
Abstract
Metabolic syndrome is associated with accelerated macrovascular and microvascular coronary disease, cardiomyopathy, and elevated inflammatory status. To determine whether metabolic syndrome-associated elevation of the inflammatory cytokine interleukin-18 (IL-18) in serum and cardiac tissue, and its potential sequelae could be attenuated pharmacologically, we studied fructose-fed rats. The fructose-fed rats exhibited increases in systolic blood pressure (SBP), body weight, heart weight, left ventricular weight, and blood insulin. Serum IL-18 levels in these rats were also elevated significantly. These changes were significantly different compared to those in control rats. Perivascular fibrosis around coronary arterioles was evident in the fructose-fed rats, accompanied by a paralleled increase in IL-18 by immunohistochemical analysis and real time polymerase chain reaction. Felodipine attenuated the increased levels in serum IL-18 and cardiac IL-18 mRNA as well as coronary perivascular fibrosis. Thus, augmented IL-18 in serum and cardiac tissue in metabolic syndrome may contribute to the coronary perivascular fibrosis; felodipine administration can attenuate the inflammatory and fibrosis process.
Similar content being viewed by others

Introduction
Accelerated coronary atherosclerosis contributes to the increased mortality associated with metabolic syndrome (1,2), a cluster of metabolic disorders including central adiposity, insulin resistance, hypertension, dyslipidemia, and proinflammatory state (3–5). In metabolic syndrome patients, these coronary microvascular changes are common, often preceded by obesity and insulin resistance (6), which are considered the most prominent pathogenic changes underlying metabolic syndrome (7,8). Perivascular arteriolar fibrosis is one of the landmark pathological changes in patients with myocardial ischemia in the absence of angiographically demonstrable stenosis (9).
Recently, activated innate immunity and chronic inflammation have been causally implicated in metabolic syndrome-associated atherogenesis (10–12). Interleukin-18 (IL-18), a member of the IL-1 cytokine superfamily, is recognized as an important regulator of innate and acquired immune responses (13,14). IL-18 is a potent proinflammatory cytokine, and plays an important role in plaque destabilization (15). Prospective studies have shown an association of circulating IL-18 levels with cardiovascular death in patients with coronary artery disease (16,17).
Recent studies suggest that IL-18 levels may be elevated in metabolic disturbances, although its relationship with metabolic syndrome has not been formally studied. Elevated IL-18 levels are found to be associated with increased adiposity and insulin resistance in obese premenopausal women (18,19). IL-18 concentrations are increased in acute hyperglycemia (20) and type 2 diabetes (21,22). Moreover, IL-18 may influence vascular remodeling (23,24). However, it is not known whether IL-18 is involved in coronary microvascular changes in metabolic syndrome.
The present study was designed to characterize IL-18 expression in serum or tissues and the relationship of that expression to coronary microvascular changes in fructose-fed rats. We established the fructose-fed rat model, which simulates the human metabolic syndrome (25,26). We further determined whether the calcium channel blockade (CCB) felodipine, known to attenuate inflammation (27,28), reduces IL-18 expression and perivascular fibrosis in the fructose-fed rats.
Materials and Methods
Experimental Animals
All rats were handled in accordance with the Animal Management Rules of the Ministry of Health, People’s Republic of China (documentation 55, 2001) and experimental protocol was approved by the Institutional Animal Care Committee of Shandong University. Male Wistar-Kyoto rats were 6 wks old, and were purchased from the Animal Research Institution of Shandong University School of Medicine. They were housed in individual cages at room temperature and a 12-h light 12-h dark cycle (12:12 L:D) (7:00 a.m. to 7:00 p.m.) was applied to the animal housing. Rats were randomly assigned to two groups: control (n = 12) and fructose (n = 18) groups. Rats in the fructose group were given 10% fructose in drinking water and standard chow diet (16% protein, 8% fat, 50% carbohydrate, 22% trace elements) ad libitum, whereas rats in the control group had free access to tap water and standard chow diet. After being fed for 8 mos, 9 rats of the fructose group were given 5 mg/kg/day felodipine (felodipine group) by intubation between 10:00 a.m. and 10:30 a.m. for 6 wks. The control group received an equal volume of drug-free vehicle (normal saline solution). The dose was adjusted each wk based on body weight calculation for each animal. Rats in the fructose and the felodipine groups were continued on 10% fructose water throughout the treatment period.
