
Abstract
Vascular endothelial growth factor (VEGF)-D, a member of the VEGF family, induces both angiogenesis and lymphangiogenesis by activating VEGF receptor-2 (VEGFR-2) and VEGFR-3 on the surface of endothelial cells. Transforming growth factor (TGF)-β1 has been shown to stimulate VEGF-A expression in human lung fibroblast via the Smad3 signaling pathway and to induce VEGF-C in human proximal tubular epithelial cells. However, the effects of TGF-β1 on VEGF-D regulation are unknown. To investigate the regulation of VEGF-D, human lung fibroblasts were studied under pro-fibrotic conditions in vitro and in idiopathic pulmonary fibrosis (IPF) lung tissue. We demonstrate that TGF-β1 downregulates VEGF-D expression in a dose- and time-dependent manner in human lung fibroblasts. This TGF-β1 effect can be abolished by inhibitors of TGF-β type I receptor kinase and Jun NH2-terminal kinase (JNK), but not by Smad3 knockdown. In addition, VEGF-D knockdown in human lung fibroblasts induces G1/S transition and promotes cell proliferation. Importantly, VEGF-D protein expression is decreased in lung homogenates from IPF patients compared with control lung. In IPF lung sections, fibroblastic foci show very weak VEGF-D immunoreactivity, whereas VEGF-D is abundantly expressed within alveolar interstitial cells in control lung. Taken together, our data identify a novel mechanism for downstream signal transduction induced by TGF-β1 in lung fibroblasts, through which they may mediate tissue remodeling in IPF.
Similar content being viewed by others


Introduction
Vascular endothelial growth factors (VEGFs) contribute to physiological and pathological angiogenesis and lymphangiogenesis during embryonic development and adult life (1). Six members of the VEGF family have been identified: VEGF-A, -B, -C, -D and -E and placenta growth factor (2). Although VEGF family members are structurally and functionally related, they possess distinct receptor binding specificities that contribute to their diversity of function (3,4). Among the six members discovered to date, VEGF-A is the most comprehensively characterized cytokine. It is a major contributor to angiogenesis and regulates multiple endothelial cell functions via VEGF receptors (VEGFRs) 1 and 2 (5). VEGF-C and -D, on the other hand, form a functional subgroup that primarily mediates lymphangiogenesis via VEGFR-3 and angiogenesis via VEGFR-2 (6).
VEGF-D is synthesized as a full-length protein with a molecular weight of ~53 kDa (7,8). It contains a central VEGF homology domain (VHD) flanked by N- and C-terminal propeptides (6). Full-length VEGF-D is secreted and is immediately proteolytically processed outside the cell by plasmin (9) and proprotein converatses (8), thereby generating a mature form (molecular weight ~21 kDa) with a much higher affinity for VEGFR-2 and -3 compared with the unprocessed form (10).
The human c-Fos-induced growth factor (FIGF) gene, which encodes for VEGF-D, maps to chromosome Xp22.31 (11). Its transcripts have been identified in a wide range of normal organs, with the most abundant expression found in the blood, colon, heart, lung and small intestine (6). Unlike VEGF-C (12), studies using VEGF-D-deficient mice suggest that embryological development of the lymphatic circulation is relatively normal in the absence of VEGF-D (13,14). However, recent findings have correlated the phenotype of adult VEGF-D-deficient mice with reduced pulmonary lymphatic density (13) and smaller dermal lymphatic caliber (15) under nonpathological conditions. Notably, when challenged with excisional wounds, these mice manifest histological evidence of pronounced tissue edema caused by poor lymphatic drainage and accelerated wound healing due to an increase in the number of fibroblasts (15).
VEGF-D expression can be induced by c-fos (16) and Fra-1 (17) activation as well as cell-cell contact mediated by cadherin-11 (18). Conversely, VEGF-D can be downregulated by interleukin (IL)-1β (19), transforming growth factor (TGF)-α, betacellulin, heregulin-β1 (20), Wnt signaling and mobilization of β-catenin from cell membrane (21). Abnormal regulation of VEGF-D has been documented in several human diseases such as lymphangioleiomyomatosis (LAM) (22) and malignancy (reviewed in [23,24]).
Idiopathic pulmonary fibrosis (IPF) is a chronic, debilitating and lethal fibrotic lung disease that is associated with aberrant wound healing (25,26). IPF is characterized by activation of the TGF-β1 pathways, an abnormal accumulation of fibroblasts/myofibroblasts, formation of fibroblastic foci, and excessive deposition of extracellular matrix (ECM) components in the alveolar interstitium (27). TGF-β1 stimulates VEGF-A expression in human fetal lung fibroblast via the Smad3 signaling pathway (28). A similar induction of VEGF-C by TGF-β1 was observed in human proximal tubular epithelial cells (29). However, the effect of TGF-β1 on VEGF-D is not well defined. The present study was undertaken to investigate the regulation and potential intracellular functions of VEGF-D.
Materials and Methods
Human Lung Tissues Collection
Under institutional review board-approved protocols, lung tissues from subjects with IPF (n = 6) undergoing lung transplantation at Brigham and Women’s Hospital were procured. IPF was diagnosed according to the American Thoracic Society guidelines (30). Control nonfibrotic lungs (n = 6) were from donor lungs rejected for transplant. Samples of pulmonary parenchyma were harvested and fixed in 10% neutral buffered formalin for 48 h and subsequently embedded with paraffin for histological analysis. The remaining lung tissue was immediately snap frozen in liquid nitrogen after excision and stored at −80°C until further processing.
