
Abstract
Endogenous hydrogen sulfide (H2S) is naturally synthesized in many types of mammalian cells from L-cysteine in the reactions catalyzed by cystathionine-β-synthase and cystathionine-γ-lyase (CSE). H2S has been demonstrated to play a proinflammatory role in various animal models of hindpaw edema, acute pancreatitis, lipopolysaccharide-induced endotoxemia and cecal ligation, and puncture-induced sepsis. Full-thickness burns that exceed 25% of the total body surface area (TBSA) produce a profound systemic inflammatory reaction characterized by leukocyte activation and plasma leakage in the microvasculature of tissues and organs remote from the wound. The aim of this study was to investigate the effect of local burn injury on induced distant organ endogenous H2S release and expression of CSE. Male BALB/c mice were subjected to 30% TBSA full-thickness burn and treated with saline (administered intraperitoneally (i.p.)); DL-propargylglycine (PAG, 50 mg/kg i.p.), which is a CSE inhibitor; or sodium hydrosulfide (NaHS, 10 mg/kg i.p.), which is an H2S donor. PAG was administered either 1 h before or 1 h after the burn injury, whereas NaHS was given at the same time as the burn injury. Measurements of liver myeloperoxidase (MPO) activities, liver H2S-synthesizing activity, plasma H2S level and liver and lung CSE mRNA expression and histological examination of tissues were performed after burn injury. Burn injury significantly increased the plasma H2S level and liver H2S synthesis 8 h after burn compared with the sham group. Burn injury also resulted in a significant upregulation of CSE mRNA in liver and lung. Prophylactic as well as therapeutic administration of PAG significantly reduced burn-associated systemic inflammation, as evidenced by MPO activity and histological changes in liver and lung. Injection of NaHS significantly aggravated burn-associated systemic inflammation. Therefore, our findings show for the first time the role of H2S in contributing to inflammatory damage after burn injury.
Similar content being viewed by others
Introduction
Patients with severe burns become susceptible to sepsis and may develop systemic inflammation and multiple organ failure, especially acute lung injury, which are common causes of morbidity and mortality (1). A profound systemic inflammatory response characterized by leukocyte activation and plasma leakage in the microvasculature of tissues or organs remote from the wound was produced in response to full-thickness burns that exceed 25% of the total body surface area (TBSA) (2). The degree of inflammatory response correlated directly with the percentage of TBSA burn (3). Systemic inflammatory-mediator release results in a massive inflammatory reaction. The development of methods for identifying and blocking specific inflammatory mediators has been a major focus of investigation (4,5).
Endogenous hydrogen sulfide (H2S) is synthesized through degradation of L-cysteine by cystathionine-γ-lyase (CSE, EC4.4.1.1) or cystathionine-β-synthase (CBS, EC4.2.1.22). Both enzymes were found to be expressed in many mammalian tissues (6). CBS converts homocysteine to cystathionine and hydrolyses L-cysteine to equimolar amounts of serine and H2S, whereas CSE converts cystathionine to L-cysteine, yielding pyruvate, ammonia and H2S (7). H2S is becoming recognized as a regulator of many physiological and pathological processes (8). For example, H2S exhibits potent vasodilator activity both in vitro and in vivo, most likely by opening vascular smooth muscle K (ATP) channels (9,10). In addition, H2S also functions as a neuromodulator in the brain. Physiological H2S can facilitate the induction of hippocampal long-term potentiation and selectively enhance excitatory postsynaptic currents mediated by the N-methyl-D-aspartate receptor (11). In addition to its functions as a neuromodulator and a vasodilator, H2S may contribute to inflammatory disease. In our laboratory, we have observed that H2S is associated with inflammation, such as hindpaw edema (12), acute pancreatitis (13), lipopolysaccharide (LPS)-induced endotoxemia (14) and cecal ligation and puncture (CLP)-induced sepsis (15).
The present study was designed to investigate the role of endogenous H2S in systemic inflammation and organ injury caused by burn injury in mice. Plasma H2S concentration, liver H2S synthesizing activity and CSE gene expression in tissues 8 hours after burn injury were analyzed to determine whether H2S has proinflammatory effects on systemic inflammation in mice subjected to burn injury. Then DL-propargylglycine (PAG) was used to further confirm the role of H2S in mice subjected to burn injury.