Blood Pressure Measurement
Systolic blood pressure (SBP) was measured every 2 wks by tail cuff plethys-mography (29,30). Measurements were obtained with the rats restrained without anesthesia in a plastic chamber. The rats were preconditioned to the experimental procedure before the actual experimental measurements. A pneumatic pulse transducer positioned on the ventral surface of the tail distal to the occlusion cuff detected the return pulse after a slow deflation of the cuff. Cuff pressure was determined by a pneumatic pulse transducer with a programmed electrosphygmomanometer (RBP1, Clinical Medicine Institute, Peking, China) and pulses were recorded on a physiograph. A minimum of three measurements were taken at each session, and the SBP registered was the average of the three readings.
Biochemical Analyses
After the rats were fasted for 14 h, blood samples were collected from the carotid sinus at baseline and at the end of the experiment period before the rats were killed. Glucose concentrations were measured by the glucose oxidase method. Aliquots of sera were assayed for cholesterol and triglyceride concentrations according to standard methods. Insulin was determined by a solid phase, sandwich immunoassay. Insulin resistance was evaluated using HOMA-IR index, defined as fasting glucose (mmol/L) × fasting insulin (µU/mL) divided by 22.5 (31). Serum IL-18 level was measured using enzyme-linked immunosorbent assay (Uscnlife, Missouri City, TX, USA) according to the instructions supplied by the manufacturer and met the quality assurance and quality control established for test validity. The intra-assay and inter-assay coefficients of variations (CVs) were 6.1% and 6.5% for IL-18.
Morphometric Analysis
All rats were killed under deep anesthetization (2.0% isoflurane) according to institutional guidelines for animal care and use. The hearts were excised and rinsed in phosphate-buffered saline (PBS). The left ventricle was separated from the right ventricle, the atria, and the great vessels, and was cut into five pieces perpendicular to the long axis. The tissue was fixed in 4% paraformaldehyde and dehydrated with graded concentrations of alcohol for embedding in paraffin. Paraffin sections (5 µm) from each heart were stained with Masson’s trichrome (32). Photomicrographs were quantified with NIH Image software to assess the area of perivascular fibrosis, which was calculated as the ratio of the fibrosis area (collagen deposition stained with aniline blue) surrounding the vessel to the total vessel area. Approximately 10 vessels were examined in each heart. The dispersion present in the fibrosis area-to-lumen or fibrosis-to-media ratios was considered.
Immunohistochemistry
Paraffin sections were cut in 5 µm widths, deparaffinized, and dehydrated. Endogenous peroxidase activity was blocked by treatment with 0.5% H2O2 in methanol for 30 min. To measure IL-18 levels in cardiac tissue, a polyclonal goat IgG anti-rat IL-18 antibody (R&D System Inc, Minneapolis, MN, USA) at a 1:200 dilution was used. After washing in phosphate-buffered saline for 10 min, the sections were incubated with secondary goat anti-mouse antibody and visualized with 3-amino-9-ethyl carbazole (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) as substrate. The nuclei were counter-stained with hematoxylin, and the sections were analyzed at a magnification of ×400 under a light microscope equipped with an image analyzer (Image Pro Plus, Media Cybernetics, PA, Italy). Quantitative assessment of intensity of IL-18 staining was performed as integrated optical density (IOD) by two observers blinded with respect to the groups of animals. Isotope IgG as the primary antibody was used as a control for comparison.
Expression of IL-18 mRNA
Total RNA was extracted from left ventricles using TRIzol reagents (Invit-rogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. After DNase (Promega, Madison, WI, USA) treatment, 1 µg of total RNA was reverse transcribed using random primers and M-MLV reverse transcriptase (Promega). A 2 µL aliquot of the cDNA was amplified by real-time polymerase chain reaction (PCR) in 20 µL of PCR mixture using SYBR Green Master Mix with the Light Cycler 2.0 (LCS4 4.0.0.23; Roche, Basel, Switzerland). The PCR primer for IL-18 amplification was forward 5′-AAA CCC GCC TGT GTT CGA-3′ and reverse 5′-ATC AGT CTG GTC TGG GAT TCG T-3′. Expression levels were normalized to the reference gene GAPDH (forward 5′-ATG TAT CCG TTG TGG ATC TGA C-3′; reverse 5′-CCT GCT TCA CCA CCT TCT TG-3′). At the end of each PCR run, the data were analyzed automatically by the system, and amplification plots were obtained. Melting curve analysis was performed to assess the specificity of the PCR products. To correct the efficiency of complementary deoxyribonucleic acid synthesis, the amounts of each measured mRNA were divided by the amounts of glyceraldehyde-3-phosphate dehydrogenase mRNA as an internal standard (33). All amplification reactions were performed in triplicate.