Cell Culture
Primary normal adult normal lung fibroblasts (NLFs) were isolated by explant culture from control nonfibrotic lungs (n = 3) rejected for transplant and were used at passages 3–6. Human fetal lung fibroblasts (MRC-5) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were used at passages 21–25. Both NLF and MRC-5 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 µg/mL streptomycin (Life Technologies, Carlsbad, CA, USA). Cells were incubated at 37°C in a humidified 5% CO2 atmosphere and were serum-starved for 24 h before they were stimulated with recombinant human TGF-β1 (5 µg/mL unless otherwise stated) (R&D Systems, Minneapolis, MN, USA) or vehicle for the indicated times. To examine the downstream signaling molecules that mediate the effect of TGF-β1 on VEGF-D transcription, MRC-5 cells were pre-treated with selective inhibitors of activin receptor-like kinase (ALK)-5 (SB431542, 10 µmol/L [inhibitors and amounts given in parentheses in this sentence]) (R&D Systems), mitogen-activated protein kinase kinase 1 and 2 (MEK1/2) (UO126, 10 µmol/L) (Cell Signaling Technology, Danvers, MA, USA), phosphatidylinositol 3-kinase (PI3K)/Akt (wortmannin, 100 nmol/L) (Cell Signaling Technology), Jun NH2-terminal kinase (JNK) (SP600125, 5 µmol/L) (Cell Signaling Technology) or p38 mitogen-activated protein kinase (p38MAPK) (SB203580, 10 µmol/L) (R&D Systems) 1 h before the addition of TGF-β1.
Cells were collected either 1 h thereafter to confirm cell signaling blockage or 48 h thereafter to determine effects of the aforementioned inhibitors on the reduction of VEGF-D expression in response to TGF-β1.
RNA Interference
The siRNA sequences for human β-catenin, Smad3, tuberous sclerosis (TSC)-2 and FIGF were designed using the web-based siRNA Selection Program from Whitehead Institute for Biomedical Research (31) and were synthesized by Sigma-Aldrich (St. Louis, MO, USA). Refer to Table 1 for siRNA sequences. MRC-5 cells were transfected with siRNA for targeted gene suppression or with MISSION siRNA Universal Negative Control (Sigma-Aldrich). Cells were treated with 1 nmol/L siRNA by using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s recommendations. RNAi-mediated knockdown efficiency was confirmed by Western blotting.
Western Blotting
Protein was extracted from human lung homogenate or cell lysate by using radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Thermo Scientific [Thermo Fisher Scientific Inc.], Rockford, IL, USA). Protein concentration was then determined by using BCA protein assay (Thermo Scientific). Equal amounts of protein samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane. The membrane was blocked with 5% nonfat dry milk or bovine serum albumin in Tris-buffered saline with Tween for 1 h at room temperature and incubated with the primary antibody overnight at 4°C. A complete list of antibodies is included in Table 2. A goat anti-mouse or goat anti-rabbit horseradish peroxidase (HRP) conjugate secondary antibody (Thermo Scientific) was applied for 1 h at room temperature. Blots were developed with SuperSignal West Pico or Femto chemiluminescent substrate (Thermo Scientific) and visualized in ChemiDoc XRS+ imaging system (Bio-Rad, Hercules, CA, USA). Density of the bands was analyzed by ImageJ software (National Institutes of Health, https://doi.org/rsbweb.nih.gov/ij).
Immunohistochemistry
Immunohistochemical staining was performed to determine the distribution of VEGF-D in lung tissue sections from IPF and control subjects. The 5-µm sections were deparaffinized in xylene and rehydrated in a graded series of alcohol. Heat-induced epitope retrieval was performed using citrate buffer (pH 6.0) for 20 min. Tissue was then stained by using VEGF-D antibody and an Anti-Mouse HRP-DAB Cell and Tissue Staining Kit (R&D Systems) according to the manufacturer’s instructions and briefly counterstained with hematoxylin. Normal mouse IgG (Cell Signaling Technology) was used in place of the primary antibody with each specimen to evaluate nonspecific staining. Images from immunohistochemical stained slides were captured with an FSX100 microscope (Olympus, Center Valley, PA, USA).
Cell Cycle Analysis
A total of 20% of control siRNA- and VEGF-D siRNA-transfected cells were used for RNAi-mediated knockdown efficiency confirmation. The remaining transfected cells were washed with ice-cold phosphate-buffered saline (PBS), resuspended in 300 µL PBS and subsequently fixed with the addition of 700 µL 100% −20°C ethanol drop-wise. Cells were stored at −20°C for 24 h. On the second day, fixed cells were washed with PBS and treated with RNase A (Qiagen, Valencia, CA, USA) and incubated with propidium iodide (Life Technologies) for 30 min at room temperature. Cells cultured in DMEM supplemented with 20% FBS were subjected to the same staining procedure and were used as positive controls. DNA contents were measured by using FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA). Data analysis was performed by using FlowJo (TreeStar, Ashland, OR, USA).
Cell Proliferation Assay
MRC-5 cells were transfected with either control siRNA or VEGF-D siRNA.
Cells cultured in DMEM supplemented with 20% FBS were used as positive controls. Cell proliferation was determined by a fluorescence-based assay (CyQUANT cell proliferation assay kit; Life Technologies) at the indicated time points after transfection.
RNA Extraction and Real-Time Polymerase Chain Reaction
RNA was extracted by using an RNeasy Mini Kit (Qiagen). Then 1 µg total RNA was reverse-transcribed by using an amfiRivert cDNA Synthesis Master Mix (GenDEPOT, Barker, TX, USA). A total of 1 µL of the resultant cDNA was subjected to quantitative real-time polymerase chain reaction (PCR) with the Applied Biosystems 7300 Real-Time PCR System (Life Technologies) by using RT2 SYBR Green qPCR Master Mix (Qiagen). Primers were designed by using Primer Premier 5 software (Premier Biosoft International, Palo Alto, CA, USA) and synthesized by Sigma-Aldrich. Refer to Table 3 for primer sequences. Thermal cycling conditions were 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Dissociation curve analysis was performed to confirm the specificity of the primers. Gene expression was determined by using the 2−ΔΔCt method normalized to the constitutively expressed housekeeping gene β2 microglobulin (B2M) (32). Relative changes were generated comparing the treated cells to control cells. Each sample was assessed in triplicate to ensure reproducibility of the quantitative measurements.
