
Abstract
Ignisphaera aggregans Niederberger et al. 2006 is the type and sole species of genus Ignisphaera. This archaeal species is characterized by a coccoid-shape and is strictly anaerobic, moderately acidophilic, heterotrophic hyperthermophilic and fermentative. The type strain AQ1.S1T was isolated from a near neutral, boiling spring in Kuirau Park, Rotorua, New Zealand. This is the first completed genome sequence of the genus Ignisphaera and the fifth genome (fourth type strain) sequence in the family Desulfurococcaceae. The 1,875,953 bp long genome with its 2,009 protein-coding and 52 RNA genes is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others


Introduction
Strain AQ1.S1T (= DSM 17230 = JCM 13409) is the type strain of the species Ignisphaera aggregans, which is the type species of the genus Ignisphaera [1], one out of nine genera in the family Desulfurococcaceae [2–5]. The generic name derives from the Latin word ‘ignis’ meaning ‘fire’, and ‘sphaera’ meaning ‘ball’, referring to coccoid cells found in the high-temperature environment such as hot springs [1]. The species epithet is derived from the Latin word ‘aggregans’ meaning ‘aggregate forming or aggregating clumping’, referring to the appearance of the cells when grown on mono-, di- or polysaccharides [1]. Strain AQ1.S1T is of particular interest because it is able to ferment quite a number of polysaccharides and complex proteinaceous substrates [1]. Here we present a summary classification and a set of features for I. aggregans AQ1.S1T, together with the description of the complete genomic sequencing and annotation.
Classification and features
Strain AQ1.S1T was isolated from a near neutral, boiling spring situated in Kuirau Park, Rotorua, New Zealand [1]. Interestingly, strains of I. aggregans could not be cultivated from pools with similar characteristics in Yellowstone National Park [1]. Only three cultivated strains are reported for the species I. aggregans in addition to AQ1.S1T, these are strains Tok37.S1, Tok10A.S1 and Tok1 [1]. The 16S rRNA sequence of AQ1.S1T is 99% identical to Tok37.S1, 98% to Tok10A.S1 and 98% to Tok1. Sequence similarities between strain AQ1.S1T and members of the family Pyrodictiaceae range from 93.0% for Pyrodictium occultum to 93.4% for P. abyssi [6] but from 89.7% for Ignicoccus islandicus to 93.5% for Staphylothermus hellenicus [6] with members of the family Desulfurococcaceae in which I. aggregans is currently classified (Table 1). Genbank [16] currently contains only three 16S rRNA gene sequences with significantly high identity values to strain AQ1.S1T: clone YNP_BP_A32 (96%, DQ243730) from hot springs of Yellowstone National Park, clone SSW_L4_A01 (95%, EU635921) from mud hot springs, Nevada, USA, and clone DDP-A02 (94%, AB462559) from a Japanese alkaline geothermal pool, which does not necessarily indicate the presence of I. aggregans but probably the presence of yet to be identified other species in the genus Ignisphaera. Environmental samples and metagenomic surveys featured in Genbank contain not a single sequence with >87% sequence identity (as of June 2010), indicating that I. aggregans might play a rather limited and regional role in the environment.
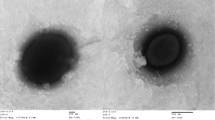
The cells of strain AQ1.S1T are regular to irregular cocci which occur singly, in pairs or as aggregates of many cells [1]. They usually have dimensions between 1–1.5 εm (Figure 1). Aggregation of cells is common when AQ1.S1T is grown on mono-, di- or polysaccharides [1]. Strain AQ1.S1T is hyperthermophilic and grows optimally between 92°C and 95°C, the temperature range for growth is 85–98°C. The pH range for growth is 5.4–7.0, with an optimum at pH 6.4. The strain grows in the presence of up to 0.5% NaCl, however, it grows optimally without NaCl. The doubling time is 7.5 h under optimal conditions [1]. I. aggregans strain AQ1.S1T is strictly anaerobic and grows heterotrophically on starch, trypticase peptone, lactose, glucose, konjac glucomannan, mannose, galactose, maltose, glycogen, and β-cyclodextrin. Growth on beef extract and glucose is weak and not observed on yeast extract, cellobiose, methanol, ethanol, trehalose, pyruvate, acetate, malate, casamino acids (0.1% w/v), carboxymethylcellulose, amylopectin (corn), xanthan gum, locust gum (bean), guar gum, dextran, xylan (oat spelts, larch or birch), xylitol, xylose or amylose (corn and potato) [1]. Mono- and disaccharides are accumulated in AQ1.S1T cultures grown in media containing konjac glucomannan, but not in sterile media that had been exposed to the same temperature as the inoculated medium or the stock of konjac glucomannan [1]. As hypothesized by Niederberger et al. [1], this most probably indicates that the konjac glucomannan is being hydrolyzed enzymatically by AQ1.S1T into sugars for metabolism. Removal of cystine from the growth medium does not affect cell density significantly. Hydrogen sulfide is also detected in AQ1.S1T cultures grown in enrichment media. Strain AQ1.S1T is resistant to novobiocin and streptomycin but sensitive to erythromycin, chloramphenicol and rifampicin [1].