Materials and Methods
Mouse Burn Injury Model
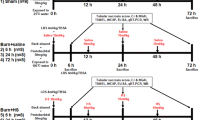
All experiments were approved by Institutional Animal Care and Use Committee of DSO National Laboratories and were conducted in accordance with their established guidelines. Groups of male BALB/c mice, 6–8 wks old, were anesthetized with ketamine (160 mg/kg) plus xylazine (4 mg/kg) and the dorsal hair was clipped. Mice were placed in an insulating mold device with an opening calculated to expose 30% TBSA. The exposed skin was immersed in 95°C water for 8 s. This procedure has been shown to produce an anesthetic full-thickness burn (16). Sham mice, which served as controls, were anesthetized, shaved, and exposed to 24°C water. After sham or burn injury, mice were resuscitated with 1 mL of 0.9% sterile normal saline solution by intraperitoneal (i.p.) injection and were individually housed. PAG (50 mg/kg, i.p.; Sigma-Aldrich, St Louis, MO, USA), an irreversible inhibitor of CSE (17), or saline was given i.p. to mice either 1 h before (“prophylactic”) or 1 h after (“therapeutic”) the burn injury, respectively. In the sodium hydrosulfide (NaHS) intervention experiment, mice subjected to burn injury were simultaneously given NaHS (10 mg/kg i.p.; Sigma-Aldrich) or saline. Eight hours after sham or burn injury, animals were killed by an i.p. injection of a lethal dose of pentobarbitone (90 mg/kg). Blood samples were drawn from the right ventricles by using heparinized syringes and were then centrifuged (1000g for 10 min, 0°C–4°C). Thereafter, plasma was aspirated and stored at −80°C for H2S measurement. Samples of lung and liver were stored at −80°C for subsequent measurement of tissue myeloperoxidase (MPO) activity, H2S-synthesizing activity and CSE mRNA expression.
Measurement of Plasma H2S
Aliquots (120 µL) of plasma were mixed with distilled water (100 µL), trichloroacetic acid (10% wt/vol, 120 µL), zinc acetate (1% wt/vol, 60 µL) and N,N-dimethyl-p-phenylenediamine sulfate (20 µmol/L; 40 µL) in 7.2 mol/L HCl and FeCl3 (30 µmol/L; 40 µL) in 1.2 mol/L HCl. The absorbance of the resulting solution (670 nm) was measured 10 min thereafter by spectrophotometry (Tecan Systems, San Jose, CA, USA) (18). H2S was calculated against a calibration curve of NaHS (3.125–100 µmol/L). Results showed plasma H2S concentrations in the micromolar range.
Assay of Liver H2S-Synthesizing Activity
H2S-synthesizing activity in liver homogenates was measured similarly as described elsewhere (14). Briefly, the assay mixture contained 100 mmol/L potassium phosphate buffer (pH 7.4), L-cysteine (20 µL, 20 mmol/L), pyridoxal 5′-phosphate (20 µL, 2 mmol/L), saline (30 µL) and 4.5% wt/vol tissue homogenate (430 µL). The reaction was performed in tightly sealed microcentrifuge tubes and initiated by transferring the tubes from ice to a water bath at 37°C. After incubation for 30 min, 1% wt/vol zinc acetate (250 µL) was added to trap evolved H2S, followed by 10% wt/vol trichloroacetic acid (250 µL) to denature the protein and stop the reaction. Subsequently, N,N-dimethyl-p-phenylenediamine sulfate (20 µmol/L; 133 µL) in 7.2 mol/L HCl was added, immediately followed by FeCl3 (30 µmol/L; 133 µL) in 1.2 mol/L HCl. The absorbance of the resulting solution at 670 nm was measured by spectrophotometry (Tecan Systems). The H2S concentration was calculated against a calibration curve of NaHS. Results were then corrected for the DNA content of the tissue sample (19) and expressed as micromoles of H2S formed per microgram of DNA.