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM) from three independent experiments. In the experiments where only two conditions are compared, analyses were made using the Student t test. Multiple comparisons were evaluated by one-way analysis of variance (ANOVA) followed by the Tukey post hoc test. Analyses were performed by using GraphPad Prism 5.0. P < 0.05 was considered significant. Additional methods are provided in the Supplementary Data.
All supplementary materials are available online at www.molmed.org .
Results
TGF-β1 Downregulates VEGF-D Expression in Human Lung Fibroblasts
TGF-β1 (5 ng/mL) induced a time-dependent decline in VEGF-D gene expression (Figure 1A) in MRC-5 cells, with the earliest significant decrease observed at 8 h after TGF-β1 exposure. Maximum suppression of VEGF-D gene expression was detected at 24 h and persisted up to 48 h after stimulation with a single dose of TGF-β1. In contrast, TGF-β1 transiently upregulated both VEGF-A and VEGF-C expression, both of which peaked at 8 h and declined toward baseline values thereafter. Activation of the TGF-β1 signaling pathway was confirmed by the robust and persistent upregulation of plasminogen activator inhibitor (PAI)-1 (Figure 1B), a well-characterized downstream target of TGF-β1 (33). Treatment with TGF-β1 yielded a concomitant reduction of full-length VEGF-D protein expression (molecular weight ~53 kDa) in a time-dependent (Figures 1C, D) and dose-dependent (Figures 1E, F) manner in MRC-5 cells. Additionally, we analyzed VEGF-D expression in primary normal adult lung fibroblasts derived from three control lungs (Figure 1G). Consistent with our data from the MRC-5 cell line, full-length VEGF-D protein expression was downregulated to a similar extent (>50%) in primary normal human lung fibroblasts (Figure 1H) when stimulated with TGF-β1 for 48 h. Consistent with previous reports demonstrating the absence of VEGF-D intracellular processing (10), full-length unprocessed VEGF-D is the only VEGF-D isoform that was detected in cell lysates (Supplementary Figure S1). Levels of secreted VEGF-D in supernatants from control and TGF-β1-treated MRC-5 cells were below the lower limit of detection of the enzyme-linked immunosorbent assay, despite trichloroacetic acid, acetone precipitation or ultrafiltration of the samples before quantification, making the detection of quantitative and molecular weight differences of VEGF-D difficult to ascertain.

TGF-β1 suppresses VEGF-D expression in lung fibroblasts. (A, B) MRC-5 fibroblasts were incubated with 5 ng/mL TGF-β1 for the indicated time points. Total RNA was isolated and reverse-transcribed, and the resultant cDNA was subjected to quantitative real-time PCR to detect gene expressions of VEGF-A (red), VEGF-C (blue), VEGF-D (black) (A) and PAI-1 (B). Real-time PCR results were normalized to β2-macroglobulin and expressed as the fold-change relative to unstimulated control cells (Ctrl). (C–F) MRC-5 fibroblasts were stimulated with TGF-β1 (5 ng/mL) for the indicated time (C, D) or stimulated with various concentrations of TGF-β1 for 48 h (E, F). Equal amounts of protein from whole cell lysates were analyzed by Western blotting with antibodies against VEGF-D and β-actin. (G, H) Primary NLFs were stimulated with TGF-β1 (5 ng/mL) for 48 h. Equal amounts of protein from whole cell lysates were analyzed by Western blotting with antibodies against VEGF-D and β-actin. (D, F, H) Ratio of VEGF-D to β-actin density was expressed as the fold change relative to unstimulated control cells in all experiments performed. Data are expressed as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, compared with control, by one-way ANOVA (A, B, D, F) or Student t test (H).
TGF-β1 Downregulates VEGF-D Expression via TGF-β Type I Receptor and JNK Signaling Pathway in MRC-5 Cells
Canonical TGF-β signal transduction involves binding of TGF-β1 to TGF-β receptor type II (TGF-βRII), which recruits and phosphorylates TGF-β receptor type I (TGF-βRI) and subsequently initiates intracellular signaling through the activation of Smad proteins to control downstream target gene expression (34). To determine whether TGF-β1 acts through its receptors and the Smad pathway to regulate VEGF-D expression, we pretreated MRC-5 cells with a selective inhibitor of TGF-β RI (SB431542) and evaluated its effect on the reduction of VEGF-D expression in response to TGF-β1. Pretreatment with SB431542 abrogated the rapid phosphorylation of Smad3 1 h after TGF-β1 exposure (Figure 2A). In a parallel set of experiments (Figures 2B, C), we found that pretreatment with SB431542 completely abolished TGF-β1-mediated downregulation of VEGF-D and concomitant upregulation of α smooth muscle actin (α-SMA), a marker of fibroblast (35,36), we also found that TGF-β1 exerted a biphasic effect on total Smad3 expression, which is demonstrated by a moderate increase in total Smad3 level 1 h after TGF-β1 exposure (Figure 2A) and a decrease of total Smad3 expression after prolonged TGF-β1 incubation (48 h, Figure 2D).

