
Abstract
Cellulomonas flavigena (Kellerman and McBeth 1912) Bergey et al. 1923 is the type species of the genus Cellulomonas of the actinobacterial family Cellulomonadaceae. Members of the genus Cellulomonas are of special interest for their ability to degrade cellulose and hemicellulose, particularly with regard to the use of biomass as an alternative energy source. Here we describe the features of this organism, together with the complete genome sequence, and annotation. This is the first complete genome sequence of a member of the genus Cellulomonas, and next to the human pathogen Tropheryma whipplei the second complete genome sequence within the actinobacterial family Cellulomonadaceae. The 4,123,179 bp long single replicon genome with its 3,735 protein-coding and 53 RNA genes is part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others

Introduction
Strain 134T (DSM 20109 = ATCC 482 = JCM 1489) is the type strain of the species Cellulomonas flavigena and was isolated from soil and first described in 1912 by Kellerman and McBeth [1], followed by a description in the first edition of Bergey’s Manual in 1923 [2].
Because of the absence of a definite proof linking the deposited strains to the original description Stackebrandt and Kandler proposed in 1979 C. flavigena and six other Cellulomonas strains as neotype strains of their respective species [3]. Here C. flavigena cells are reported as Gram-positive, non-motile and coryneform with snapping divisions [3].
In addition to the type species C. flavigena, the five Cellulomonas species, C. biazotea, C. cellasea, C. gelida, C. fimi and C. uda have been members of the genus since their original description in the first edition of Bergey’s Manual in 1923 [2]. Because of the phenetic resemblance of the different species to each other C. flavigena was recognized as the only species in the genus Cellulomonas in the eighth edition of Bergey’s Manual. This reduction to a single species was questioned by Braden and Thayer based on serological studies in 1976 [4] and by Stackebrandt and Kandler based on DNA reassociation studies in 1979 [3]. In 1980 the Approved Lists of Bacterial Names already listed six species: C. flavigena, C. biazotea, C. gelida, C. uda, C. fimi and C. cellasea [5]. Currently, 17 species belonging to the genus Cellulomonas are noted in the actual version of the List of Procaryotic names with Standing in Nomenclature [6]. Due to the cellulolytic activity of these organisms, their preferred habitats are cellulose enriched environments such as soil, bark, wood, and sugar fields, but they were also successfully isolated from rumen and from activated sludge. Here we present a summary classification and a set of features for C. flavigena 134T, together with the description of the complete genomic sequencing and annotation.
Classification and features
The 16S rRNA genes of the 16 other type strains in the genus Cellulomonas share between 92.2% (C. bogoriensis [7]) and 98.1% (C. persica [8]) sequence identity with strain 124T, whereas the other type strains from the family Cellulomonadaceae, which belong to the genera Actinotalea, Oerskovia, Paraoerskovia and Tropheryma, share less than 95.6% sequence identity [9]. Cultivated strains with highest sequence similarity include a so far unpublished strain 794 (Y09565) from human clinical specimen (99.7% sequence identity) and Everest-gws-44 (EU584517) from glacial meltwater at 6,350 m height on Mount Everest (98.1% sequence identity). The only reported uncultured clone with high sequence similarity (98.5%) originated from a diet-related composition of the gut microbiota of the earthworm Lumbricus rubellus [10]. Metagenomic surveys and environmental samples based on 16S rRNA gene sequences delivered no indication for organisms with sequence similarity values above 93–94% to C. flavigena, indicating that members of this species are not abundant in the so far screened habitats. The majority of these 16S rRNA gene sequences with similarity between 88% and 93% originate from marine metagenomes (status June 2010).
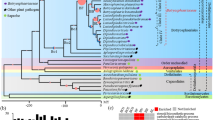
Figure 1 shows the phylogenetic neighborhood of C. flavigena 134T in a 16S rRNA based tree. The sequences of the two 16S rRNA gene copies in the genome differ by two nucleotides from each other and by up to four nucleotides from the previously published sequence generated from NCIMB 8073 (Z79463).

Phylogenetic tree highlighting the position of C. flavigena 134T relative to the other type strains within the family Cellulomonadaceae. The tree was inferred from 1,393 aligned characters [11,12] of the 16S rRNA gene sequence under the maximum likelihood criterion [13] and rooted with the type strain of the suborder Micrococcineae. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 1,000 bootstrap replicates [14] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [15] are shown in blue, published genomes in bold.