MPO Estimation
Tissue samples were thawed, homogenized in 20 mmol/L phosphate buffer (pH 7.4) and centrifuged (13,000g, 10 min, 4°C), and the resulting pellets were resuspended in 50 mmol/L phosphate buffer (pH 6.0) containing 0.5% wt/vol hexadecyltrimethylammonium bromide (Sigma-Aldrich). The suspension was subjected to four cycles of freezing and thawing and further disrupted by sonication (40 s). The samples were then centrifuged (13,000g, 5 min, 4°C), and the supernatants were used for the MPO assay. The reaction mixture consisted of the supernatant (50 µL), 1.6 mmol/L tetramethylbenzidine (Sigma-Aldrich), 80 mmol/L sodium phosphate buffer (pH 5.4) and 0.3 mmol/L hydrogen peroxide (reagent volume: 50 µL). This mixture was incubated at 37°C for 110 s. The reaction was terminated with 50 µL of 0.18 mol/L H2SO4, and the absorbance was measured at 405 nm. This absorbance was then corrected for the DNA content of the tissue sample (19), and the results were expressed as fold increase over normal control.
Morphological Examination
Samples of lung and liver were fixed in 10% vol/vol neutral phosphate-buffered formalin and subsequently dehydrated through a graded ethanol series as described previously (13,20). After impregnation in paraffin wax, tissue samples were sectioned. Liver and lung sections (4–5 µm) were stained with hematoxylin-eosin and examined with light microscopy by using a Carl Zeiss microscope (objective lens magnification 40×; eyepiece magnification 10×).
Reverse Transcription-Polymerase Chain Reaction Analysis of Liver and Lung CSE mRNA
Total RNA from liver and lung was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol (13). The concentration of isolated nucleic acids was determined spectrophotometrically by measuring the absorbance at 260 nm, and the integrity was verified by GelRedTM nucleic acid gel staining (Biotium, Hayward, CA, USA) of 18S and 28S rRNA bands on a denaturing agarose gel. All samples were thereafter stored at −80°C until required for analysis. RNA (1 µg) was reverse transcribed by using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) at 25°C for 5 min and 42°C for 30 min, followed by 85°C for 5 min. The cDNA was used as a template for polymerase chain reaction (PCR) amplification by iQ Supermix (Bio-Rad). The primer sequences for detection of CSE and 18S were as follows: CSE (forward) 5′-GACCTCAATAGTCGG CTTCGTTTC-3′, CSE (reverse) 5′-CAGTTCTGCGTATGCTCCGTAATG-3′; 18S (forward) 5′ -GTAACCCGTTGAACC CCATT-3′, 18S (reverse) 5′-CCATC CAATC GGTAGTAGCG-3′. The PCR product sizes for CSE and 18S were 618 and 150 bp, respectively. PCR amplification was conducted in a MyCycler (Bio-Rad). The reaction mixture was first subjected to 95°C for 3 min, followed by an optimal cycle (CSE 30 cycles, 18S 20 cycles) of amplifications, consisting of 95°C for 50 s, optimal annealing temperature (CSE 63°C, 18S 59°C) for 60 s, and 72°C for 60 s. Final extension was at 72°C for 7 min. PCR products were analyzed on 1.5% (wt/vol) agarose gels containing GelRedTM nucleic acid gel stain (Biotium).
Statistical Analysis
Data were expressed as mean ± the standard error of mean (SEM). The significance of differences among groups was evaluated by ANOVA with post hoc Tukey test when comparing three or more groups. A P value < 0.05 was considered to indicate a statistically significant difference.
Results
Burn Injury Is Associated with Increase in Plasma H2S Level and H2S-Synthesizing Activity in Liver
Burn injury resulted in a significant increase by 1.31-fold in the plasma H2S level (P < 0.01; Figure 1A) compared with the sham group. The amounts of the formation of H2S in lung were undetectable. Therefore only liver H2S-synthesizing activity was measured. As expected, the amounts of H2S formed in liver after burn injury were significantly enhanced by 1.23-fold compared with the sham group (P <0.01; Figure 1B). In addition, those mice subjected to burn injury showed a significant systemic inflammation, which was indicated by the lung and liver MPO activity, a marker of tissue neutrophil infiltration. The lung MPO activity (fold increase over normal control) was significantly increased by 3.06-fold 8 h after burn injury compared with that in the sham group (P <0.01; Figure 1C). Similarly, in the liver, a significant elevation (1.48-fold burn versus sham) in MPO activity, but smaller than that in the lung, was observed 8 h after burn injury (P <0.01; Figure 1D) compared with that in the sham group.