TGF-β1 downregulates VEGF-D expression in MRC-5 cells via a TGF-β receptor type I (TGF-βRI)-dependent, Smad3-independent mechanism. (A, B) MRC-5 cells were treated for 1 h with SB431542 (10 µmol/L), an inhibitor of activin receptor-like kinase (ALK)-5, followed by the addition of TGF-β1 (5 ng/mL) for the indicated time points. Equal amounts of protein from whole cell lysates were analyzed by Western blotting with antibodies against phospho-Smad3 and total Smad3 (A) and VEGF-D, α-SMA and β-actin (B). (C) Ratio of VEGF-D to β-actin density was expressed as the fold change relative to unstimulated control in (B). (D, E) MRC-5 cells were transfected with control siRNA (Ctrl) or Smad3 siRNA for 24 h, serum-starved for 24 h and then incubated in serum-free medium in the absence or presence of TGF-β1 (5 ng/mL) for 48 h. Equal amounts of protein from whole cell lysates were analyzed by Western blot with antibodies against VEGF-D, Smad3, α-SMA and β-actin (D). Ratio of VEGF-D to β-actin density was expressed as the fold-change relative to control siRNA alone (E). Data represent means ± SEM of three independent experiments. *P < 0.05, ***P < 0.001, NS, not significant, by one-way ANOVA.
Binding of TGF-β1 to its receptor also activates various noncanonical signaling pathways to modulate downstream cellular responses (37). Therefore, additional studies were pursued to decipher the role of noncanonical TGF-β signal transduction in VEGF-D regulation. In the first set, we determined whether PI3K, MEK, p38MAPK and JNK pathways in MRC-5 cells were activated in the presence of TGF-β1 and validated the functional activities of several chemical inhibitors of the aforementioned signaling pathways. As expected, TGF-β1 stimulation for 1 h initiated rapid intracellular signaling of PI3K, MEK, p38MAPK and JNK pathways in MRC-5 cells, demonstrated by increased phosphorylation of Akt (downstream target of PI3K; Figure 3A), p44/42MAPK (downstream target of MEK; Figure 3D), HSP27 (downstream target of p38MAPK; Figure 3G) and c-Jun (downstream target of JNK; Figure 3J). Pretreatment with wortmannin (PI3K inhibitor; Figure 3A), UO126 (MEK inhibitor; Figure 3D), SB203580 (p38MAPK inhibitor; Figure 3G) and SP600125 (JNK inhibitor; Figure 3J) profoundly inhibited both basal and TGF-β1-induced kinase activity for each individual noncanonical signaling pathway. We also conducted a parallel set of experiments to explore which noncanonical signaling pathways are responsible for downregulation of VEGF-D mediated by TGF-β1. Dissection of these signaling cascades demonstrated that TGF-β1-induced downregulation of VEGF-D was substantially abrogated by inhibition of JNK signaling by using SP600125 (Figures 3K, L) and not by PI3K, MEK or p38MAPK inhibition (Figures 3B, C; Figures E, F; and Figures 3H, I, respectively).

TGF-β1 downregulates VEGF-D expression in MRC-5 cells via the JNK signaling pathway. (A, B, D, E, G, H, J, K) MRC-5 fibroblasts were pretreated for 1 h with wortmannin (100 nmol/L) (A, B), UO126 (10 µmol/L) (D, E), SB203580 (10 µmol/L) (G, H) or SP600125 (5 µmol/L) (J and K), specific chemical inhibitors for PI3K/Akt, MEK1/2, p38MAPK and JNK, respectively. Subsequently, cells were treated with TGF-β1 (5 ng/mL) for the indicated time. (A, D, G, J) MRC-5 fibroblasts were collected 1 h after TGF-β1 stimulation. Equal amounts of protein from whole cell lysates were analyzed by Western blotting with antibodies against phosphorylated and total Akt (A), phospho-p44/42 and total p44/42MAPK (D), phospho-HSP27 and total HSP27 (G), and phosphorylated and total c-Jun (J). (B, E, H, K) MRC-5 fibroblasts were collected 48 h after TGF-β1 stimulation. Equal amounts of protein from whole cell lysates were analyzed by Western blotting with antibodies against VEGF-D, α-SMA and β-actin. Ratio of VEGF-D to β-actin density was expressed as the fold-change relative to unstimulated control (C, F, I, L). Data represent meads ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, NS, 3ot significant, by one-way ANOVA.
VEGF-D Knockdown Induces G1/S Transition and Promotes Cell Proliferation but Does Not Affect Fibroblast Activation, Collagen Synthesis and Fibroblast Contractility
Previous studies have shown that fibroblasts derived from IPF lung demonstrate increased proliferation (38,39), contraction (40) and collagen production (41) compared with normal fibroblasts. To explore whether VEGF-D is associated with the IPF fibroblast phenotype, we performed a series of experiments to evaluate the biological behavior of VEGF-D-depleted lung fibroblasts. Transfection of siRNA targeting human VEGF-D led to an increase in proliferating cell nuclear antigen (PCNA) expression but had no effects on α-SMA expression (Figures 4A, B). Next, we investigated the consequences of VEGF-D depletion on cell cycle and cell proliferation. Lung fibroblasts transfected with VEGF-D siRNA exhibited a significant increase in the percentage of S-phase cells compared with control siRNA transfection, indicating that VEGF-D knockdown promoted the transition from G1 to S phase in both MRC-5 cells (Figures 4C, D) and NLFs (Figures 4E, F). Furthermore, siRNA-mediated silencing of VEGF-D augmented the proliferative capacity of MRC-5 cells (Figure 4G). Western blot analysis of MRC-5 protein lysates did not show any immunoreactive bands with anti-VEGFR-2 and anti-VEGFR-3 antibodies (Supplementary Figure S2), which excludes potential autocrine effects of VEGF-D through its known canonical receptors. In accordance with published literature (42), we also found that stimulating cells with 20% FBS (positive control) induced a clear enrichment of S phase in lung fibroblasts (Figures 4C–F) and accelerated cell proliferation (Figure 4G). Interestingly, however, siRNA-induced VEGF-D silencing did not result in significant differences in soluble collagen production (Supplementary Figure S3A) and gel contraction area (Supplementary Figures S3B, C), as measured by Sircol and cell contraction assay, respectively.