Statistical Analysis
Data are expressed as mean ± SD. Statistical significance of differences between three or more groups was determined by one-way analysis of variance (ANOVA) followed by Bonferroni multiple-comparison correction. Levels of serum IL-18 and myocardium IL-18 mRNA expression were correlated with perivascular fibrosis using the Pearson correlation coefficient. A two-tailed probability of < 0.05 was considered statistically significant. SPSS version 13.0 for Windows was used in analysis.
Results
Blood Pressure Changes
At baseline, SBP was similar in the control and the fructose groups (111.31 ± 6.22 compared with 113.24 ± 7.07 mmHg). However, SBP increased significantly (138.66 ± 6.43 compared with 115.60 ± 3.30 mmHg, P < 0.01) in fructose-fed rats during the first 8 months. When rats were treated with felodipine, SBP was significantly reduced (115.20 ± 10.66 compared with 137.95 ± 6.01 mmHg, P < 0.01) to a level similar to the level in the control group.
Body Weight, Heart Weight, and Left Ventricular Weight
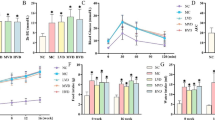
Fructose-fed rats exhibited an increase in body weight compared with that of controls (Figure 1). Body weights of fructose-fed rats treated with felodipine were not significantly different from those of untreated fructose-fed rats. Heart weights and left ventricular weights also were increased significantly in fructose-fed rats. Heart weights and left ventricular weights in fructose-fed rats treated with felodipine were significantly less than those in untreated fructose-fed rats.

Body weight, heart weight, and left ventricular weight in the control (n = 12), fructose (n = 9), and felodipine (n = 9) groups. *P < 0.05, **P < 0.01 compared with the control group; §P < 0.05, §§P < 0.01 compared with the fructose group.
Biochemical Parameters and IL-18 Level in Blood
Insulin (17.34 ± 3.08 compared with 8.94 ± 2.03, P < 0.01), triglycerides (1.34 ± 0.74 compared with 0.81 ± 0.24, P < 0.05) and HOMA-IR index (4.15 ± 1.45 compared with 2.13 ± 0.66, P < 0.05) were increased in the fructose group (Table 1). However, glucose and cholesterol levels were not different between fructose-fed and control rats. While felodipine significantly attenuated the increases in insulin level (8.98 ± 2.91 compared with 17.34 ± 3.08, P < 0.01) and HOMA-IR index (2.16 ± 0.89 compared with 4.15 ± 1.45, P < 0.05), it had no effect on triglycerides. Serum IL-18 levels (59.53 ± 6.46 compared with 35.47 ± 6.58, P < 0.01) were significantly higher in fructose-fed rats compared to controls. Felodipine treatment attenuated the increase in serum IL-18 level in fructose-fed rats (50.76 ± 5.66 compared with 59.53 ± 6.46, P < 0.05).
Morphological and Immunohistochemical Changes
Considerable fibrosis was observed around coronary arterioles in fructosefed rats (Figure 2), with a significant increase in the fibrosis-to-lumen (2.75 ± 1.35 compared with 1.39 ± 0.67, P < 0.01) and fibrosis-to-wall (1.47 ± 0.65 compared with 0.71 ± 0.34, P < 0.01) ratios compared with values in controls. Treatment with felodipine attenuated these increases (fibrosis-to-lumen: 1.50 ± 0.46 compared with 2.75 ± 1.35, P < 0.01; fibrosis-to-wall: 0.71 ± 0.21 compared with 1.47 ± 0.65, P < 0.01).