Plasma hydrogen sulfide (H2S) concentration (A), liver H2S-synthesizing activity (B) and lung (C) and liver (D) MPO activity 8 h after burn injury. Results shown are mean ± SEM (n = 9 animals/group). *P < 0.05 and **P < 0.01 when mice subjected to burn injury were compared with sham group.
Liver and Lung CSE mRNA Expression Were Upregulated by Burn Injury
Burn injury resulted in a significant upregulation of CSE gene expression in both liver and lung. The expression level of CSE mRNA was significantly increased by 4.06-fold in the liver of mice subjected to burn injury compared with that in the sham group (P < 0.01; Figure 2A). In addition, a significant upregulation of CSE gene expression was found in the lungs of mice subjected to burn injury. Compared with the expression level of CSE mRNA in the lung from the sham group, CSE gene expression was significantly elevated by 1.83-fold in the lungs of mice subjected to burn injury (P < 0.01; Figure 2B).

Liver (A) and lung (B) CSE mRNA expression in normal, sham and burn group (n = 5 animals/group). Mouse 18S RNA is shown as a control. The bar graph shows the quantification of band intensity. Data were normalized to controls and expressed as fold-change ratio to control levels. ** P< 0.01 when mice subjected to burn injury were compared with sham group.
PAG Blocks H2S Formation and Attenuates Systemic Inflammation Caused by Burn Injury
Administration of PAG either 1 h before (prophylactic) or 1 h after (therapeutic) burn injury almost completely abolished the H2S-synthesizing activity in the liver (P < 0.01; Figure 3A). In addition, both prophylactic and therapeutic administration of PAG not only reduced the plasma H2S level significantly 8 h after burn injury (P < 0.05; Figure 3B) compared with saline injection group, but also recovered it to a level comparable to that of mice in the sham group. Therefore, PAG prophylactic and therapeutic treatment abolished H2S synthesizing activity in the liver and restored the plasma H2S to a similar level of that in sham group and did not eliminate the circulatory H2S. These results indicate that CSE plays an important role in endogenous H2S formation after burn injury.

Effect of PAG administration on liver H2S synthesizing activity (A), plasma H2S concentration (B) and lung (C) and liver (D) MPO activity 8 h after burn injury. Mice with burn injury were randomly given PAG (50 mg/kg i.p.) or saline 1 h before (PAG + burn or saline + CLP) or 1 h after (burn + PAG or burn + saline) burn injury. Eight hours after burn injury, plasma H2S concentration and H2S-synthesizing activity in liver were measured as described in Materials and Methods. Results shown are mean ± SEM (n = 9 animals/group). +P< 0.05, ++P<0.01 when PAG treated animals were compared with saline treated animals.
Prophylactic and Therapeutic Administration of PAG Mitigated the Systemic Inflammation and Multiple Organ Damage
MPO activity 8 h after burn injury in the lungs from mice pretreated or post-treated with PAG was significantly reduced compared with MPO activity in the lungs of saline-injected mice (both P < 0.01; Figure 3C). In addition, with PAG pretreatment or posttreatment, a significant reduction in liver MPO activity was also seen consistently (P < 0.05; Figure 3D). It seems that CSE may be involved in the systemic inflammation induced by burn injury. Furthermore, histological examination of lung and liver tissues revealed significant alveolar thickening, severe leukocyte infiltration in the interstitium and alveoli, hepatocyte swelling and marginated, pavemented and transmigrated neutrophils in mice subjected to burn injury compared with the sham group. These morphological changes in both lung and liver suggested the presence of systemic inflammation and multiple organ damage. Both prophylactic and therapeutic administration of PAG attenuated the histological damage in lung and liver tissue after burn injury (Figure 4A–J), providing evidence (in addition to MPO activity and H2S measurement) that CSE may be involved in the systemic inflammation and multiple organ damage induced by burn injury.

