallida Worochin 1927 [5] which was previously described as a cyanobacterium and later as a yeast. Here we present a summary classification and a set of features for I. pallida strain IS1BT, together with the description of the complete genomic sequencing and annotation.
Classification and features
A representative genomic 16S rRNA sequence of strain IS1BT was compared using NCBI BLAST under default values (e.g., considering only the best 250 hits) with the most recent release of the Greengenes database [6] and the relative frequencies, weighted by BLAST scores, of taxa and keywords (reduced to their stem [7]) were determined. The five most frequent genera were Isosphaera (35.4%), Nostocoida (26.4%; a genus with Candidatus status [8]), Singulisphaera (20.4%), ‘Isophaera’ (15.9%; a misspelling of Isosphaera) and Planctomyces (1.9%). The species yielding the highest score was Candidatus Nostocoida limicola [8]. The five most frequent keywords within the labels of environmental samples which yielded hits were ‘skin’ (3.9%), ‘soil’ (3.0%), ‘fossa’ (2.2%), ‘adult/zebrafish’ (2.2%) and ‘microbi’ (1.9%). The two most frequent keywords within the labels of environmental samples which yielded hits of a higher score than the highest scoring species were ‘adult, zebrafish’ (10.0%) and ‘conventionally-rais, digest, gender, germ-fre, gut, habitat, host, mice, micro-biota, mix, pool, recipi, reciproc, select, tract, transplant’ (5.0%), i.e. many ties occurred, rendering it difficult to ecologically interpret this outcome.
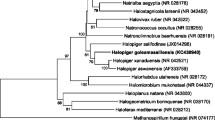
Figure 1 shows the phylogenetic neighborhood of I. pallida IS1BT in a 16S rRNA based tree. The sequences of the three copies in the genome do not differ from each other, and differ by two nucleotides from the previously published 16S rRNA sequence (AJ231195).

Phylogenetic tree highlighting the position of I. pallida relative to the other type strains within the class family Planctomycetacia. The tree was inferred from 1,362 aligned characters [9–10] of the 16S rRNA gene sequence under the maximum likelihood criterion [11] and rooted in with members of the class Phycisphaerae. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 450 bootstrap replicates [12] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [13] are shown in blue, published genomes in bold [14–15].
Cells of strain IS1BT are spherical with 2.5 to 3 µm in diameter (Figure 2 and Table 1), with cell growth and division occurring by intercalary budding, resulting in filaments [1]. The cells are salmon-colored (caused by carotenoids), contain gas vesicles and resemble Isocystis pallida Worochin 1927 [5]. Ultra-thin sections observed by TEM revealed pit-like ultrastructural features in the cell wall [1–24]. The cells contain numerous pili (not visible in Figure 2) but no flagella, and form motile phototactic “comets” in liquid cultures or on media containing Gelrite® as the solidifying agent [1].

Photomicrograph (1000 x) of I. pallida IS1BT (provided by ATCC)
Chemotaxonomy
Muramic acid and diaminopimelic acid are absent from the cell wall [1–24], like in other members of the Planctomycetes. Cells stain Gram-negative but lack an outer membrane [1]. Cells possess a proteinaceous cell wall structure without cysteine, methionine, proline and tryptophan [24]. Ester-linked lipids with predominantly unbranched C14 and C18 fatty acids, traces of C18:1 acids, no hydroxyl-fatty acids [24].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [25], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [26]. The genome project is deposited in the Genomes OnLine Database [13] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
I. pallida IS1BT, ATCC 43644, has been in the American Type Culture Collection since July 1987. The culture used at ATCC to prepare genomic DNA (gDNA) for sequencing was only two transfers away from the original deposit. The purity of the culture was determined by growth in ATCC medium 1962 Broth [27] at 45oC under aerobic conditions. Cells were harvested by centrifugation after 72 hours of incubation. The cell pellet exhibited a salmon color. Genomic DNA was extracted from lysozyme-treated cells using a standard CTAB and phenol-chloroform protocol. The purity, quality and size of the bulk gDNA preparation were assessed according to DOE-JGI guidelines. Amplification and partial sequencing of the 16S rRNA gene confirmed the isolate as I. pallida. The quantity of the DNA was determined on a 1% agarose using gel mass markers of known concentration supplied by JGI. The average fragment size of the purified gDNA determined to be ∼43 kb by pulsed-field gel electrophoresis.
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [28]. Pyrosequencing reads were assembled using the Newbler assembler version 2.0.00.20-PostRelease-11-05-2008-gcc-3.4.6 (Roche). The initial Newbler assembly, consisting of 36 contigs in 1 scaffold, was converted into a phrap assembly by making fake reads from the consensus [29], to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (461 Mb) was assembled with Velvet [30] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 172.7 Mb of 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [29] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [28], Dupfinisher, or sequencing cloned bridging PCR fragments with subcloning or transposon bombing (Epicentre Biotechnologies, Madison, WI) [31]. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F.Chang, unpublished). A total of 411 additional reactions and 14 shatter libraries were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [32]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 140.7 × coverage of the genome. The final assembly contained 764,175 pyrosequence and 16,816,247 Illumina reads.
Genome annotation
Genes were identified using Prodigal [33] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [34]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [35].
Genome properties
The genome consists of a 5,472,964 bp long chromosome with a 62% GC content and a 56,340 bp plasmid with 67% GC content (Table 3 and Figures 3a and 3b). Of the 3,823 genes predicted, 3,763 were protein-coding genes, and 60 RNAs; 41 pseudogenes were identified. The majority of the protein-coding genes (59.7%) were assigned with a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the chromosome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.