Cells of C. flavigena stain Gram-positive with a very fast rate of decolorization [3]. Cells in young broth cultures are typically coryneform with a snapping division (Table 1). In week old cultures a transformation to short rods can occur (Figure 2) [3]. On yeast extract-glucose agar C. flavigena forms smooth, glistening, yellow colonies about 5 mm in diameter. C. flavigena is described as non-motile [3,28], but according to Thayer et al. (1984) C. flavigena cells possess polar multitrichous flagella [31] (not visible in Figure 2). C. flavigena grows under aerobic conditions with an optimal growth temperature of 30°C [2] and an optimal pH of 7 [32].

Scanning electron micrograph of C. flavigena 134T.
Strain 134T is able to ferment glucose, maltose, sucrose, xylose and dextrin, but no fermentation of mannitol was observed [3]. While ribose, acetate and gluconate are utilized, there is no utilization of raffinose and L(+)-lactate [3]. It was shown by Kim et al. (1987) that gluconate is catabolized via the Entner-Doudoroff pathway and hexose monophosphate shunt [33]. C. flavigena produces catalase but no urease [3]. Esculin and gelatin are hydrolyzed and nitrate is not reduced to nitrite [3].
Chemotaxonomy
The peptidoglycan of C. flavigena contains as the diagnostic amino acid in position 3 of the peptide subunit ornithine with the interpeptide bridge containing D-aspartic acid. The major cell wall sugar is rhamnose, whereas mannose and ribose occur in minor amounts [34]. The major components of the fatty acid profile of C. flavigena are 12-methyltetradecanoic (ai-C15:0) and hexadecanoic (C16:0) acids; i-C15:0, ai-C17:0, C14:0 and C15:0 occur in lower amounts [35]. Menaquinone MK-9(H4) is the predominant isoprenoid quinone; minor amounts of MK-9(H2), MK-8(H4) and MK-7(H4) were detected [36]. The polar lipids consist of diphosphatidylglycerol, phosphatidylinositol and two so far unidentified phosphoglycolipids [37].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [38], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [39]. The genome project is deposited in the Genome OnLine Database [15] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
C. flavigena 134T, DSM 20109, was grown in DSMZ medium 92 (Trypticase-Soy-Yeast Extract Medium) [40] at 30°C. DNA was isolated from 0.5–1 g of cell paste using Qiagen Genomic 500 DNA Kit (Qiagen, Hilden, Germany) following the standard protocol as recommended by the manufacturer.
Genome sequencing and assembly
The genome was sequenced using a combination of Sanger and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website (http://www.jgi.doe.gov/). Pyrosequencing reads were assembled using the Newbler assembler version 1.1.02.15 (Roche). Large Newbler contigs were broken into 4,499 overlap ping fragments of 1,000 bp and entered into assembly as pseudo-reads. The sequences were assigned quality scores based on Newbler consensus q-scores with modifications to account for overlap redundancy and adjust inflated q-scores. A hybrid 454/Sanger assembly was made using PGA assembler. Possible mis-assemblies were corrected and gaps between contigs were closed by primer walks off Sanger clones and bridging PCR fragments and by editing in Consed. A total of 704 Sanger finishing reads were produced to close gaps, to resolve repetitive regions, and to raise the quality of the finished sequence. 12,171,379 Illumina reads were used to improve the final consensus quality using an in-house developed tool (the Polisher [41]). The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Sanger and 454 sequencing platforms provided 65.38× coverage of the genome. The final assembly contains 46,659 Sanger reads and 601,307 pyrosequencing reads.
Genome annotation
Genes were identified using Prodigal [42] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [43]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [44].