Morphological changes in lung and liver 8 h after induction of burn injury with or without PAG treatment in sections stained with hematoxylin and eosin. (A) Lung from sham group; (B) lung from mice subjected to burn injury with saline pretreatment; (C) lung from mice subjected to burn injury with PAG prophylactic treatment; (D) lung from mice subjected to burn injury with saline posttreatment; (E) lung from mice subjected to burn injury with PAG therapeutic treatment; (F) liver from sham group; (G) liver from mice subjected to burn injury with saline pretreatment; (H) liver from mice subjected to burn injury with PAG prophylactic treatment; (I) liver from mice subjected to burn injury with saline posttreatment; (J) liver from mice subjected to burn injury with PAG therapeutic treatment. The scale bar is 2 µm. Mice subjected to burn injury demonstrated alveolar thickening and severe leukocyte infiltration in the interstitium (arrowed) in the lung (B,D) and hepatocyte swelling and transmigrated neutrophils (arrow) in the liver (G,I).
CSE mRNA Expression Was Reduced in the Liver and Lung
Consistently, administration of prophylactic or therapeutic PAG significantly reduced the increased liver CSE gene expression induced by burn injury (P < 0.05; Figure 5A). In addition, the upregulated lung CSE gene expression induced by burn injury was reduced by both prophylactic and therapeutic administration of PAG (P < 0.01; Figure 5B). However, both CSE expression in liver and lung in mice subjected to burn injury treated with PAG were reduced only to a level comparable to that of mice in the sham group. It appears that PAG has an inhibitory effect on both liver and lung CSE gene expression induced by burn injury.

Effect of PAG administration on liver (A) and lung (B) CSE mRNA expression in mice with burn injury (n = 5 animals/group). Mice with burn injury were randomly given PAG (50 mg/kg i.p.) or saline 1 h before (PAG + burn or saline + CLP) or 1 h after (burn + PAG or burn + saline) burn injury. Mouse 18S RNA is shown as a control. The bar graph shows the quantification of band intensity. Data were normalized to controls and expressed as fold change ratio to control levels. +P< 0.05, ++P< 0.01 when PAG-treated animals were compared with saline-treated animals.
Administration of H2S Donor Aggravates Systemic Inflammation Induced by Burn Injury
To directly investigate the potential proinflammatory role of H2S in mice subjected to burn injury, the H2S donor drug NaHS was applied. The injection of NaHS at the time of burn injury significantly aggravated the systemic inflammatory response and increased multiple organ damage. NaHS injection caused a further and pronounced increase in lung MPO activity in mice subjected to burn injury (P < 0.01; Figure 6A). However, NaHS injection only slightly increased the liver MPO activity in mice subjected to burn injury (Figure 6B). In addition, NaHS also resulted in histological evidence of more severe organ injury in the lung. Obvious epithelial disorganization, the presence of airway exudates, leukocyte infiltration and alveolar wall thickness could be easily detected in the lung from NaHS-injected mice compared with saline-treated mice (Figure 6C, D). However, histological images of liver sections from the mice subjected to burn injury and NaHS injection were similar to those of mice subjected to burn injury and saline injection (Figure 6E, F).

Effect of H2S donor NaHS administration on MPO activity in lung (A) and liver (B) from mice subjected to burn injury. Mice subjected to burn injury were randomly given NaHS (10 mg/kg i.p.) or saline at the same time of burn injury. Eight hours after burn injury, MPO activity in lung and liver was measured as described in Materials and Methods. Results shown are mean ± SEM (n = 9 animals/group). ##P <0.01 when NaHS-treated mice were compared with saline-treated mice. (C–F) Morphological changes in lung and liver from mice subjected to burn injury with NaHS administration in hematoxylin and eosin-stained sections. (C) Lung from mice subjected to burn injury with saline simultaneously; (D) lung from mice subjected to burn injury with NaHS simultaneously; leukocyte infiltration and alveolar wall thickness (arrow) can be easily detected. (E) Liver from mice subjected to burn injury with saline simultaneously; (F) liver from mice subjected to burn injury with NaHS simultaneously. The scale bar is 2 µm.
Discussion
The gas H2S is increasingly recognized to be a novel regulator of important physiologic functions and to be involved in the pathogenesis of various diseases. Most endogenous H2S is generated during L-cysteine metabolism in many types of mammalian cells in reactions catalyzed by the pyridoxal phosphate-dependent enzymes CSE and CBS (21).