Graphical circular map of the plasmid (not drown to scale with chromosome). From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
References
Giovannoni SJ, Schabtach E, Castenholtz RW. Isosphaera pallida, gen. and comb. nov., a gliding, budding eubacterium from hot springs. Arch Microbiol 1987; 147:276–284. doi:10.1007/BF00463488
Validation List No. 54. Int J Syst Bacteriol 1995; 45:619–620. doi:10.1099/00207713-45-3-619
Schlesner H, Stackebrandt E. Assignment of the genera Planctomyces and Pirella to a new family Planctomycetaceae fam. nov. and description of the order Planctomycetales ord. nov. Syst Appl Microbiol 1986; 8:174–176.
Euzéby JP. List of Bacterial Names with Standing in Nomenclature: a folder available on the internet. Int J Syst Bacteriol 1997; 47:590–592. PubMed doi:10.1099/00207713-47-2-590
Woronichin NN. Materiali kagologitscheskoj flore i rastitjelnosti mineralnich istotschnikov gruppi Kaukaskich mineralnich wod. Trav Inst Balneol aux Eaux Miner du Caucase 1927; 5:90–121.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie E, Keller K, Huber T, Dalevi D, Hu P, Andersen G. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed doi:10.1128/AEM.03006-05
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130–137.
Backall LL, Seviour EM, Bradford D, Rossetti S, Tandoi V, Sviour RJ. ‘Candidatus Nostocoida limicola’, a filamentous bacterium from activated sludge. Int J Syst Evol Microbiol 2000; 50:703–709. PubMed
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-7_13
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354; doi:10.1093/nar/gkp848. PubMed
Clum A, Tindall BJ, Sikorski J, Ivanova N, Mavromatis K, Lucas S, Glavina Del Rio T, Nolan M, Chen F, Tice H, et al. Complete genome sequence of Pirellula staleyi type strain (ATCC 27377T). Stand Genomic Sci 2009; 1:308–316. PubMed doi:10.4056/sigs.51657
LaButti K, Sikorski J, Schneider S, Nolan M, Lucas S, Glavina Del Rio T, Tice H, Cheng JF, Goodwin L, Pitluck S, et al. Complete genome sequence of Planctomyces limnophilus type strain (290T). Stand Genomic Sci 2010; 3:47–56. PubMed doi:10.4056/sigs.1052813
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Woese CR, Kandier O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Springer, New York, 2001, p. 119–169.
Garrity GM, Holt JG. Taxonomic Outline of the Archaea and Bacteria. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 155–166.
List Editor. Validation List no. 23. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1987; 37:179. doi:10.1099/00207713-37-2-179
Euzéby JP, Tindall BJ. Nomenclatural type of orders: corrections necessary according to Rules 15 and 21a of the Bacteriological Code (1990 Revision), and designation of appropriate nomenclatural types of classes and subclasses. Request for an opinion. Int J Syst Evol Microbiol 2001; 51:725–727. PubMed
Judicial Commission of the International Committee on Systematics of Prokaryotes. The nomenclatural types of the orders Acholeplasmatales, Halanaerobiales, Halobacteriales, Methanobacteriales, Methanococcales, Methanomicrobiales, Planctomycetales, Prochlorales, Sulfolobales, Thermococcales, Thermoproteales and Verrucomicrobiales are the genera Acholeplasma, Halanaerobium, Halobacterium, Methanobacterium, Methanococcus, Methanomicrobium, Planctomyces, Prochloron, Sulfolobus, Thermococcus, Thermoproteus and Verrucomicrobium, respectively. Opinion 79. Int J Syst Evol Microbiol 2005; 55:517–518. PubMed doi:10.1099/ijs.0.63548-0
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Giovannoni SJ, Godchaux W, III, Schabtach E, Castenholtz RW. Cell wall and lipid composition of Isosphaera pallida, a budding eubacterium from hot springs. J Bacteriol 1987; 169:2702–2707. PubMed
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
The DOE Joint Genome Institute. http://www.jgi.doe.gov
Phrap and Phred for Windows. MacOS, Linux, and Unix. http://www.phrap.com
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed doi:10.1101/gr.074492.107
Han C, Chain P. 2006. Finishing repeat regions automatically with Dupfinisher. in Proceeding of the 2006 international conference on bioinformatics & computational biology. Edited by Hamid R. Arabnia & Homayoun Valafar, CSREA Press. June 26–29, 2006: 141–146.
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A gene prediction improvement pipeline for microbial genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Acknowledgements
This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle, and Oak Ridge National Laboratory under contract DE-AC05-00OR22725.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Göker, M., Cleland, D., Saunders, E. et al. Complete genome sequence of Isosphaera pallida type strain (IS1BT). Stand in Genomic Sci 4, 63–71 (2011). https://doi.org/10.4056/sigs.1533840
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.1533840