
Abstract
Background
The major aim of this study was to develop a strategy for predicting human pharmacokinetics using physiologically based pharmacokinetic (PBPK) modelling. This was compared with allometry (of plasma concentration-time profiles using the Dedrick approach), in order to determine the best approaches and strategies for the prediction of human pharmacokinetics.
Methods
PBPK and Dedrick predictions were made for 19 F. Hoffmann-La Roche compounds. A strategy for the prediction of human pharmacokinetics using PBPK modelling was proposed in this study. Predicted values (pharmacokinetic parameters, plasma concentrations) were compared with observed values obtained after intravenous and oral administration in order to assess the accuracy of the prediction methods.
Results
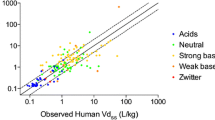
By following the proposed strategy for PBPK, a prediction would have been made prospectively for approximately 70% of the compounds. The prediction accuracy for these compounds in terms of the percentage of compounds with an average-fold error of <2-fold was 83%, 50%, 75%, 67%, 92% and 100% for apparent oral clearance (CL/F), apparent volume of distribution during terminal phase after oral administration (Vz/F), terminal elimination half-life (t½), peak plasma concentration (Cmax), area under the plasma concentration-time curve (AUC) and time to reach Cmax (tmax), respectively. For the other 30% compounds, unacceptable prediction accuracy was obtained in animals; therefore, a prospective prediction of human pharmacokinetics would not have been made using PBPK. For these compounds, prediction accuracy was also poor using the Dedrick approach. In the majority of cases, PBPK gave more accurate predictions of pharmacokinetic parameters and plasma concentration-time profiles than the Dedrick approach.
Conclusions
Based on the dataset evaluated in this study, PBPK gave reasonable predictions of human pharmacokinetics using preclinical data and is the recommended approach in the majority of cases. In addition, PBPK modelling is a useful tool to gain insights into the properties of a compound. Thus, PBPK can guide experimental efforts to obtain the relevant information necessary to understand the compound’s properties before entry into human, ultimately resulting in a higher level of prediction accuracy.

















Similar content being viewed by others


Notes
The use of trade names is for product identification purposes only and does not imply endorsement.
References
Reigner BG, Williams PEO, Patel JH, et al. An evaluation of the integration of pharmacokinetic and pharmacodynamic principles in clinical drug development: experience within Hoffmann La Roche. Clin Pharmacokinet 1997; 33: 142–52
Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm 1982; 10: 201–27
Boxenbaum H. Interspecies scaling and the evolutionary comparative paradigm. Drug Metab Rev 1984; 15: 1071–121
Bernareggi A, Rowland M. Physiologic modelling of cyclosporine kinetics in rat and man. J Pharm Sci 1991; 19(1): 21–50
Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies II: generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci 2002; 91: 1358–70
Dedrick RL. Animal scale-up. J Pharmacokinet Biopharm 1973; 1(5): 435–61
Mordenti J. Man vs beast: pharmacokinetic scaling in mammals. J Pharm Sci 1986; 75: 1028–40
Dedrick RL, Bischoff KB, Zaharko DS. Interspecies correlation of plasma concentration history of methotrexate (NSC-740). Cancer Chemother Rep 1970; 54: 95–101
Mahmood I, Balian JD. Interspecies scaling: predicting pharmacokinetic parameters of antiepileptic drugs in humans from animals with special emphasis on clearance. J Pharm Sci 1996; 85: 411–4
Sanwald-Ducray P, Dow J. Prediction of the pharmacokinetic parameters of reduced-dolasetron in man using in vitro-in vivo and interspecies allometric scaling. Xenobiotica 1997; 27(2): 189–201
Mahmood I. Interspecies scaling of renally secreted drugs. Life Sci 1998; 63: 2365–71
Lavé T, Portmann R, Schenker G, et al. Interspecies pharmacokinetic comparisons and allometric scaling of napsagatran, a low molecular weight thrombin inhibitor. J Pharm Pharmacol 1999; 51: 85–91
Lavé T, Coassolo P, Reigner B. Prediction of hepatic metabolic clearance based on interspecies allometric scaling techniques and in vitro-in vivo correlations. Clin Pharmacokinet 1999; 36(3): 211–31
Zuegge J, Schneider G, Coassolo P, et al. Prediction of hepatic metabolic clearance: comparison and assessment of prediction methods. Clin Pharmacokinet 2001; 40(7): 553–63
Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol 1994; 47: 1469–79
Iwatsubo T, Hirota N, Ooie T, et al. Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol Ther 1997; 73(2): 147–71
Poulin P, Theil FP. A priori prediction of tissue:plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J Pharm Sci 2000; 89: 16–35
Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies: I. Mechanism-based prediction of volume of distribution. J Pharm Sci 2002; 91: 129–56
Poulin P, Schoenlein K, Theil FP. Prediction of adipose tissue:plasma partition coefficients for structurally unrelated drugs. J Pharm Sci 2000; 90: 436–47
Houston JB, Cariile DJ. Prediction of hepatic clearance from microsomes, hepatocytes and liver slices. Drug Metab Rev 1997; 29: 891–922
Ito K, Houston JB. Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm Res 2004; 21(5): 785–92
Jones HM, Houston JB. Use of the substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab Dispos 2004; 32(9): 973–82
Lavé T, Dupin S, Schmitt C, et al. The use of human hepatocytes to select compounds based on their expected hepatic extraction ratios in humans. Pharm Res 1997; 14(2): 152–5
Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos 1999; 27(11): 1350–9
Luttringer O, Theil FP, Poulin P, et al. Physiologically based pharmacokinetic (PBPK) modelling of disposition of epiroprim in humans. J Pharm Sci 2003; 92: 1990–2007
Kansy M, Senner F, Gubernator K. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J Med Chem 1998; 41(7): 1007–10
Alsenz J, Haenel E. Development of a 7-day, 96-well Caco-2 permeability assay with high-throughput direct UV compound analysis. Pharm Res 2003; 20(12): 1961–9
Brown RP, Delp MD, Lindstedt SL, et al. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 1997; 13: 407–84
Yokogawa K, Nakashima E, Ichimura F. Effect of fat tissue volume on the distribution kinetics of biperiden as a function of age in rats. Drug Metab Dispos 1990; 18(2): 258–63
Blakey GE, Nestorov IA, Arundel PA, et al. Quantitative structure-pharmacokinetics relationships: I. Development of a whole-body physiologically based model to characterize changes in pharmacokinetics across a homologous series of barbiturates in the rat. J Pharmacokinet Biopharm 1997; 25(3): 277–312
Austin RP, Barton P, Cockroft SL, et al. The influence of non-specific microsomal binding on apparent intrinsic clearance, and its prediction from physiochemical properties. Drug Metab Dispos 2002; 30: 1497–501
Cariile DJ, Zomorodi K, Houston JB. Scaling factors to relate drug metabolic clearance in hepatic microsomes, isolated hepatocytes and the intact liver: studies with induced livers involving diazepam. Drug Metab Dispos 1997; 25(8): 903–11
Naritomi Y, Terashita S, Kimura S, et al. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab Dispos 2001; 29(10): 1316–24
Wilson ZE, Rostami-Hodjegan A, Burn JL, et al. Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br J Clin Pharmacol 2003; 56: 433–40
Lin JH. Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab Dispos 1998; 26(12): 1202–12
Ellmerer M, Schaupp L, Brunner GA, et al. Measurement of interstitial albumin in human skeletal muscle and adipose tissue by open-flow microperfusion. Am J Physiol Endocrinol Metab 2000; 278: E352–6
Arundel PH. A multi-compartmental model generally applicable to physiologically-based pharmacokinetics (AstraZeneca, UK) [poster]. 3rd IFAC symposium: modelling and control in biomedical systems; 1997 March 23–26; Warwick
Sawada Y, Hanano M, Sugiyama Y, et al. Prediction of the volume of distribution of basic drugs in human based on data from animals. J Pharmacokinet Biopharm 1984; 12: 587–96
Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev 2001; 50: S41–67
Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm 1999; 186: 119–25
Gabrielsson J, Weiner D. Interspecies scaling. In: Gabrielsson J, Weiner D, editors. PK/PD data analysis: concepts and applications. Stockholm: Swedish Pharmaceutical Press, 2000: 153–174
Bischoff KB, Dedrick RL. Thiopental pharmacokinetics. J Pharm Sci 1968; 57: 1347–57
Lin JH, Sugiyama Y, Awazu S, et al. Physiological pharmacokinetics of ethoxybenzamide based on biochemical data obtained in vitro as well as on physiological data. J Pharmacokinet Biopharm 1982; 10: 649–61
Igari Y, Sugiyama Y, Sawada Y, et al. Prediction of diazepam disposition in the rat and man by a physiologically based pharmacokinetic model. J Pharmacokinet Biopharm 1983; 11(6): 577–93
Theil FP, Haddad S, Guentert TW, et al. Utility of physiologically based pharmacokinetic models to drug development and rational drug discovery candidate selection. Toxicol Lett 2003; 138: 29–49
Bonate PL, Howard D. Critique of prospective allometric scaling: does the emperor have new clothes? J Clin Pharmacol 2000; 40: 335–40
Obach RS, Baxter JG, Liston TE, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther 1997; 283: 46–58
Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog and monkey to humans: I. Clearance. Drug Metab Dispos 2004; 32(6): 603–11
Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog and monkey to humans: II. Volume of distribution and mean residence time. Drug Metab Dispos 2004; 32(6): 612–9