The present study investigated the role of H2S in mice subjected to burn injury. As demonstrated in this study and previous studies (22,23), burn injury resulted in a significant systemic inflammatory response, and multiple organ damage characterized by results of both biochemical (MPO activity) and histological (hematoxylin-eosin-stained section) analysis were observed in lung and liver from the mice subjected to burn injury. In addition, this study demonstrated that burn injury caused a significant increase in both plasma H2S concentrations and liver H2S synthesizing activity, and a significant upregulation of both liver and lung CSE mRNA expression 8 hours after burn injury. These findings are similar to those observed in our previous studies on other animal models. Our group previously reported that CLP-induced sepsis upregulates liver CSE mRNA expression and causes a significant increase in plasma H2S concentration (15). A similar proinflammatory effect of H2S has also been reported in other animal models of inflammatory diseases, such as hindpaw edema (12), acute pancreatitis (13) and LPS-induced endotoxemia (14). Altogether, with the well-established burn model, our findings indicated an important proinflammatory role of endogenous H2S in the postburn inflammatory response.
Mice subjected to burn injury had significantly elevated plasma H2S levels, CSE gene expression levels, and H2S-synthesizing enzyme activity in liver. It is reasonable to conclude that burn injury may upregulate CSE expression and activity in liver and consequently raise the plasma levels of H2S. However, the mechanism by which burn injury promotes the expression levels of CSE mRNA and CSE activity in liver is not clear. Results of our previous studies showed that blockage of H2S formation by administration of PAG, an inhibitor of CSE enzyme activity, reduced the severity and mortality of sepsis, whereas exogenous H2S resulted in augmented MPO activity as well as histological changes in lung and liver (15). Hence, to further confirm the proinflammatory role of H2S in the postburn inflammatory response and to investigate any role of H2S in burn-injury-induced CSE mRNA expression, we added the CSE inhibitor PAG.
We demonstrated in mice subjected to burn injury that prophylactic as well as therapeutic administration of PAG significantly alleviated the systemic inflammation characterized by reduced lung and liver MPO activity and multiple organ damage compared with saline-treated mice subjected to burn injury. In addition, PAG administration significantly reversed the liver H2S-synthesizing activity and consequently reduced the plasma H2S concentration. Furthermore, both prophylactic and therapeutic PAG administration resulted in a significant downregulation of liver and lung CSE gene expression compared with saline-treated mice subjected to burn injury, but still higher than those in sham group. Inhibition of the gene expression and activity of CSE was found in WKY rats when the endogenous production of H2S was deprived by administration of PAG (24). Therefore, inhibition of the activity and expression of CSE by PAG could significantly alleviate burn injury-associated inflammation and multiple organ damage. PAG reduced the hepatocellular, pancreatic and neuromuscular injury and abolished the increase in MPO activity and in the formation of H2S in the liver caused by endotoxemia (25). Inhibition of H2S biosynthesis by PAG also reduced hemorrhagic shock-induced inflammatory responses and organ injury (26). These findings combined with results from the present study indicate that PAG could partially reverse the proinflammatory function of H2S. These data also indicate that H2S-synthesis inhibitors may be useful in the treatment of the systemic inflammatory response and organ injury associated with burn injury, sepsis and shock, and suggest a new approach to the development of novel drugs for this condition.
To directly investigate the potential proinflammatory role of H2S in mice subjected to burn injury, H2S donor NaHS was also applied. Our previous studies in other animal models showed that exogenous H2S from injected NaHS aggravated the systemic inflammation characterized by elevated MPO activity in lung and liver, and the tissue damage characterized by increased neutrophil infiltration in liver and lung tissue samples (14,15). This study also showed that NaHS injection produces an elevated inflammatory response in mice subjected to burn injury treated with NaHS, a response characterized by a significant increase in lung MPO activity and tissue damage to the lung. However, exogenous H2S from injected NaHS only slightly, but not significantly, increased the liver MPO activity and tissue damage to the liver. The increase in liver MPO activity was not as high as that in lung tissue, a finding that is consistent with the previous data from the literature and attributable to a tissue-specific difference (27).
