
Abstract
Leukocyte infiltration of the lung is a characteristic feature of allergic asthma and it is thought that these cells are selectively recruited by chemokines. Extensive research has confirmed that chemokine receptors are expressed on the main cell types involved in asthma, including eosinophils, T helper type 2 cells, mast cells and even neutrophils. Moreover, animal experiments have outlined a functional role for these receptors and their ligands. Chemokines signal via seven-transmembrane spanning G-protein coupled receptors, which are favored targets of the pharmaceutical industry due to the possibility of designing small-molecule inhibitors. In fact, this family represents the first group of cytokines where small-molecule inhibitors have been designed. However, the search for efficient antagonists of chemokine/chemokine receptors has not been easy; a particular feature of the chemokine system is the number of molecules with overlapping functions and binding specificities, as well as the difficulty in reconciling the in vivo biologic functional validation of chemokines in rodent models with the development of antagonists which bind the human receptor, because of the lack of species cross-reactivity. The chemokines and their receptors that are active during allergic reactions are reviewed. Possible points of interaction that may be a target for development of new therapies, as well as the progress to date in developing inhibitors of key chemokine receptors for asthma therapy, are also discussed.


Similar content being viewed by others

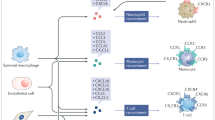
References
Thelen M. Dancing to the tune of chemokines. Nat Immunol 2001; 2: 129–34
Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Mol Med Today 2000; 6: 365–72
Ying S, Robinson DS, Meng Q, et al. C-C chemokines in allergen-induced latephase cutaneous responses in atopic subjects: association of eotaxin with early 6-hour eosinophils, and of eotaxin-2 and monocyte chemoattractant protein-4 with the later 24-hour tissue eosinophilia, and relationship to basophils and other C- C chemokines (monocyte chemoattractant protein-3 and RANTES). J Immunol 1999; 163: 3976–84
Robinson DS, Hamid Q, Ying S, et al. Predominant Th2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med 1992; 326: 298–304
Gonzalo JA, Lloyd CM, Wen D, et al. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airways hyperresponsiveness. J Exp Med 1998; 188: 157–67
Coyle AJ, Lloyd C, Tian J, et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med 1999; 190: 895–902
Brightling CE, Bradding P, Symon FA, et al. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med 2002; 346: 1699–705
Tillie-Leblond I, Gosset P, Tonnel AB. Inflammatory events in severe acute asthma. Allergy 2005; 60: 23–9
Lloyd C. Chemokines in allergic lung inflammation. Immunology 2002; 105: 144–54
D’Ambrosio D. Targeting chemoattractant receptors in allergic inflammation. Curr Drug Targets Inflamm Allergy 2005; 4: 163–7
Palframan RT, Collins PD, Severs NJ, et al. Mechanisms of acute eosinophil mobilization from the bone marrow stimulated by interleukin 5: the role of specific adhesion molecules and phosphatidylinositol 3-kinase. J Exp Med 1998; 188: 1621–32
Gu L, Tseng S, Horner RM, et al. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 2000; 404: 407–11
Jamieson T, Cook DN, Nibbs RJB, et al. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol 2005; 6: 403–11
Miller AL, Bowlin TL, Lukacs NW. Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J Infect Dis 2004; 189: 1419–30
Openshaw PJM, Tregoning JS. Immune responses and disease enhancement during respiratory syncytial virus infection. Clin Microbiol Rev 2005; 18: 541–55
Miller AL, Strieter RM, Gruber AD, et al. CXCR2 Regulates respiratory syncytial virus-induced airway hyperreactivity and mucus overproduction. J Immunol 2003; 170: 3348–56
Lamblin C, Gosset P, Tillie-Leblond I, et al. Bronchial neutrophilia in patients with noninfectious status asthmaticus. Am J Respir Crit Care Med 1998; 157: 394–402