VEGF-D knockdown promotes G1/S transition and cell proliferation but does not affect fibroblast activation. (A, B) MRC-5 cells were transfected with control siRNA (Ctrl) or VEGF-D siRNA for 48 h. Equal amounts of protein from whole cell lysates were analyzed by Western blotting with antibodies against VEGF-D, PCNA, α-SMA and β-actin (A). Ratio of PCNA to β-actin density was expressed as the fold-change relative to control siRNA alone (B). (C, D, E, F) Effects of siRNA-mediated VEGF-D silencing on cell cycle distribution of MRC-5 cells and NLFs. MRC-5 cells (C) and NLFs (E) were transfected with control siRNA or VEGF-D siRNA for 48 h. Cells cultured in DMEM containing 20% FBS were used as positive controls. Cells were collected, stained with propidium iodide and analyzed for DNA content. The number of cells in the G1, S and G2/M phases was calculated and expressed as a percentage of the total MRC-5 (D) and NLF (F) population. (G) Effects of siRNA-mediated VEGF-D silencing on the proliferative capacity of MRC-5 cells. MRC-5 cells were transfected with control siRNA (black) or VEGF-D siRNA (red). Cells cultured in DMEM containing 20% FBS were used as positive controls (blue). Cell proliferation was determined at the indicated time points after transfection. Data represent mean ± SEM of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 (versus control siRNA); #P < 0.05; ###P < 0.001 (versus VEGF-D siRNA), by the Student t test (B) and one-way ANOVA (D, FF G).
Knockdown of TSC2 or β-Catenin Is Not Sufficient to Block TGF-β1-Induced Suppression of VEGF-D in MRC-5 Cells
TGF-β1 has been shown to signal through the mammalian target of rapamycin (mTOR) (43). In addition, elevated serum VEGF-D levels have been described in lymphangioleiomyomatosis (LAM), a rare progressive lung disease characterized by genetic mutations in tuberous sclerosis complex (TSC)-1 or, more commonly, TSC2 (22,44). We therefore investigated whether TSC2 is associated with TGF-β1-induced VEGF-D downregulation. As shown in Figures 5A and B, transfection of MRC-5 cells with TSC2 siRNA led to significant knockdown of tuberin, the target gene product, but failed to modify either basal VEGF-D expression or TGF-β1-induced VEGF-D downregulation in MRC-5 cells.

Knockdown of TSC2 or β-catenin is not sufficient to block TGF-β1-induced suppression of VEGF-D in MRC-5 cells. (A, B) MRC-5 cells were transfected with control siRNA (Ctrl) or TSC2 siRNA for 24 h, serum starved for 24 h and then incubated in serum-free medium in the absence or presence of TGF-β1 (5 ng/mL) for 48 h. (A) Equal amounts of protein from whole cell lysates were analyzed by Western blotting with antibodies against VEGF-D, tuberin, α-SMA and β-actin. (B) Ratio of VEGF-D to β-actin density was expressed as the fold-change relative to control siRNA alone. (C) β-Catenin (β-cat) knockdown efficiency in MRC-5 cells 48 h after transfection, as determined by Western blot analysis. (D) Real-time PCR analysis of total RNA extracted from MRC-5 cells transfected as in (C) for 24 h, serum-starved for 24 h and then stimulated with TGF-β1 (5 ng/mL) for 48 h. VEGF-D mRNA levels were normalized to β2-macroglobulin mRNA and expressed as the fold-change relative to control siRNA alone. Data represent means ± SEM of three independent experiments. ***P < 0.001, NS, not significant, by one-way ANOVA.
In mouse 3T3 cells and human embryonic kidney 293 cells, it was shown that β-catenin negatively regulates VEGF-D mRNA stability (21). To determine whether a similar mechanism is involved in our findings, we performed transfection of β-catenin siRNA into MRC-5 cells to substantially reduce β-catenin protein expression (Figure 5C). Consistent with the previous report (21), we observed an approximately twofold elevation of VEGF-D gene expression associated with β-catenin knockdown (Figure 5D). However, silencing β-catenin did not abrogate TGF-β1-induced VEGF-D downregulation (Figure 5D).
VEGF-D Expression Is Decreased in Human IPF Lung Tissues
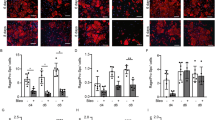
Given our findings of reduced VEGF-D expression in human lung fibroblasts treated with TGF-β1 and the pivotal role TGF-β1 plays in the pathogenesis of pulmonary fibrosis, we performed Western blotting (Figure 6A) to evaluate VEGF-D expression in IPF lung homogenates. Densitometric analysis (Figure 6B) of the immunoblot (Figure 6A and Supplementary Figure S4) showed that the mature form of VEGF-D (~21 kDa) was ~40% lower in IPF lung lysates (n = 6) compared with controls (n = 6). Because VEGF-D is not processed in the intracellular space (10), but is immediately cleaved in the extracellular space (8,9), our finding is consistent with the relative abundance of extracellular VEGF-D. We then sought to localize VEGF-D expression in control and fibrotic lungs by using immunohistochemical staining (Figures 6C–H). As demonstrated in Figures 6C, D, F and G, VEGF-D immunoreactivity was primarily observed in alveolar macrophages and epithelial cells in both control and IPF lungs. In IPF lung sections (Figure 6G), fibroblastic foci (area enclosed by black dotted lines) showed very weak VEGF-D immunoreactivity, whereas VEGF-D was expressed by alveolar interstitial cells (black arrow) in control lung (Figure 6D). No staining was observed when the primary VEGF-D antibody was replaced by normal mouse IgG (Figures 6E, H).