Consequently, based on these results, we conclude that H2S is one of the pivotal factors that determines the severity of inflammation and organ injury induced by burns. However, the precise mechanism by which H2S modulates the systemic inflammation remains to be elucidated. Significant increases in dermal interleukin 6 (IL-6), tumor necrosis factor-α (TNF-α) and IL-1β expression induced by burn injury have been reported (28). In addition, H2S caused a significant increase in expression of IL-6, TNF-α and IL-1β in vitro and in vivo (29,30). Therefore, it is necessary to further investigate the possibility that H2S modulates the development of the inflammatory response to burn injury via association with chemokines and cytokines. Furthermore, the interaction between H2S and other gaseous mediators, such as nitric oxide (NO), suggests another possible way that H2S promotes inflammation. A significant increase of NO and its metabolites was discovered in noninjured skin and skin wounds in burned sheep (31). In addition, inhibition of NO production resulted in decreased damage to the postburn intestinal mucosal barrier function (32). H2S can inhibit NO production in LPS-stimulated macrophages through a mechanism that involves the action of hemeoxygenase-1 (33). Anuar et al. reported that the NO donor nitroflurbiprofen can downregulate the biosynthesis of proinflammatory H2S in LPS-treated rats (34). Thus, the possibility of interaction between H2S and NO in the inflammatory response to burn injury warrants further study. Moreover, our group demonstrated that substance P contributed to the exaggerated pulmonary inflammatory damage after burn injury via activation of neurokinin-1-receptor signaling (35). The acute lung injury and inflammatory response were significantly attenuated in mice deficient in the preprotachykinin-A gene, which encodes for the classical bioactive neuropeptide substance P, whereas the exogenous administration of substance P to preprotachykinin-A gene-deficient mice restored the inflammatory response and lung injury (36). Our group also found that H2S upregulated the generation of substance P in a CLP-induced sepsis model (37). Thus there is much evidence to indicate that multifactorial and numerous mediators other than H2S are involved in the inflammatory response to burn injury. The interaction between H2S and other mediators of the proinflammatory effect requires further study.
In summary, burn injury upregulates CSE gene expression and leads to enhanced H2S biosynthesis, which promotes inflammation, whereas administration of an inhibitor of H2S synthesis substantially attenuates the inflammation and multiple organ injury caused by burn injury. In future studies we will investigate the molecular mechanism linked to H2S and burn injury and burn-injury-associated organ injury.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Moore FA, Moore EE. (1995) Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg. Clin. North. Am. 75:257–77.
Cioffi WG. (2001) What’s new in burns and metabolism. J. Am. Coll. Surg. 192:241–54.
Barber RC, Maass DL, White DJ, Horton JW. (2008) Increasing percent burn is correlated with increasing inflammation in an adult rodent model. Shock. 30:388–93.
Bone RC. (1996) Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit. Care Med. 24:163–72.
Arturson G. (2000) Forty years in burns research: the postburn inflammatory response. Burns. 26:599–604.
Brancaleone V, et al. (2008) Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice. Br. J. Pharmacol. 155:673–80.
Navarra P, Dello Russo C, Mancuso C, Preziosi P, Grossman A. (2000) Gaseous neuromodulators in the control of neuroendocrine stress axis. Ann. N. Y. Acad. Sci. 917:638–46.
Wang R. (2002) Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 16:1792–8.
Zhao W, Zhang J, Lu Y, Wang R. (2001) The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 20:6008–16.
Bhatia M. (2005) Hydrogen sulfide as a vasodilator. IUBMB Life. 57:603–6.
Kimura H. (2000) Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 267:129–33.
Bhatia M, Sidhapuriwala J, Moochhala SM, Moore PK. (2005) Hydrogen sulphide is a mediator of carrageenan-induced hindpaw oedema in the rat. Br. J. Pharmacol. 145:141–4.
Bhatia M, et al. (2005) Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 19:623–5.
Li L, et al. (2005) Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 19:1196–8.
Zhang H, Zhi L, Moore PK, Bhatia M. (2006) Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell Mol. Physiol. 290: L1193–201.