Schuh JM, Blease K, Hogaboam CM. CXCR2 is necessary for the development and persistence of chronic fungal asthma in mice. J Immunol 2002; 168: 1447–56
Ponath PD, Qin S, Post TW, et al. Molecular cloning and characterization of a human eotaxin receptor expressed selectively on eosinophils. J Exp Med 1996; 183: 2437–48
Daugherty BL, Siciliano SJ, DeMartino J, et al. Cloning, expression and characterization of the human eosinophil eotaxin receptor. J Exp Med 1996; 183: 2349–54
Sallusto F, Lenig D, Mackay CR, et al. Flexible programs of chemokine receptor expression on human polarised T helper 1 and 2 lymphocytes. J Exp Med 1998; 187: 875–83
Teran LM, Davies DE. The chemokines: their potential role in allergic inflammation. Clin Exp Allergy 1996; 26: 1005–19
Sabroe I, Peck MJ, Jan Van Keulen B, et al. A small molecule antagonist of the chemokine receptors CCR1 and CCR3: potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J Biol Chem 2000; 275: 25985–92
De Lucca GV, Kim UT, Vargo BJ, et al. Discovery of CC chemokine receptor-3 (CCR3) antagonists with picomolar potency. J Med Chem 2005; 48: 2194–211
Zhang L, Soares MP, Guan Y, et al. Functional expression and characterization of macaque C-C chemokine receptor 3 (CCR3) and generation of potent antagonistic anti-macaque CCR3 monoclonal antibodies. J Biol Chem 2002; 277: 33799–810
Leckie MJ, ten Brinke A, Khan J, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 2000; 356: 2144–8
Humbles AA, Lloyd CM, McMillan SJ, et al. A critical role for eosinophils in allergic airways remodeling. Science 2004; 305: 1776–9
Flood-Page P, Menzies-Gow A, Phipps S, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest 2003; 112: 1029–36
Gonzalo JA, Pan Y, Lloyd CM, et al. Mouse monocyte-derived chemokine is involved in airway hyperreactivity and lung inflammation. J Immunol 1999; 163: 403–11
Kawasaki S, Takizawa H, Yoneyama H, et al. Intervention of thymus and activation-regulated chemokine attenuates the development of allergic airway inflammation and hyperresponsiveness in mice. J Immunol 2001; 166: 2055–62
Chvatchko Y, Hoogewerf AJ, Meyer A, et al. A key role for CC chemokine receptor 4 in lipopolysaccharide-induced endotoxic shock. J Exp Med 2000; 191: 1755–64
Pilette C, Francis JN, Till SJ, et al. CCR4 ligands are up-regulated in the airways of atopic asthmatics after segmental allergen challenge. Eur Respir J 2004; 23: 876–84
Purandare AV, Gao A, Wan H, et al. Identification of chemokine receptor CCR4 antagonist. Bioorg Med Chem Lett 2005; 15: 2669–72
Newhouse B, Allen S, Fauber B, et al. Racemic and chiral lactams as potent, selective and functionally active CCR4 antagonists. Bioorg Med Chem Lett 2004; 14: 5537–42
Chensue SW, Lukacs NW, Yang TY, et al. Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in cc chemokine receptor 8 knockout mice. J Exp Med 2001; 193: 573–84
Goya I, Villares R, Zaballos A, et al. Absence of CCR8 does not impair the response to ovalbumin-induced allergic airway disease. J Immunol 2003; 170: 2138–46
Chung CD, Kuo F, Kumer J, et al. CCR8 is not essential for the development of inflammation in a mouse model of allergic airway disease. J Immunol 2003; 170: 581–7
Bochner BS, Hudson SA, Xiao HQ, et al. Release of both CCR4-active and CXCR3-active chemokines during human allergic pulmonary late-phase reactions. J Allergy Clin Immunol 2003; 112: 930–4
Liu L, Jarjour NN, Busse WW, et al. Enhanced generation of helper T type 1 and 2 chemokines in allergen-induced asthma. Am J Respir Crit Care Med 2004; 169: 1118–24
Brightling CE, Ammit AJ, Kaur D, et al. The CXCL10/CXCR3 axis mediates human lung mast cell migration to asthmatic airway smooth muscle. Am J Respir Crit Care Med 2005; 171: 1103–8
Heise CE, Pahuja A, Hudson SC, et al. Pharmacologic characterization of CXC chemokine receptor 3 ligands and a small molecule antagonist. J Pharmacol Exp Ther 2005; 313: 1263–71
Gonzalo JA, Lloyd CM, Peled A, et al. Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1 alpha in the inflammatory component of allergic airway disease. J Immunol 2000; 165: 499–508
