
Abstract
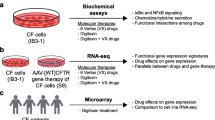
Cystic fibrosis (CF) is caused by a mutation in the CFTR gene, encoding a chloride channel. For the most common mutation, ΔF508, the basis of the deficit is the failure of the mutant CFTR channel protein to traffic properly to the apical plasma membrane of the affected epithelial cell. The trafficking failure results in loss of the cyclic adenosine monophosphate (cAMP)-activated chloride channel function of the CFTR protein in the plasma membrane. The lung is the principal site affecting patient morbidity and mortality in CF. The main reason is that the CF airway epithelial cells also secrete high levels of the proinflammatory cytokine interleukin (IL)-8, resulting in massive cellular inflammation, infection, tissue damage and lung destruction. The relationship between the trafficking defect, the loss of chloride channel activity, and inflammation is not known. However, gene therapy of CF lung epithelial cells with the wild-type CFTR gene can repair the chloride channel defect, as well as suppress the intrinsic hypersecretion of IL-8. Repair of both defective channels and high IL-8 secretion can also be effected by treatment with the candidate CF drug CPX, which is in clinical trials in CF patients. CPX acts by binding to the mutant CFTR protein, and helps the protein to mature and gain access to the plasma membrane. CPX also suppresses the synthesis and secretion of IL-8 from CF epithelial cells, presumably by virtue of its repair of the trafficking defect of mutant CFTR. To guide pharmacogenomic experiments we have therefore hypothesized that the genomic signature of CF epithelial cells treated with CPX should resemble the signature of the same cells repaired by gene therapy. We have developed two algorithms for identifying genes modified by repair of CFTR defects. The GRASP algorithm uses a statistical test to identify the most profoundly changing genes. The GENESAVER algorithm allows us to identify those genes whose pattern of expression changes in-phase or out-of-phase with IL-8 secretion by CF cells. For the latter algorithm we modified IL-8 secretion from CF cells by treatment with wild-type CFTR, with CPX, or by exposure to bacteria. The results have supported the hypothesis, and have provided a basis for considering the common pharmacogenomic expression signature as a surrogate endpoint for CF drug discovery. Significantly, the nature of the hypothesis, as well as the algorithm developed for this study, can be easily applied to pharmacogenomic studies with other goals.






Similar content being viewed by others
References
Welsh MJ, Ramsey BW, Accurso B, et al. Cystic fibrosis. In: Scriver CL, Beaudet AL, Valle D, et al., editors. The metabolic and molecular bases of inherited diseases. 8th ed. New York: McGraw-Hill, 2001: 5121–88
Rommens JM, Lannuzzi MC, Karem B-S, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245(1): 1059–65
Riordan JR, Rommens JM, Karem B-S, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066–73
Kerem B-S, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073–80
Shak S, Capon DJ, Hellmiss R, et al. Recombinant human DNAse I reduces viscosity of cystic fibrosis sputum. Proc Nat Acad Sci U S A 1990; 87: 9188–92
Konstan MW, Byard PJ, Hoppel CL et al. Effect of high dose ibuprofen in patients with cystic fibrosis. N Eng J Med 1995; 332: 848–54
Bonfield TL, Konstan MW, Burfeind P, et al. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin 10 which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol 1995; 13: 257–61
Bonfield TL, Panuska JR, Konstan MW, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med 1995; 152: 2111–18
Dean TP, Dai Y, Shute JK, et al. Interleukin-8 concentrations are elevated in bronchoalveolar lavage sputum and sera of children with cystic fibrosis. Pediatr Res 1993; 34: 159–61