Scanning electron micrograph of I. aggregans AQ1.S1T
Chemotaxonomy
No chemotaxonomic data are currently available for I. aggregans strain AQ1.S1T. Also, chemotaxonomic information for the family Desulfurococcaceae is scarce. What is known is that the type species of this family, Desulfurococcus mucosus, lacks a murein cell wall and contains phytanol and polyisopreonoid dialcohols as major components of the cellular lipids [3].
Figure 2 shows the phylogenetic neighborhood of I. aggregans AQ1.S1T in a 16S rRNA based tree. The sequence of the single 16S rRNA gene copy in the genome of strain AQ1.S1 does not differ from the previously published 16S rRNA sequence from DSM 17230 (DQ060321).

Phylogenetic tree highlighting the position of I. aggregans AQ1.S1T relative to the type strains of the other genera within the order Desulfurococcales. The tree was inferred from 1,329 aligned characters [17,18] of the 16S rRNA gene sequence under the maximum likelihood criterion [19] and rooted with the type strains of the genera of the neighboring order Acidilobales. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 250 bootstrap replicates [20], if greater than 60%. Lineages with type strain genome sequencing projects registered in GOLD [21] are shown in blue, published genomes in bold ([22–25], CP000504 and CP000852).
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [26], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [27]. The genome project is deposited in the Genome OnLine Database [21] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
I. aggregans AQ1.S1T, DSM 17230, was grown anaerobically in DSMZ medium 1043 (Ignisphaera medium) [28] at 92°C. DNA was isolated from 0.5–1 g of cell paste using MasterPure Gram Positive DNA Purification Kit (Epicentre MGP04100). One µl lysozyme and five µl mutanolysin and lysostaphine, each, were added to the standard lysis solution for one hour at 37°C followed by 30 min incubation on ice after the MPC-step.
Genome sequencing and assembly
The genome of strain AQ1.S1T was sequenced using a combination of Illumina and 454 technologies. An Illumina GAii shotgun library with reads of 152 Mb, a 454 Titanium draft library with average read length of 320 bases, and a paired end 454 library with average insert size of 15 kb were generated for this genome. All general aspects of library construction and sequencing can be found at http://www.jgi.doe.gov/. Illumina sequencing data was assembled with VELVET and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. Draft assemblies were based on 177 Mb 454 draft data, and 454 paired end data. Newbler parameters are -consed - a 50 -l 350 -g -m -ml 20. The initial assembly contained 20 contigs in 1 scaffold. The initial 454 assembly was converted into a phrap assembly by making fake reads from the consensus, collecting the read pairs in the 454 paired end library. The Phred/Phrap/Consed software package (http://www.phrap.com) was used for sequence assembly and quality assessment [29] in the following finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution (http://www.jgi.doe.gov), Dupfinisher [29], or sequencing cloned bridging PCR fragments with subcloning or transposon bombing (Epicentre Biotechnologies, Madison, WI). Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chan, unpublished). A total of 32 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to improve the final consensus quality using an in-house developed tool (the Polisher [30]). The error rate of the final genome sequence is less than 1 in 100,000
Genome annotation
Genes were identified using Prodigal [31] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [32]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [33].