Lukacs NW, Berlin A, Schols D, et al. AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol 2002; 160: 1353–60
Proudfoot AE, Power CA, Hoogewerf AJ, et al. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem 1996; 271: 2599–603
Chvatchko Y, Proudfoot AEI, Buser R, et al. Inhibition of airway inflammation by amino-terminally modified RANTES/CC chemokine ligand 5 analogues is not mediated through CCR3. J Immunol 2003; 171: 5498–506
Simmons G, Clapham PR, Picard L, et al. Potent inhibition of HIV-1 infectivity in macrophage and lymphocytes by a novel CCR5 antagonist. Science 1997; 276: 276–9
Nibbs RJ, Salcedo TW, Campbell JD, et al. C-C chemokine receptor 3 antagonism by the beta-chemokine macrophage inflammatory protein 4, a property strongly enhanced by an amino-terminal alanine-methionine swap. J Immunol 2000 Feb 1; 164(3): 1488–97
Eisner J, Petering H, Hochstetter R, et al. The CC chemokine antagonist Met-RANTES inhibits eosinophil effector functions through the chemokine receptors CCR1 and CCR3. Eur J Immunol 1997; 27: 2892–8
Eisner J, Mack M, Bruhl H, et al. Differential activation of CC chemokine receptors by AOP-RANTES. J Biol Chem 2000; 275: 7787–94
Homey B, Zlotnik A. Chemokines in allergy. Curr Opin Immunol 1999; 11: 626–34
Heath H, Qin S, Wu L, et al. Chemokine receptor usage by human eosinophils: the importance of CCR3 demonstrated using an antagonistic monoclonal antibody. J Clin Invest 1997; 99: 178–84
Justice JP, Borchers MT, Crosby JR, et al. Ablation of eosinophils leads to a reduction of allergen-induced pulmonary pathology. Am J Physiol Lung Cell Mol Physiol 2003; 284: LI 69–78
Ding C, Li J, Zhang X. Bertilimumab Cambridge Antibody Technology Group. Curr Opin Investig Drugs 2004; 5: 1213–8
Boshoff C, Endo Y, Collins PD, et al. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 1997; 278: 290–4
Sozzani S, Allavena P, Vecchi A, et al. Chemokine receptors: interaction with HIV-1 and viral-encoded chemokines. Pharm Acta Helv 2000; 74: 305–12
Gao JL, Murphy PM. Human cytomegalovirus open reading frame US28 encodes a functional beta chemokine receptor. J Biol Chem 1994; 269: 28539–42
Johnson Z, Kosco-Vilbois MH, Herren S, et al. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J Immunol 2004; 173: 5776–85
Johnson Z, Schwarz M, Power CA, et al. Multi-faceted strategies to combat disease by interference with the chemokine system. Trends Immunol 2005; 26: 268–74
Wymann MP, Bjorklof K, Calvez R, et al. Phosphoinositide 3-kinase gamma: a key modulator in inflammation and allergy. Biochem Soc Trans 2003; 31: 275–80
Wymann MP, Marone R. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol 2005; 17: 141–9
Ward SG, Finan P. Isoform-specific phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr Opin Pharmacol 2003; 3: 426–34
Barnes PJ. New drugs for asthma. Nat Rev Drug Discov 2004; 3: 831–44
Kumar S, Boehm J, Lee JC. P38 map kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2003; 2: 717–26
Scandella E, Men Y, Gillessen S, et al. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood 2002; 100: 1354–61
Wong M, Uddin S, Majchrzak B, et al. RANTES activates Jak2 and Jak3 to regulate engagement of multiple signaling pathways in T cells. J Biol Chem 2001; 276: 11427–31
Roshak AK, Callahan JF, Blake SM. Small-molecule inhibitors of NF-[kappa]B for the treatment of inflammatory joint disease. Curr Opin Pharmacol 2002; 2: 316–21
Gutierrez-Ramos JC, Lloyd C, Kapsenberg ML, et al. Non-redundant functional groups of chemokines operate in a coordinate manner during the inflammatory response in the lung. Immunol Rev 2000; 177: 31–42
Acknowledgments
CML is a Wellcome Senior Research Fellow and her work is funded by the Wellcome Trust (Ref 05774); ZB is employed by Novartis. No sources of funding were used in the preparation of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lloyd, C.M., Brown, Z. Chemokine Receptors. Treat Respir Med 5, 159–166 (2006). https://doi.org/10.2165/00151829-200605030-00002
Published:
Issue Date:
DOI: https://doi.org/10.2165/00151829-200605030-00002