Ruef C, Jefferson DM, Schlegel-Haueter SE, et al. Regulation of cytokine secretion by cystic fibrosis airway epithelial cells. Eur Resp J 1993; 6: 1429–36
Anderson MP, Rich DP, Gregory RJ, et al. Generation of cAMP activated chloride currents by expression of CFTR. Science 1991; 2516: 679–682
Collins FS. Cystic fibrosis: Molecular biology and therapeutic implications. Science 1992; 256774–9
Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev 1999; 79: S215–55
Griesenbach U, Scheid P, Hillery E, et al. Anti-inflammatory gene therapy directed at the airway epithelium. Gene Ther 2000; 7: 306–13
Pollard HB, Eidelman O, Jacobson KA, et al. Pharmacogenomics of cystic fibrosis. Mol Interventions 2001; 1: 54–63
Eidelman O, Srivastava M, Zhang J, et al. Control of the proinflammatory state in cystic fibrosis lung epithelial cells by genes from the TNFαR/NFκB pathway. Mol Med 2001. In press
Bailey DS, Bondar A, Furness LM. Pharmacogenomics — it’s not just pharmacogenetics. Curr Opin Biotechnol 1998; 6: 595–601
Ferrar P. Pharmacogenomics: a new approach to individual therapy of hypertension? Curr Opin Nephrol Hypertens 1998; 7: 217–21
Graever G, Shoemaker DD, Jones TW, et al. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet 1999; 3: 278–83
Eidelman O, Guay-Broder C, van Galen PJM, et al. A1-adenosine-receptor antagonists activate chloride efflux from cystic fibrosis cells. Proc Nat Acad Sci U S A 1992; 89: 5562–6
Guay-Broder C, Jacobson KA, Sarnoy S, et al. A-1 receptor antagonist ‘cyclopentyll,3-dipropylxanthine selectively activates chloride efflux from human epithelial and mouse fibroblast cell lines expressing the cystic fibrosis transmembrane regulator ΔF508 mutation. Biochemistry 1995; 34: 9079–87
Jacobson KA, Guay-Broder C, van Galen PJM, et al. Stimulation by alkylxanthines of chloride efflux in CFPAC-1 cells does not involve A1 adenosine receptors. Biochemistry 1995; 34: 9088–94
Kartner N, Hanrahan JW, Jensen TJ, et al. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produce a regulated anion conductance. Cell 1991; 64: 681–91
Berger HA, Anderson MP, Gregory RJ, et al. Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J Clin Invest 1991; 88: 1422–31
Rommens JM, Dho S, Bear CE, et al. cAMP-inducible chloride conductance in mouse fibroblast lines stably expressing the human cystic fibrosis transmem-brane conductance regulator. Proc Natl Acad Sci U S A 1991; 88: 7500–4
Bear CE, Reyes EF. cAMP-activated chloride conductance in the colonic cell line Caco-2. Am J Physiol 1992; 262: C251–6
Denning GM, Ostergaard LS, Cheng SH, et al. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J Clin Inv 1992; 89: 339–49
Tsui L-C. Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the Cystic Fibrosis Genetic Analysis Consortium. Hum Mutat 1992; 1: 197–203
Tsui L-C. The spectrum of cystic fibrosis mutations. Trends Genet 1992; 8: 392–8
Hung LW, Wang IX, Nikaido K, et al. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature 1998; 396: 703–7
Diederichs K, Diez J, Greller G, et al. Crystal structure of MalK the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus Litoralis. EMBO J 2000; 19: 5951–61
Drumm ML, Wilkinson DJ, Smit LS, et al. Chloride conductance expressed by DF508 and other mutant CFTRs in xenopus oocytes. Science 1991; 254: 1797–9
Arispe N, Rojas F, Hartman J, et al. Intrinsic anion channel activity of the recombinant first nucleotide binding fold domain to cystic fibrosis transmembrane conductance regulator protein. Proc Nat Acad Sci U S A 1992; 89: 1539–43
Cheng SH, Gregory RJ, Marshall J, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990; 63: 827–34
Yang Y, Janich S, Cohn JA, et al. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-golgi non-lysosomal compartment. Proc Nat Acad Sci U S A 1993; 90: 9480–4
Lukacs GL, Mohamed A, Kartner N, et al. Conformational maturation of CFTR but not its mutant counterpart (ΔF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J 1994; 13: 6076–86
Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator inefficient processing and rapid degradation of wildtype and mutant protein. J Biol Chem 1994; 269: 25710–18
Denning GM, Anderson MP, Amara JF, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992; 358(6389): 761–4
Denning GM, Ostedgaard LS, Welsh MJ. Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol 1992; 118(3): 551–9
Engelhardt JF, Yankaskas JR, Ernst SA, et al. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet 1992; 2: 240–7
Kartner N, Augustinas O, Jensen TJ, et al. Mislocalization of ΔF508 CFTR in cystic fibrosis sweat gland. Nat Genet 1992; 1: 321–7
Srivastava M, Eidelman O, Pollard HB. Pharmacogenomics of the cystic fibrosis transmembrane conductance regulator (CFTR) and the cystic fibrosis drug CPX using genome microarray analysis. Mol Med 1999; 5: 753–67
Abman SH, Ogle JW, Harbeck RJ, et al. Early bactriologic immunologic and clinical courses of young infants with cystic fibrosis identified by neonatal screening. J Pediatr 1991; 119: 211–17
Khan TZ, Wagene JS, Bost T, et al. Early pulmonary inflammation in infants with cystic fbrosis. Am J Respir Crit Care Med 1995; 151: 1075–82
Cruse JM, Lewis RE. Illustrated dictionary of immunology. Boca Raton: CRC Press Boca, 1995: Appendix 3
Richman-Eisenstat JB, Jorens PG, Hebert CA, et al.Interleukin-8: an important chemoattractant in sputum of patients with chronic inflammatory airway diseases. Am J Physiol 1993; 264: L413–8
Francoeur C, Denis M. Nitric oxide and interleukin-8 as inflammatory components of cystic fibrosis. Inflammation 1995; 19: 587–98
Armstrong DS, Grimwood K, Carlin JB, et al. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med 1997; 156: 1197–204
Briars GL, Dean TP, Murphy JL, et al. Interleukin-8 and tumour necrosis factor-alpha concentrations in cystic fibrosis. Arch Dis Child 1995; 73: 74–6
Tabary O, Zahm JM, Hinnrasky J, et al. Selective upregulation of the chemokine IL8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am JPathol 1998; 153: 921–30
Bedard M, McClure CD, Schiller NL, et al. Release of interleukin-8 interleukin-6 and colony-stimulating factors by upper airways epithelial cells: implications for cystic fibrosis. Am J Resp Cell Mol Biol 1993; 9: 455–62
DiMango E, Ratner AJ, Bryan R, et al. Activation of NF-kappaB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest 1998; 101: 2598–605
Black HR, Yankaskas JR, Johnson LG, et al. Interleukin-8 production by cystic fibrosis nasal epithelial cells after tumor necrosis factor-alpha and respiratory syncytial virus stimulation. Am J Respir Cell Mol Biol 1998; 19: 210–15
Massion PP, Inoue H, Richman-Eisenstat J, et al. Novel Pseudomonas product stimulates interleukin-8 production in airway epithelial cells in vitro. J Clin Invest 1994; 93: 26–32
Schwiebert LM, Estell K, Probst SM. Chemokine expression in CF epithelia: implications for the role of CFTR in RANTES expression. Am J Physiol 1999, 276: C700–10
Griesenbach U, Scheid P, Hillery E, et al. Towards anti-inflammatory lung gene therapy. Pediatr Pulmonol 1999; S19: 237
Arispe N, Ma J, Jacobson KA, et al. Direct activation of cystic fibrosis transmembrane conductance regulator by 8-cyclopentyl-1,3-dipropylxanthine and 1,3-diallyl-8-cyclohexylxanthine. J Biol Chem 1998; 273: 5724–34
Pollard HB. Role of CPX in promoting trafficking and chloride channel activity of wildtype and mutant CFTR. Pediatr Pulmonol 1997; S14: 128–31
Cohen BE, Lee G, Jacobson KA, et al. CPX (1,3-dipropyl-8-cyclopentyl xanthine) and other alkyl-xanthines differentially bind to the wild type and ΔF508 mutant first nucleotide binding fold (NBF-1) domains of the cystic fibrosis transmembrane conductance regulator (CFTR). Biochemistry 1997; 36: 6455–61
Schultz BD, Frizzel RA, Bridges RJ. IBMX stabilizes the ATP-bound state of ΔF508CFTR. J Gen Physiol 1994; 104: 35a