Genome properties
The genome consists of a 1,875,953 bp long chromosome with a 35.7% G+C content (Table 3 and Figure 3). Of the 2,061 genes predicted, 2,009 were protein-coding genes, and 52 RNAs; 79 pseudogenes were also identified. The majority of the protein-coding genes (56.2%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the genome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Insights from the genome sequence
Even though the tree depicted in Figure 1 is not particularly well resolved, the fact that I. aggregans does not cluster with the Desulfurococcaceae in 16S rRNA gene sequence-based phylogenies calls for a more detailed whole-genome-based analysis [34]. Both, in Figure 1 and in the All-Species-Living-Tree [35], I. aggregans is located deep on the branch leading to the Thermoproteaceae (and Sulfolobaceae). By circumstance, the class Thermoprotei within the phylum Crenarchaeota already offers a reasonably large set of reference genomes required for such an analysis. We thus assembled a dataset comprising all publicly available genomes from the set of organisms represented in the 16S rRNA tree (Fig. 1). Pairwise distances were calculated using the GBDP algorithm [36,37], which has recently been used to mimic DNA-DNA-hybridization values [37,38]. Here we applied the logarithmic version of formula (3) in [34,38]. The NeighborNet algorithm as implemented in SplitsTree version 4.10 [39] was used to infer a phylogenetic network from the distances, which is shown in Fig. 4.
The results indicate that the placement of I. aggregans as sister group of Thermoproteales (Fig. 1) is an artifact of the 16S rRNA analysis. The whole-genome network, while showing some conflicting signal close to the backbone, is in agreement with the splitting of the considered genera into the orders Desulfurococcales and Thermoproteales. However, the analysis provides some evidence that Aeropyrum pernix (Desulfurococcaceae) is more closely related to Pyrodictiaceae (represented by Hyperthermus and Pyrolobus) than to the remaining Desulfurococcaceae. The numerous additional type strain genome sequencing projects in the Desulfurococcales (Fig. 1) are likely to shed even more light on the phylogenetic relationships within this group by enabling future whole-genome phylogenies based on many more taxa.
A separate status of I. aggregans within the Desulfurococcaceae is supported by a lack of genes encoding membrane-bound multienzyme complexes that are thought to participate in the energy metabolism of members of this group. Operons encoding a MBX-related ferredoxin-NADPH oxidoreductase and a dehydrogenase-linked MBX complex are lacking in I. aggregans, although both are present in the completed genome sequences of Thermosphaera aggregans [24], Staphylothermus marinus [25] and Desulfurococcus kamchatkensis. The genome of A. pernix also lacks genes for the MBH-related energy-coupling hydrogenase, which are found in most members of the Desulfurococcaceae including I. aggregans (Igag_1902 - Igag_1914).

Phylogenetic network inferred from whole-genome (GBDP) distances, showing the relationships between Desulfurococcaceae (Aeropyrum, Ignisphaera, Staphylothermus and Thermosphaera), Pyrodictiaceae (Hyperthermus and Pyrolobus), Thermoproteaceae (Caldivirga, Pyrobaculum, Thermoproteus and Vulcanisaeta) and Thermofilaceae (Thermofilum).
References
Niederberger TD, Götz DK, McDonald IR, Ronimus RS, Morgan HW. Ignisphaera aggregans gen. nov., sp. nov., a novel hyperthermophilic crenarchaeote isolated from hot springs in Rotorua and Tokaanu, New Zealand. Int J Syst Evol Microbiol 2006; 56:965–971. PubMed doi:10.1099/ijs.0.63899-0
Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List no. 10. Int J Syst Bacteriol 1983; 33:438–440. doi:10.1099/00207713-33-2-438
Zillig W, Stetter KO, Prangishvilli D, Schäfer W, Wunderl S, Janekovic D, Holz I, Palm P. Desulfurococcaceae, the second family of the extremely thermophilic, anaerobic, sulfur-respiring Thermoproteales. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg Abt 1982; 1 Orig. C:304–317
List Editor. Validation List no. 10. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1983; 33:438–440. doi:10.1099/00207713-33-2-438
Burggraf S, Huber H, Stetter KO. Reclassification of the crenarchael orders and families in accordance with 16S rRNA sequence data. Int J Syst Bacteriol 1997; 47:657–660. PubMed doi:10.1099/00207713-47-3-657
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YW. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 2007; 57:2259–2261. PubMed doi:10.1099/ijs.0.64915-0
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Holt JG. 2001. The Road Map to the Manual In G. M. Garrity, D. R. Boone, and R. W. Castenholz (ed.), Bergey’s Manual of Systematic Bacteriology, 2 ed, vol. 1. Springer, New York.
List Editor. Validation List no. 85. Validation of publication of new names and new combinations previously effectively published outside the IJSEM. Int J Syst Evol Microbiol 2002; 52:685–690. PubMed doi:10.1099/ijs.0.02358-0
Reysenbach AL. 2001. Class I. Thermoprotei class. nov., p. 169. In GM Garrity, DR Boone, and RW Castenholz (eds), Bergey’s Manual of Systematic Bacteriology, 2 ed, vol. 1. Springer, New York.