Genome properties
The genome is 4,123,179 bp long and comprises one main circular chromosome with a 74.3% G+C content (Table 3 and Figure 3). Of the 3,788 genes predicted, 3,735 were protein-coding genes, and 53 RNAs; 57 pseudogenes were also identified. The majority of the protein-coding genes (71.1%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the genome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Insights from genome sequence
A closer look on the genome sequence of C. flavigena revealed a set of genes which are probably responsible for the yellowish color of C. flavigena cells by encoding enzymes that are involved in the synthesis of carotenoids. Carotenoids are produced by the action of geranylgeranyl pyrophosphate synthase (Cfla_2893), squalene/phytoene synthase (Cfla_2892), phytoene desaturase (Cfla_2891), lycopene cyclase (Cfla_2890, Cfla_2889) and lycopene elongase (Cfla_2888). Cfla_2893 is declared as a pseudo gene, but when ignoring the frame shift the deduced amino acid sequence shows significant similarity to geranylgeranyl pyrophosphate synthases. Geranylgeranyl pyrophosphate synthases start the biosynthesis of carotenoids by combining farnesyl pyrophosphate with C5 isoprenoid units to C20-molecules, geranylgeranyl pyrophosphate. The phytoene synthase catalyzes the condensation of two geranylgeranyl pyrophosphate molecules followed by the removal of diphosphate and a proton shift leading to the formation of phytoene. Sequential desaturation steps are conducted by the phytoene desaturase followed by cyclisation of the ends of the molecules catalyzed by the lycopene cyclase [45]. It is remarkable that the genes belonging to the putative carotenoid biosynthesis clusters of Beutenbergia cavernae (Bcav_3492-Bcav_3488) [46], Leifsonia xyli subsp. xyli (crtE, crtB, crtI, crtYe, lctB, crtEb) and Sanguibacter keddieii (Sked_12750-Sked_12800) [47] have a similar size and show the same organization as in the genome of C. flavigena.
In the eighth edition of Bergey’s manual the members of the genus Cellulomonas are described as motile by one or a few flagella or non-motile, even within the genus both characteristics occur [32]. Regarding the motility of C. flavigena there are different observations described. Thayer et al. (1984) report the existence of polar multitrichous flagella [31], whereas Stackebrandt et al. (1979) and Schaal (1986) reported C. flavigena as non-motile [3,48]. In contrast to Thayer’s observation we found no genes coding proteins belonging to the category ‘flagellum structure and biogenesis’ in the genome sequence. Kenyon et al. (2005) report for the genus Cellulomonas a coherency between the production of curdlan, a β-1,3-glucan, and non-motility. They observed that the production of curdlan EPS by the non-motile C. flavigena leads to a closer adherence to cellulose and hemicellulose. In contrast, cells of the motile Cellulomonas strain C. gelida produce no curdlan EPS and are not directly attached to the cellulose fibers [28]. The production of curdlan by C. flavigena is consistent with the observation of 17 glycosyl transferases (GT) belonging to family 2, as β-1,3-glucan synthases are often found in this GT family.
The characteristic attribute of C. flavigena and the other members of the genus Cellulomonas is the ability to degrade cellulose, xylan and starch. The most molecular work has been done on cellulase and xylanase genes from C. fimi, but also cellulases, xylanases and chitinases of C. flavigena were identified and characterized [49–52]. The genome sequence and the subsequent annotation revealed that 9.6% of encoded proteins are classified into the COG category ‘carbohydrate transport and metabolism’. Among them several genes coding for xylan degrading enzymes; 14 genes coding for putative endo-1,4-β-xylanases belonging to glycoside hydrolase family 10 and five genes encoding β-xylosidases. For the hydrolysis of cellulose the concerted action of endo-1,4-β-glucanases, 1,4-β-cellobiohydrolases and β-glucosidases is necessary. Endo-1,4-β-glucanases randomly cleave within the cellulose molecule and increase the number of non-reducing ends which are attacked by 1,4-β-cellobiohydrolases. The released cellobiose is cleaved by β-glucosidases. In the genome of C. flavigena two genes coding endo-1,4-β-glucanases (Cfla_0016, Cfla_1897), three genes encoding 1,4-β-cellobiohydrolases (Cfla_1896, Cfla_2912, Cfla_2913) and three genes coding β-glucosidases (Cfla_1129, Cfla_3027, Cfla_2913) were identified.
References
Kellermann FK, McBeth FM. The fermentation of cellulose. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg Abt 2. 1912; 39:502–552.
Bergey DH, Breed RS, Hammer RW, Harrison FC, Huntoon FM. Bergey’s manual of determinative bacteriology, 1st ed. The Willimas and Wilkins Co., Baltimore 1923.