Stutts MJ, Canessa CM, Olsen JC, et al. CFTR as a cAMP-dependent regulator of sodium channels. Science 1995; 269: 847–50
Ismailov II, Awayda MS, Jovov B. Regulation of epithelial sodium channel by the cystic fibrosis transmembrane conductance regulator. J Biol Chem 1996; 271: 4725–32
Egan M, Flotte T, Affione S, et al. Defective regulation of outwardly rectifying Cl channels by protein kinase A corrected by insertion of CFTR. Nature 1992; 358: 581–4
Gabriel SE, Clarke LL, Boucher FCC, et al. CFTR and outward rectifying chloride channel are distinct proteins with a regulatory relationship. Nature 1993; 363: 263–8
Schwiebert EM, Flotte T, Cutting GR, et al. Both CFTR and outwardly rectifying chloride channels contribute to cAMP-stimulated whole cell chloride currents. Am J Physiol 1994; 266: C1464–7
Jovov B, Ismailov II, Benos DJ. Cystic fibrosis transmembrane conductance regulator is required for protein kinase A activation of an outwardly rectified anion channel purified from bovine tracheal epithelia. J Biol Chem 1995; 270: 1521–8
Fulmer SB, Schwiebert EM, Morales MM, et al. Two cystic fibrosis transmembrane conductance regulator mutations have different effects on both pulmonary phenotype and regulation of outwardly rectified chloride currents. Proc Nat Acad Sci USA 1995; 92: 6832–6
Naren AP, Nelson DJ, Xie W, et al. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature 1997; 390: 302–5
Naren AP, Quick MW, Collawn JF, et al. Syntaxin 1A inhibits CFTR chloride channels by means of domain-specific protein-protein interactions. Proc Nat Acad Sci U S A 1998; 95: 10972–7
Moyer BD, Denton J, Karlson K, et al. Interacting domain in CFTR is required for apical polarization and export from the endoplasmic reticulum. Pediatr Pulmonol 1999a; S19: 164
Moyer BD, Denton J, Karlson K, et al. A PDZ interacting domain in CFTR is an apical membrane polarization signal. J Clin Invest 1999b; 104: 1353–61
Raghuram V, Hallows KR, Foskett JK. Multiple PDZ domains involved in the association of NHERF with CFTR. Pediatr Pulmonol 1999; S19: 178
Casavola V, Turner RJ, Guay-Boder C, et al. CPX: a selective A1-adenosine-receptor antagonist regulates intracellular pH in cystic fibrosis cells. Am J Physiol 1995; 269: C226–33
Sun F, Hug MJ, Yun CH, et al. The PDZ domain-containing protein E3KARP couples EZRIN to cystic fibrosis transmembrane conductance regulator (CFTR). Pediatr Pulmonol 1999; S19: 164
Short DB, Trotter KW, Reczek D, et al. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem 1998; 273: 19797–801
Bretscher A. Regulation of cortical structure by the Ezrin-Radaxin-Moesin protein family. Curr Opin Cell Biol 1999; 11: 109–16
Pearson MA, Reczek D, Bretscher A, et al. Structure of the ERM protein Moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 2000; 101: 259–70
Lamprecht G, Weinman EJ, Yun CH. The role of NHERF and E3KARP in the cAMP-mediated inhibition of NHE3. J Biol Chem 1998; 273: 29972–8
DiMango E, Zar HJ, Bryan R et al. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest 1995; 96: 2204–10
Steffan NM, Bren GD, Frantz B, et al. Regulation of IκkB alpha phosphorylation by PKC-and Ca2+ dependent signal transduction pathways. J Immunol 1995; 155: 4685–91
Sun L, Carpenter G. Epidermal growth factor activation of NF-κB is mediated through IκBa degradation and intracellular free calcium. Oncogene 1998; 16: 2095–105
Trushin SA, Pennington KN, Algeciras-Schimnich A, et al. Protein kinase C and calcineurin synergize to activate IκB kinase and NF-κB in T lymphocytes. J Biol Chem 1999; 274: 22923–31
Bren GD, Pennington KN, Paya CV. PKC-zeta-associated CK2 participates in the turnover of free IκBa. J Mol Biol 2000; 297: 1245–58
Taylor JA, Bren GD, Pennington KN, et al. Serine 32 and serine 36 of IκBa are directly phosphorylated by protein kinase CKII in vitro. J Mol Biol 1999; 290: 839–50