VEGF-D expression is decreased in human IPF lung tissues. (A) Equal amounts of protein from human lung homogenates were analyzed by Western blotting with antibodies against VEGF-D and β-actin. (B) Densitometric analysis of (A). Ratio of VEGF-D to β-actin density was expressed as the fold-change relative to control lung. Data are presented as means ± SEM (n = 6). *P < 0.05, compared with control, by Student t test. (C–H) Immunoreactivity for VEGF-D visualized with diaminobenzidine (DAB) is observed in alveolar macrophages (red arrow) and epithelial cells (blue arrow) in both control (C) and IPF lungs (F). In IPF lung sections (G), fibroblastic foci (area enclosed by black dotted lines) showed very weak VEGF-D immunoreactivity, whereas VEGF-D was present within alveolar interstitial cells (black arrow) in control lungs (D). Absence of immunoreactivity with nonimmune IgG in control (E) and IPF (H) lungs. One representative image from control (n = 6) and IPF (n = 6) lungs was shown. Scale bars: 50 µm.
Discussion
Individual members of the VEGF family have distinct biological functions and unique mechanisms of regulation. For example, the expressions of both VEGF-A and -C are significantly elevated in colorectal cancer tissue, whereas VEGF-D expression is down-regulated (45,46). Furthermore, TGF-α, betacellulin and heregulin-β1 have all been shown to upregulate both VEGF-A and -C but simultaneously downregulate VEGF-D expression in cancer cells (20). The underlying mechanisms for differential regulation of VEGF family members and the biological implications of this differential regulation are not completely understood.
TGF-β1 is a highly pleiotropic cytokine critical to fibrogenesis in the lung (47). In light of the stimulating effects of TGF-β1 on VEGF-A and -C (28,29), we examined the possibility that TGF-β1 might also modulate VEGF-D expression and result in altered VEGF-D expression in IPF lung tissue. Similar to the findings of previously published reports (28,29), our current study shows that TGF-β1 significantly upregulates fibroblast expression of VEGF-A and -C, but conversely, treatment with TGF-β1 suppresses VEGF-D expression in a time- and dose-dependent fashion.
TGF-β1 signals through both the canonical Smad pathway and Smad-independent noncanonical pathways (34,48). In the present study, we used selective kinase inhibitors and siRNA-based gene knockdown to delineate the pathways that mediate VEGF-D down-regulation in response to TGF-β1. Our study shows that the kinase activity of TGF-β RI and JNK are required for TGF-β1-induced reduction of VEGF-D, whereas Smad3, PI3K, TSC2, MEK and p38MAPK are not essential for this specific signal transduction process. TGF-β1 activation of JNK signaling depends on TGF-β RI kinase activity and is Smad3 independent (49,50), which explains our results. Consistent with these findings, we show that the down-regulation of VEGF-D can be abrogated by TGF-β RI kinase inhibitor SB431542, but not by Smad3 knockdown. Importantly, JNK has been shown to be a key mediator of fibrosis, since JNK activation is persistently elevated in fibrotic lung fibroblasts in vitro and in IPF lung tissue (51,52).
JNK activation could lead to VEGF-D downregulation through two potential mechanisms: (a) transcriptional suppression, as observed in the case of gap junction protein in cardiomyocytes (53), or (b) an increase in the adenylateuridylate (AU)-rich element RNA-binding protein 1 (AUF1) and the consequent impairment of target mRNA stabilization (54). Interestingly, analysis of the 3′-untranslated region of human VEGF-D mRNA (NCBI Reference Sequence: NM_004469) using an online database (https://doi.org/utrdb.ba.itb.cnr.it) revealed the presence of several AU-rich elements, which are known binding sites for AUF1 (55).
VEGF-D is mitogenic for endothelial cells in vitro and promotes both angiogenesis and lymphangiogenesis in vivo. However, the roles of intracellular VEGF-D have not been extensively examined. Our data show that VEGF-D knockdown in lung fibroblasts promotes G1/S phase transition and enhances their proliferative potential. Consistent with our findings, a recent report demonstrates aberrant fibroblast proliferation in VEGF-D-deficient mice during cutaneous wound healing (15). In contrast, VEGF-D knockdown has been generally associated with a reduction of proliferation in cancer cells (56,57). The cause for this potential contradiction is not clear, although it might simply reflect the biological variations among different cell types. Further studies are warranted to elucidate the underlying mechanisms for VEGF-D-associated cell cycle regulation and identify the downstream molecular targets of endogenous VEGF-D.
VEGF-D levels are increased in LAM and serve as a potential diagnostic and prognostic biomarker (58). LAM cells, characterized most commonly by mutation of the TSC2 gene, are thought to be the source of increased VEGF-D. Our data show that, in fibroblasts, silencing TSC2 and inhibiting the PI3K/AKT pathway do not result in increased VEGF-D levels at baseline or after TGF-β1 stimulation. These data suggest that the mechanisms controlling VEGF-D expression may differ in fibroblasts and LAM cells.