Stackebrandt E, Kandler O. Taxonomy of the genus Cellulomonas, based on phenotypic characters and deoxyribonucleic acid — deoxyribonucleic acid homology, and proposal of seven neotype strains. Int J Syst Bacteriol 1979; 29:273–282. doi:10.1099/00207713-29-4-273
Braden AR, Thayer DW. Serological study of Cellulomonas. Int J Syst Bacteriol 1976; 26:123–126. doi:10.1099/00207713-26-2-123
Skerman VBD, Mc Gowan V, Sneath PHA, eds. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225–420. doi:10.1099/00207713-30-1-225
Euzeby JP. List of Bacterial Names with Standing in Nomenclature: a folder available on the Internet. [List of Prokaryotic names with Standing in Nomenclature. http://www.bacterio.net]. Int J Syst Bacteriol 1997; 47:590–592. PubMed doi:10.1099/00207713-47-2-590
Jones BE, Grant WD, Duckworth AW, Schumann P, Weiss N, Stackebrandt E. Cellulomonas bogoriensis sp. nov., an alkaliphilic cellulomonad. Int J Syst Evol Microbiol 2005; 55:1711–1714. PubMed doi:10.1099/ijs.0.63646-0
Elberson MA, Malekzadeh F, Yazdi MT, Kameranpour N, Noori-Daloii MR, Matte MH, Shahamat M, Colwell RR, Sowers KR. Cellulomonas persica sp. nov. and Cellulomonas iranensis sp. nov., mesophilic cellulose-degrading bacteria isolated from forest soils. Int J Syst Evol Microbiol 2000; 50:993–996. PubMed
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YW. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 2007; 57:2259–2261. PubMed doi:10.1099/ijs.0.64915-0
Knapp BA, Podmirseg SM, Seeber J, Meyer E, Insam H. Diet-related composition of the gut microbiota of Lumbricus rubellus as revealed by a molecular fingerprinting technique and cloning. Soil Biol Biochem 2009; 41:2299–2307. doi:10.1016/j.soilbio.2009.08.011
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Stamatakis A, Hoover P, Rougemont J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst Biol 2008; 57:758–771. PMID:18853362 doi:10.1080/10635150802429642.
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How Many Bootstrap Replicates Are Necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-713
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed doi:10.1093/nar/gkp848
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Holt J. Taxonomic outline of the Archaea and Bacteria. In: Bergey’s Manual of Systematic Bacteriology, 2nd ed. vol. 1. The Archaea, Deeply Branching and Phototrophic Bacteria. Garrity GM, Boone DR and Castenholz RW (eds). 2001; 155–166.
Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol 1997; 47:479–491. doi:10.1099/00207713-47-2-479
Buchanan RE. Studies in the nomenclature and classification of bacteria. II. The primary subdivisions of the Schizomycetes. J Bacteriol 1917; 2:155–164. PubMed
Zhi XY, Li WJ, Stackebrandt E. An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the higher taxa. Int J Syst Evol Microbiol 2009; 59:589–608. PubMed doi:10.1099/ijs.0.65780-0
Stackebrandt E, Prauser H. The family Cellulomonadaceae. In: The prokaryotes. Balows A, Trüper HG, Dworkin M, Harder W, Schleifer KH (eds). 1991; 1323–1345.
Stackebrandt E, Schumann P. Description of Bogoriellaceae fam. nov., Dermacoccaceae fam. nov., Rarobacteraceae fam. nov. and Sanguibacteraceae fam. nov. and emendation of some families of the suborder Micrococcineae. Int J Syst Evol Microbiol 2000; 50:1279–1285. PubMed
Stackebrandt E, Prauser H. Assignment of the genera Cellulomonas, Oerskovia, Promicromonospora, and Jonesia to Cellulomonadaceae fam. nov. Syst Appl Microbiol 1991; 14:261–265.
List Editor. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List no. 39. Int J Syst Bacteriol 1991; 41:580–581. PubMed doi:10.1099/00207713-41-4-580
Stackebrandt E, Seiler H, Schleifer KH. Union of the genera Cellulomonas Bergey et al. and Oerskovia Prauser et al. in a redefined genus Cellulomonas. [Orig A]. Zentralbl Bakteriol 1982; 3:401–409.
Bergey DH, Harrison FC, Breed RS, Hammer BW, Huntoon FM. In: Bergey DH, Harrison FC, Breed RS, Hammer BW, Huntoon FM (eds), Bergey’s manual of determinative bacteriology, First Edition, The Williams and Wilkins Co., Baltimore, 1923, p. 1–442.