Brightman FA, Leahy DE, Searle GE, et al. Application of a generic physiologically-based pharmacokinetic model to the estimation of xenobiotic levels in human plasma. Drug Metab Dispos 2005; 34(1): 94–101
Wenlock MC, Austin RP, Barton P, et al. A comparison of physicochemical property profiles of development and marketed oral drugs. J Med Chem 2003; 46: 1250–6
Lapka R, Rejholec V, Sechser T, et al. Interspecies pharmacokinetic scaling of metazosin, a novel alpha-adrenergic antagonist. Biopharm Drug Dispos 1989; 10(6): 581–9
Lavé T, Saner A, Coassolo P, et al. Animal pharmacokinetics and interspecies scaling from animals to man of lamifiban, a new platelet aggregation inhibitor. J Pharm Sci 1996; 48(6): 573–7
Hutchaleelaha A, Chow HH, Mayersohn M. Comparative pharmacokinetics and interspecies scaling of amphotericin B in several mammalian species. J Pharm Pharmacol 1997; 49: 178–83
Chapell WR, Mordenti J. Extrapolation of toxicological and pharmacological data from animals to humans. In: Testa B, editor. Advances in drug research. London: Academic Press, 1991: 1–116
Poggesi I. Predicting human pharmacokinetics from preclinical data. Curr Opin Drug Discov Devel 2004; 7(1): 100–11
Smith RL. Excretion of drugs in bile. In: Brodie BB, Gillette JR, editors. Handbook of experimental pharmacology. Concepts in biochemical pharmacology. Vol. XXVIII. Berlin: Springer-Verlag, 1971: 354–389
Smith RL, editor. The excretory function of bile: the elimination of drugs and toxic substances in bile. London: Chapman and Hall, 1973
Liu X, Chism JP, LeCluyse EL, et al. Correlation of biliary excretion in sandwich-cultured rat hepatocytes and in vivo in rats. Drug Metab Dispos 1999; 27(6): 637–44
Paine MF, Khalighi M, Fisher JM, et al. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J Pharmacol Exp Ther 1997; 283: 1552–62
Obach RS. The importance of nonspecific binding in in vitro matrices, its impact on enzyme kinetic studies of drug metabolism reactions, and implications for in vitro-in vivo correlations. Drug Metab Dispos 1996; 24(10): 1047–9
Obach RS. Nonspecific binding to microsomes: impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab Dispos 1997; 25(12): 1359–69
Raub TJ, Barsuhn CL, Williams LR, et al. Use of a biophysical-kinetic model to understand the roles of protein binding and membrane partitioning on passive diffusion of highly lipophilic molecules across cellular barriers. J Drug Target 1993; 1(4): 269–86
Sawada GA, Barsuhn CL, Lutzke BS, et al. Increased lipophilicity and subsequent cell partitioning decrease passive transcellular diffusion of novel, highly lipophilic antioxidants. J Pharmacol Exp Ther 1999; 288(3): 1317–26
Yata N, Toyoda T, Murakami T, et al. Phosphatidylserine as a determinant for the tissue distribution of weakly basic drugs in rats. Pharm Res 1990; 7: 1019–25
Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 2005; 94(6): 1259–76
Nicolaides E, Galia E, Efthymiopoulos C, et al. Forecasting the in vivo performance of four low solubility drugs from their in vitro dissolution data. Pharm Res 1998; 16(12): 1876–82
Nicolaides E, Symillides M, Dressman JB, et al. Biorelevant dissolution testing to predict the plasma profile of lipophilic drugs after oral administration. Pharm Res 2001; 18(3): 380–8
de Zwart LL, Rompelberg CJM, Sips AJAM, et al. Anatomical and physiological differences between various species used in studies on the pharmacokinetics and toxicology of xenobiotics: a review of the literature. Bilthoven: Research for Man and Enviroment, National Institute of Public Health and the Environment; 1999 Oct. RIVM report no. 623860 010
Mithani SD, Bakatselou V, Ten Hoor CN, et al. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharm Res 1996; 13(1): 163–7
Nestorov I. Whole body pharmacokinetic models. Clin Pharmacokinet 2003; 42(10): 883–908
Nestorov I, Gueorguieva I, Jones HM, et al. Incorporating measures of variability and uncertainty into the prediction of in vivo hepatic clearance from in vitro. Drug Metab Dispos 2002; 30: 276–82
Jones HM, Hallifax D, Houston JB. Quantitative prediction of the in vivo inhibition of diazepam metabolism by omeprazole using rat liver microsomes and hepatocytes. Drug Metab Dispos 2004; 32(5): 572–80
Acknowledgements
The authors would like to thank Patrick Poulin, Alex MacDonald and Frank-Peter Theil for their assistance in this study. This work was supported by F. Hoffman La Roche. The authors have no conflicts of interest directly relevant to the content of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jones, H.M., Parrott, N., Jorga, K. et al. A Novel Strategy for Physiologically Based Predictions of Human Pharmacokinetics. Clin Pharmacokinet 45, 511–542 (2006). https://doi.org/10.2165/00003088-200645050-00006
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200645050-00006