IPF is a disease characterized by increased TGF-β1 activity and aberrant fibroblast proliferation (59). Our analysis of IPF lung tissue homogenates shows a decrease in the mature form of VEGF-D compared with controls. Immunohistochemical analysis of IPF and normal lung sections shows that IPF lung fibroblasts have a marked decrease in VEGF-D immunoreactivity, whereas alveolar epithelial cells and alveolar macrophages show intense immunoreactivity for VEGF-D. The decreased VEGF-D expression by IPF lung fibroblasts is consistent with our previous findings that lymphatic vessels are absent within fibroblastic foci in IPF (60). Previous reports have shown that concentrations of VEGF-D in bronchoalveolar lavage fluid (60) and serum (61) are not significantly different between subjects with IPF and controls. Taken together, these data suggest that there are distinct variations among pulmonary interstitial, alveolar and systemic distribution patterns of VEGF-D in patients with pulmonary fibrosis.
Conclusion
In summary, we present evidence that, in human lung fibroblasts, TGF-β1 downregulates VEGF-D expression through the TGF-β receptor and JNK signaling pathway. We demonstrate the functional roles of intracellular VEGF-D in fibroblast biology, and we correlate our in vitro findings with the distribution of VEGF-D expression in pulmonary fibrosis. Further delineation of the mechanisms through which VEGF-D exerts its intracellular functions may lead to the development of novel therapeutic targets for fibrotic lung disorders.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Adams RH, Alitalo K. (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 8:464–78.
Man XY, Yang XH, Cai SQ, Yao YG, Zheng M. (2006) Immunolocalization and expression of vascular endothelial growth factor receptors (VEGFRs) and neuropilins (NRPs) on keratinocytes in human epidermis. Mol. Med. 12:127–36.
Yamazaki Y, Morita T. (2006) Molecular and functional diversity of vascular endothelial growth factors. Mol. Divers. 10:515–27.
Jia H, et al. (2004) Vascular endothelial growth factor (VEGF)-D and VEGF-A differentially regulate KDR-mediated signaling and biological function in vascular endothelial cells. J. Biol. Chem. 279:36148–57.
Ferrara N, Gerber HP, LeCouter J. (2003) The biology of VEGF and its receptors. Nat. Med. 9:669–76.
Achen MG, et al. (1998) Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. U. S. A. 95:548–53.
Stacker SA, et al. (2001) VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 7:186–91.
McColl BK, et al. (2007) Proprotein convertases promote processing of VEGF-D, a critical step for binding the angiogenic receptor VEGFR-2. FASEB J. 21:1088–98.
McColl BK, et al. (2003) Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J. Exp. Med. 198:863–8.
Stacker SA, et al. (1999) Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J.Biol. Chem. 274:32127–36.
Yamada Y, Nezu J, Shimane M, Hirata Y. (1997) Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics. 42:483–8.
Karkkainen MJ, et al. (2004) Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 5:74–80.
Baldwin ME, et al. (2005) Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol. Cell. Biol. 25:2441–9.
Koch M, et al. (2009) VEGF-D deficiency in mice does not affect embryonic or postnatal lymphangiogenesis but reduces lymphatic metastasis. J. Path. 219:356–64.
Paquet-Fifield S, et al. (2013) Vascular endothelial growth factor-d modulates caliber and function of initial lymphatics in the dermis. J. Invest. Dermatol. 133:2074–84.
Orlandini M, Marconcini L, Ferruzzi R, Oliviero S. (1996) Identification of a c-fos-induced gene that is related to the platelet-derived growth factor/vascular endothelial growth factor family. Proc. Natl. Acad. Sci. U. S. A. 93:11675–80.
Debinski W, et al. (2001) VEGF-D is an X-linked/AP-1 regulated putative onco-angiogen in human glioblastoma multiforme. Mol. Med. 7:598–608.
Orlandini M, Oliviero S. (2001) In fibroblasts Vegf-D expression is induced by cell-cell contact mediated by cadherin-11. J. Biol. Chem. 276:6576–81.
Mountain DJ, Singh M, Singh K. (2008) Downregulation of VEGF-D expression by interleukin-1beta in cardiac microvascular endothelial cells is mediated by MAPKs and PKCalpha/beta1. J. Cell. Physiol. 215:337–43.
P Oc, Rhys-Evans P, Modjtahedi H, Eccles SA. (2000) Vascular endothelial growth factor family members are differentially regulated by c-erbB signaling in head and neck squamous carcinoma cells. Clin. Exp. Metastasis. 18:155–61.
Orlandini M, Semboloni S, Oliviero S. (2003) Beta-catenin inversely regulates vascular endothelial growth factor-D mRNA stability. J. Biol. Chem. 278:44650–6.
Glasgow CG, Avila NA, Lin JP, Stylianou MP, Moss J. (2009) Serum vascular endothelial growth factor-D levels in patients with lymphangioleiomyomatosis reflect lymphatic involvement. Chest. 135:1293–300.
Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. (2002) Lymphangiogenesis and cancer metastasis. Nat. Rev. Cancer. 2:573–83.
Achen MG, Stacker SA. (2012) Vascular endothelial growth factor-D: signaling mechanisms, biology, and clinical relevance. Growth Factors. 30:283–96.
Selman M, Pardo A, Kaminski N. (2008) Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med. 5: e62.
Ju W, et al. (2012) Inhibition of alpha-SMA by the ectodomain of FGFR2c attenuates lung fibrosis. Mol. Med. 18:992–1002.
Gross TJ, Hunninghake GW. (2001) Idiopathic pulmonary fibrosis. N. Engl. J. Med. 345:517–25.
Kobayashi T, et al. (2005) Smad3 mediates TGF-beta1 induction of VEGF production in lung fibroblasts. Biochem. Biophys. Res. Commun. 327:393–8.
Suzuki Y, et al. (2012) Transforming growth factor-beta induces vascular endothelial growth factor-C expression leading to lymphangiogenesis in rat unilateral ureteral obstruction. Kidney Int. 81:865–79.
Raghu G, et al. (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183:788–824.
Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. (2004) siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 32:W130–4.
Schmittgen TD, Livak KJ. (2008) Analyzing realtime PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–8.
Bonniaud P, et al. (2005) Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am. J. Respir. Crit. Care Med. 171:889–98.
Derynck R, Zhang YE. (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 425:577–84.
Bauge C, Cauvard O, Leclercq S, Galera P, Boumediene K. (2011) Modulation of transforming growth factor beta signalling pathway genes by transforming growth factor beta in human osteoarthritic chondrocytes: involvement of Sp1 in both early and late response cells to transforming growth factor beta. Arthritis Res. Ther. 13: R23.
Poncelet AC, Schnaper HW, Tan R, Liu Y, Runyan CE. (2007) Cell phenotype-specific down-regulation of Smad3 involves decreased gene activation as well as protein degradation. J. Biol. Chem. 282:15534–40.
Zhang YE. (2009) Non-Smad pathways in TGF-beta signaling. Cell Res. 19:128–39.
Jordana M, et al. (1988) Heterogeneous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am. Rev. Respir. Dis. 137:579–84.
Raghu G, Chen YY, Rusch V, Rabinovitch PS. (1988) Differential proliferation of fibroblasts cultured from normal and fibrotic human lungs. Am. Rev. Respir. Dis. 138:703–8.
Miki H, et al. (2000) Fibroblast contractility: usual interstitial pneumonia and nonspecific interstitial pneumonia. Am. J. Respir. Crit. Care Med. 162:2259–64.
Shoda H, et al. (2007) Overproduction of collagen and diminished SOCS1 expression are causally linked in fibroblasts from idiopathic pulmonary fibrosis. Biochem. Biophys. Res. Commun. 353:1004–10.
Chen M, et al. (2012) Serum starvation induced cell cycle synchronization facilitates human somatic cells reprogramming. PLoS One. 7: e28203.
Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. (2012) TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 125:1259–73.
Carsillo T, Astrinidis A, Henske EP. (2000) Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. U. S. A. 97:6085–6090.
Hanrahan V, et al. (2003) The angiogenic switch for vascular endothelial growth factor (VEGF)-A, VEGF-B, VEGF-C, and VEGF-D in the adenoma-carcinoma sequence during colorectal cancer progression. J. Pathol. 200:183–94.
George ML, et al. (2001) VEGF-A, VEGF-C, and VEGF-D in colorectal cancer progression. Neoplasia. 3:420–7.
Leask A, Abraham DJ. (2004) TGF-beta signaling and the fibrotic response. FASEB J. 18:816–27.
Shin EH, Basson MA, Robinson ML, McAvoy JW, Lovicu FJ. (2012) Sprouty is a negative regulator of transforming growth factor beta-induced epithelial-to-mesenchymal transition and cataract. Mol. Med. 18:861–73.
Itoh S, et al. (2003) Elucidation of Smad requirement in transforming growth factor-beta type I receptor-induced responses. J. Biol. Chem. 278:3751–61.
Yamashita M, et al. (2008) TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell. 31:918–24.
Shi-Wen X, et al. (2006) Constitutive ALK5-independent c-Jun N-terminal kinase activation contributes to endothelin-1 overexpression in pulmonary fibrosis: evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Mol. Cell. Biol. 26:5518–27.
Wygrecka M, et al. (2012) TGF-beta1 induces tissue factor expression in human lung fibroblasts in a PI3K/JNK/Akt-dependent and AP-1-dependent manner. Am. J. Respir. Cell Mol. Biol. 47:614–27.
Petrich BG, et al. (2002) c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ. Res. 91:640–7.
Glaser ND, Lukyanenko YO, Wang Y, Wilson GM, Rogers TB. (2006) JNK activation decreases PP2A regulatory subunit B56alpha expression and mRNA stability and increases AUF1 expression in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 291:H1183–92.
Zucconi BE, Wilson GM. (2013) Assembly of functional ribonucleoprotein complexes by AU-rich element RNA-binding protein 1 (AUF1) requires base-dependent and -independent RNA contacts. J. Biol. Chem. 288:28034–48.
Liu YH, et al. (2008) Up-regulation of vascular endothelial growth factor-D expression in clear cell renal cell carcinoma by CD74: a critical role in cancer cell tumorigenesis. J. Immunol. 181:6584–94.
Majumder M, et al. (2012) Co-expression of alpha9beta1 integrin and VEGF-D confers lymphatic metastatic ability to a human breast cancer cell line MDA-MB-468LN. PLoS One. 7:e35094.
Young LR, et al. (2013) Serum VEGF-D concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir. Med. 1:445–52.
Wynn TA. (2011) Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 208:1339–50.
El-Chemaly S, et al. (2009) Abnormal lymphangiogenesis in idiopathic pulmonary fibrosis with insights into cellular and molecular mechanisms. Proc. Natl. Acad. Sci. U. S. A. 106:3958–63.
Lara AR, et al. (2012) Increased lymphatic vessel length is associated with the fibroblast reticulum and disease severity in usual interstitial pneumonia and nonspecific interstitial pneumonia. Chest. 142:1569–76.
Acknowledgments
This work was supported by National Institutes of Health Grant K22HL092223-A1 (to S El-Chemaly) and in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
The authors would like to thank Mark Perrella and Gustavo Pacheco-Rodriguez for helpful discussions and advice.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Cui, Y., Osorio, J.C., Risquez, C. et al. Transforming Growth Factor-β1 Downregulates Vascular Endothelial Growth Factor-D Expression in Human Lung Fibroblasts via the Jun NH2-Terminal Kinase Signaling Pathway. Mol Med 20, 120–134 (2014). https://doi.org/10.2119/molmed.2013.00123
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2013.00123