Han Y, Meng T, Murray NR, et al. Interleukin-1-induced nuclear factor-κB-IκBa autoregulatory feedback loop in hepatocytes. A role for protein kinase Ca in post-transcriptional regulation of IκBa resynthesis. J Biol Chem 1999; 274: 939–47
Vertegaal AC, Kuiperj HB, Yamaoka S, et al. Protein kinase C-alpha is an upstream activator of the IκBa complex in the TPA signal transduction pathway to NF-κB in U2OS cells. Cell Signal 2000; 12: 759–68
Khoshnan A, Bae D, Tindell CA, et al. The physical association of protein kinase C-theta with a lipid raft-associated inhibitor of KB factor kinase (IKK) complex plays a role in the activation of the NF-κB cascade by TCR and CD28. J Immunol 2000; 165: 6933–40
Wang G, Siow YL, O K. Homocystein stimulates nuclear factor κB activity and monocyte chemoattractant protein-1 expression in vascular smooth-muscle cells: a possible role for protein kinase C. Biochem J 2000; 352: 817–26
Maiuri L, Raia V, De Marco G, et al. DNA fragmentation is a feature of cystic fibrosis epithelial cells: a disease with inappropriate apoptosis? FEBS Lett 1997; 408: 225–31
Durieu I, Amsellem C, Paulin C, et al. Fas and Fas ligand expression in cystic fibrosis airway epithelium. Thorax 1999; 54: 1093–8
Gottlieb RA, Dasanjh A. Mutant cystic fibrosis transmembrane conductance regulator inhibits acidification and apoptosis in C127 cell: possible relevance to cystic fibrosis. Proc Nat Acad Sci U S A 1996; 93: 3587–91
DeFranco AL. Macrophage signaling in response to bacterial lipopolysaccharides. Pediatr Pulmonol 1999; S19: 124
Prince A. Pulmonary pathogens activate Ca2+-dependent MAPK expression in respiratory epithelial cells. Pediatr Pulmonol 1999; S19: 100–1
Laub MT, McAdams HH, Feldblyum T, et al. Global analysis of the genetic network controlling a bacterial cell cycle. Science 2000; 290: 2144–8
Chu S, DeRisi J, Eisen M, Mulholland J, et al. The transcriptional program of sporulation in budding yeast. Science 1998; 282: 699–705
Wen X, Fuhrman S, Michaels GS, et al. Large-scale temporal gene expression mapping of central nervous system development. Proc Nat Acad Sci U S A 1998; 95: 334–9
van Someren EP, Wessels LF, Reinders MJ. Linear modeling of genetic networks from experimental data. Proc Int Conf Intell Syst Mol Biol 2000; 8: 355–66
Akutsu T, Miyano S, Kuhara S. Inferring qualitative relations in genetic networks and metabolic pathways. Bioinformatics 2000; 16: 727–34
Szallasi Z. Genetic network analysis in light of massively parallel biological data acquisition. Pac Symp Biocomput 1999; 16(2 Suppl.): 5–16
Wahde M, Hertz J. Coarse-grained reverse engineering of genetic regulatory networks. Biosystems 2000; 55: 129–36
D’haeseleer P, Liang S, Somogyi R. Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics 2000; 16: 707–26
Eisen MB, Spellman PT, Brown PO, et al. Cluster analysis and display of genome-wide expression patterns. Proc Nat Acad Sci USA 1998; 95: 14863–8
Alizaheh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–11
Perous CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000; 406: 747–52
Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Gen 2000; 24: 227–35
Srivastava M, Eidelman O, Pollard HB. cDNA microarrays for pharmacogenomic analysis of cystic fibrosis. Methods in Molecular Medicine 2001. In press
Zeitlin PL, Lu L, Hwang T-C, et al. A cystic fibrosis bronchial epithelial cell line: immortalization by adeno12-SV40 infection. Am J Respir Cell Mol Biol 1991; 4: 313–9
Acknowledgements
This work was supported in part by grants from the Cystic Fibrosis Foundation and the National Institutes of Health (R01-DK53051).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eidelman, O., Zhang, J., Srivastava, M. et al. Cystic Fibrosis and the Use of Pharmacogenomics to Determine Surrogate Endpoints for Drug Discovery. Am J Pharmacogenomics 1, 223–238 (2001). https://doi.org/10.2165/00129785-200101030-00006
Published:
Issue Date:
DOI: https://doi.org/10.2165/00129785-200101030-00006