Kenyon WJ, Esch SW, Buller CS. The curdlan-type exopolysaccharide produced by Cellulomonas flavigena KU forms part of an extracellular glycocalyx involved in cellulose degradation. Antonie Van Leeuwenhoek 2005; 87:143–148. PubMed doi:10.1007/s10482-004-2346-4
Classification of bacteria and archaea in risk groups. www.baua.de TRBA 466
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Thayer DW. Motility and flagellation of cellulomonads. Int J Syst Bacteriol 1984; 34:218–219. doi:10.1099/00207713-34-2-218
Keddie RM. Genus III. Cellulomonas Bergey et al. 1923, 154, emend. mut. char. Clark 1952, 50AL. In: Bergey’s Manual of Systematic Bacteriology. Williams and Wilkins. Baltimore, MD. Sneath PHA, Mair NS, Sharpe ME, Holt JG (eds). 1974; 629–631.
Kim BH. Carbohydrate catabolism in cellulolytic strains of Cellulomonas, Pseudomonas, and Nocardia. Kor J Microbiol 1987; 25:28–33.
Fieldler F, Kandler O. Die Mureintypen in der Gattung Cellulomonas Bergey et al. Arch Microbiol 1973; 89:41–50. doi:10.1007/BF00409398
Funke G, Ramos CP, Collins MD. Identification of some clinical strains of CDC coryneform group A-3 and A-4 bacteria as Cellulomonas species and proposal of Cellulomonas hominis sp. nov. for some group A-3 strains. J Clin Microbiol 1995; 33:2091–2097. PubMed
Collins MD, Jones D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol Rev 1981; 45:316–354. PubMed
Mimmikin DE, Collins MD, Goodfellow M. Fatty acid and polar lipid composition in the classification of Cellulomonas, Oerskovia and related taxa. J Appl Bacteriol 1979; 47:87–95.
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php.
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A gene prediction improvement pipeline for microbial genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Sandmann G. Carotenoid biosynthesis and biotechnological application. Arch Biochem Biophys 2001; 385:4–12. PubMed doi:10.1006/abbi.2000.2170
Land M, Pukall R, Abt B, Göker M, Rohde M, Glavina Del Rio T, Tice H, Copeland A, Cheng JF, Lucas S, et al. Complete genome sequence of Beutenbergia cavernae type strain (HKI 0122T). Stand Genomic Sci 2009; 1:21–28. doi:10.4056/sigs.1162
Ivanova N, Sikorski J, Sims D, Brettin T, Detter JC, Han C, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, et al. Complete genome sequence of Sanguibacter keddieii type strain (ST-74T). Stand Genomic Sci 2009; 1:110–118. doi:10.4056/sigs.16197
Schaal KP. Genus Actinomyces Harz 1877, 133AL. In: Bergey’s Manual of Systematic Bacteriology. Williams and Wilkins. Baltimore, MD. Sneath PHA, Mair NS, Sharpe ME, Holt JG (eds). 1986; 1383–1418.
Gutiérrez-Nava A, Herrera-Herrera A, Mayorga-Reyes L, Salgado LM, Ponce-Noyola T. Expression and characterization of the celcflB gene from Cellulomonas flavigena encoding an endo-ß-1,4-glucanase. Curr Microbiol 2003; 47:359–363. PubMed doi:10.1007/s00284-002-4016-y
Mayorga-Reyes L, Morales Y, Salgado LM, Ortega A, Ponce-Noyola T. Cellulomonas flavigena: characterization of an endo-1,4-xylanase tightly induced by sugarcane bagasse. FEMS Microbiol Lett 2002; 214:205–209. PubMed doi:10.1111/j.1574-6968.2002.tb11348.x
Chen HC, Hsu MF, Jiang ST. Purification and characterization of an exo-N,N’-diacetylchitobiohydrolase-like enzyme from Cellulomonas flavigena NTOU 1. Enzyme Microb Technol 1997; 20:191–197. doi:10.1016/S0141-0229(96)00111-1
Horcasitas CM, López JO, Plaza IM. Xylanases from Cellulomonas flavigena: purification and characterization. Biotechnol Tech 1998; 12:663–666. doi:10.1023/A:1008852618130
Acknowledgements
We would like to gratefully acknowledge the help of Gabriele Gehrich-Schröter for growing C. flavigena cultures and Susanne Schneider for DNA extraction and quality analysis (both at DSMZ). This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-2.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Abt, B., Foster, B., Lapidus, A. et al. Complete genome sequence of Cellulomonas flavigena type strain (134T). Stand in Genomic Sci 3, 15–25 (2010). https://doi.org/10.4056/sigs.1012662
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.1012662