Stevenson JM, Gamelli RL, Shankar R. (2003) A mouse model of burn wounding and sepsis. Methods Mol. Med. 78:95–105.
Washtien W, Abeles RH. (1977) Mechanism of inactivation of gamma-cystathionase by the acetylenic substrate analogue propargylglycine. Biochemistry. 16:2485–91.
Chunyu Z, et al. (2003) The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem. Biophys. Res. Commun. 302:810–6.
Labarca C, Paigen K. (1980) A simple, rapid, and sensitive DNA assay procedure. Anal. Biochem. 102:344–52.
Bhatia M, Sidhapuriwala JN, Sparatore A, Moore PK. (2008) Treatment with H2S-releasing diclofenac protects mice against acute pancreatitis-associated lung injury. Shock. 29:84–8.
Hosoki R, Matsuki N, Kimura H. (1997) The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237:527–31.
Cetinkale O, Konukoglu D, Senel O, Kemerli GD, Yazar S. (1999) Modulating the functions of neutrophils and lipid peroxidation by FK506 in a rat model of thermal injury. Burns. 25:105–12.
Iseri SO, et al. (2008) Oxytocin ameliorates skin damage and oxidant gastric injury in rats with thermal trauma. Burns. 34:361–9.
Yan H, Du J, Tang C. (2004) The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem. Biophys. Res. Commun. 313:22–7.
Collin M, et al. (2005) Inhibition of endogenous hydrogen sulfide formation reduces the organ injury caused by endotoxemia. Br. J. Pharmacol. 146:498–505.
Mok YY, Moore PK. (2008) Hydrogen sulphide is pro-inflammatory in haemorrhagic shock. Inflamm. Res. 57:512–8.
Iseri SO, Ersoy Y, Gedik N, Ercan F, Alican I. (2008) Protective role of adrenomedullin in burn-induced remote organ damage in the rat. Regul. Pept. 146:99–105.
Ipaktchi K, et al. (2006) Topical p38MAPK inhibition reduces dermal inflammation and epithelial apoptosis in burn wounds. Shock. 26:201–9.
Zhi L, Ang AD, Zhang H, Moore PK, Bhatia M. (2007) Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappaB pathway. J. Leukoc. Biol. 81:1322–32.
Zhang H, Zhi L, Moochhala S, Moore PK, Bhatia M. (2007) Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-kappaB. Am. J. Physiol. Lung Cell Mol. Physiol. 292:L960–71.
Oliveira GV, et al. (2004) Skin nitric oxide and its metabolites are increased in nonburned skin after thermal injuries. Shock. 22:278–82.
Chen LW, Hsu CM, Wang JS, Chen JS, Chen SC. (1998) Specific inhibition of iNOS decreases the intestinal mucosal peroxynitrite level and improves the barrier function after thermal injury. Burns. 24:699–705.
Oh GS, et al. (2006) Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic. Biol. Med. 41:106–19.
Anuar F, et al. (2006) Nitric oxide-releasing flurbiprofen reduces formation of proinflammatory hydrogen sulfide in lipopolysaccharide-treated rat. Br. J. Pharmacol. 147:966–74.
Sio SW, Puthia MK, Lu J, Moochhala S, Bhatia M. (2008) The neuropeptide substance P is a critical mediator of burn-induced acute lung injury. J. Immunol. 180:8333–41.
Sio SW, Moochhala S, Lu J, Bhatia M. (2010) Early protection from burn-induced acute lung injury by deletion of preprotachykinin-A gene. Am. J. Respir. Crit. Care Med. 181:36–46.
Zhang H, et al. (2007) Hydrogen sulfide up-regulates substance P in polymicrobial sepsis-associated lung injury. J. Immunol. 179:4153–60.
Acknowledgments
We thank ML Shoon (Department of Pharmacology, National University of Singapore), C Lim, LL Tan and J Yeo (DSO Laboratories, Singapore) for excellent technical assistance and P Rajagopal and CC Wah (DSO Laboratories, Singapore) for animal care and management.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, J., Sio, S.W.S., Moochhala, S. et al. Role of Hydrogen Sulfide in Severe Burn Injury-Induced Inflammation in Mice. Mol Med 16, 417–424 (2010). https://doi.org/10.2119/molmed.2010.00027
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2010.00027