Huber H, Stetter O. 2001. Order II. Desulfurococcales ord. nov., p. 179–180. In G. M. Garrity, D. R. Boone, and R. W. Castenholz (ed.), Bergey’s Manual of Systematic Bacteriology, 2 ed, vol. 1. Springer, New York.
Classification of Bacteria and Archaea in risk groups. http://www.baua.de TRBA 466.
Niederberger TD, Ronimus RS, Morgan HW. The microbial ecology of a high-temperature near-neutral spring situated in Rotorua, New Zealand. Microbiol Res 2008; 163:594–603. PubMed doi:10.1016/j.micres.2006.09.001
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res 2009; 37:D26–D31. PubMed doi:10.1093/nar/gkn723
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Stamatakis A, Hoover P, Rougemont J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-713
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markovitzz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed doi:10.1093/nar/gkp848
Brügger K, Chen L, Stark M, Zibat A, Redder P, Ruepp A, Awayez M, She Q, Garrett RA, Klenk HP. The genome of Hyperthermus butylicus: a sulfur-reducing, peptide fermenting, neutrophilic crenarchaeote growing up to 108°C. Archaea 2007; 2:127–135. PubMed doi:10.1155/2007/745987
Kawarabayasi Y, Hino Y, Horikawa H, Yamazaki S, Haikawa Y, Jinno K, Takahashi M, Sekine M, Baba S, Ankai A, et al. Complete genome sequence of an aerobic hyperthermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res 1999; 6:83–101, 145–152. PubMed doi:10.1093/dnares/6.2.83
Spring S, Rachel R, Lapidus A, Davenport K, Tice H, Copeland A, Cheng JF, Lucas S, Chen F, Nolan M, et al. Complete genome sequence of Thermosphaera aggregans type strain (M11TLT). Stand Genomic Sci 2010; 2:245–259. doi:10.4056/sigs.821804
Anderson IJ, Dharmarajan L, Rodriguez J, Hooper S, Porat I, Ulrich LE, Elkins JG, Mavromatis K, Sun H, Land M, et al. Complete genome sequence of Staphylothermus marinus reveals differences in sulfur metabolism among heterotrophic Crenarchaeota. BMC Genomics 2009; 10:145. PubMed doi:10.1186/1471-2164-10
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php.
Sims D, Brettin T, Detter J, Han C, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Chen F, Lucas S, et al. Complete genome sequence of Kytococcus sedentarius type strain (541T). Stand Genomic Sci 2009; 1:12–20. doi:10.4056/sigs.761
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A gene prediction improvement pipeline for microbial genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Auch AF, Klenk HP, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2010; 2:142–148. doi:10.4056/sigs.541628
Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH, Ludwig W, Glöckner FO, Rosselló-Móra R. The All-Species Living Tree project: A 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol 2008; 31:241–250. PubMed doi:10.1016/j.syapm.2008.07.001
Henz SR, Huson DH, Auch AF, Nieselt-Struwe K, Schuster SC. Whole-genome prokaryotic phylogeny. Bioinformatics 2005; 21:2329–2335. PubMed doi:10.1093/bioinformatics/bth324
Auch AF, Henz SR, Holland BR, Göker M. Genome BLAST distance phylogenies inferred from whole plastid and whole mitochondrion genome sequences. BMC Bioinformatics 2006; 7:350. PubMed doi:10.1186/1471-2105-7-350
Auch AF, von Jan M, Klenk HP, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2010; 2:117–134. doi:10.4056/sigs.531120
Huson DH, Bryant D. Application of Phylogenetic Networks in Evolutionary Studies. Mol Biol Evol 2005; 23:254–267. PubMed doi:10.1093/molbev/msj030
Acknowledgements
We would like to gratefully acknowledge the help of Maren Schröder (DSMZ) for growth of I. aggregans and the help of Alexander Auch (Tübingen, Germany) in creating a local version of the GBDP software. This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-2 and SI 1352/1-2 and Thailand Research Fund Royal Golden Jubilee Ph.D. Program No. PHD/0019/2548 for MY.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Göker, M., Held, B., Lapidus, A. et al. Complete genome sequence of Ignisphaera aggregans type strain (AQ1.S1T). Stand in Genomic Sci 3, 66–75 (2010). https://doi.org/10.4056/sigs.1072907
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.1072907