Fibrosis-to-lumen and fibrosis-to-wall ratios of coronary arteries in the control (n = 12), fructose (n = 9), and felodipine (n = 9) groups. Top, representative photograph of coronary arteries in control (A), fructose-fed rat (B), and felodipine-treated fructose-fed rat (C). *P < 0.05, **P< 0.01 compared with the control group; §P < 0.05, §§P < 0.01 compared with the fructose group. Bar = 50 µm.
While only weak immunoreactivity of IL-18 was detected around microvessels of the control rats, strong signals were detected in endothelium of the microvessels in fructose-fed rats (Figure 3). Furthermore, the intensity of the immunoreactivity of IL-18 was reduced in specimens from fructosefed rats treated with felodipine comparing to those fructose-fed rats not treated with felodipine. Quantitative results were consistent with these observations visually. Samples from fructose-fed rats exhibited significantly higher IL-18 expression than those from controls (400.83 ± 92.36 compared with 59.87 ± 11.25, P < 0.01), which is consistent with the morphological changes (see Figure 3). However, felodipine significantly attenuated the increase (380.33 ± 103.14 compared with 400.83 ± 92.36, P < 0.05).

Representative photographs of cardiac cross-sections stained immunohistochemically for IL-18. Sections were counterstained with hematoxylin to show nuclei in dark blue. A) IL-18 stain of cardiac tissue from control mouse. B) IL-18 stain of cardiac tissue from fructose-fed rat. C) IL-18 stain of cardiac tissue from fructose-fed rat treated with felodipine for 6 wks. D) Isotope IgG stain of cardiac tissue used as the primary antibody and as a control for comparison. Arrows indicate staining for IL-18. Bar = 50 µm. Bottom, results of quantitative image analysis for detection of IL-18 in the control (n= 12), fructose (n = 9), and felodipine (n = 9) groups. *P < 0.05, **P < 0.01 compared with the control group; §P < 0.05, §§P < 0.01 compared with the fructose group.
IL-18 mRNA Expression
The abundance of IL-18 relative to GAPDH in the hearts of fructose-fed rats and controls is shown in Figure 4. IL-18 mRNA was increased markedly in fructose-fed rats compared with controls. Treatment with felodipine significantly reduced the expression of IL-18 mRNA in fructose-fed rats. Expression of GAPDH mRNA was not altered.

IL-18 mRNA in cardiac tissue in the control (n = 12), fructose (n = 9) and felodipine (n = 9) groups. *P < 0.05, **P < 0.01 compared with the control group; §P < 0.05, §§P < 0.01 compared with the fructose group.
Notably, in the present study, serum IL-18 levels and IL-18 mRNA expression were highly and significantly correlated with the perivascular fibrosis in the fructose group but not in the other two groups (Figure 5A–5D).

The correlation between fibrosis-to-lumen ratio and serum IL-18 level (A) or IL-18 mRNA expression (B) in data taken from the fructose group (n = 9). The correlation between fibrosis-to-wall ratio and serum IL-18 level (C) or IL-18 mRNA expression (D) in data taken from the fructose group (n = 9).
Discussion
High dietary intake of fructose has rapidly become an important causative factor in the development of metabolic syndrome (34). Inflammation and activated innate immunity are thought to play an important role in the development of atherosclerosis and diabetes, which may represent a unifying link between metabolic syndrome, type 2 diabetes, and cardiovascular disease (10–12). Several cross-sectional studies have shown that acute-phase reactants such as C-reactive protein (CRP), and cytokines such as IL-6 and tumor necrosis factor-α are associated with features of the metabolic syndrome such as body mass index, measures of insulin resistance, hypertension, and dyslipidemia (35–38). We found that IL-18 levels were increased in rats with metabolic syndrome, consistent with observations by others (39) who demonstrated a strong association between IL-18 and metabolic syndrome in humans. However the association diminished after accounting for traditional cardiovascular risk factors (40).
The mechanisms for the link between inflammation and metabolic syndrome are not fully understood. One explanation is that adipose tissue in obese persons with the metabolic syndrome releases increased amounts of cytokines into the circulation (41). Another possibility is that insulin resistance per se is responsible for a higher production of cytokines (42).
We found that fructose-fed rats exhibit increased perivascular fibrosis, which may be associated with altered IL-18 expression. The increased IL-18 is hypothesized to have induced fibroblast activity, thereby stimulating the synthesis of extracellular matrix (43). Other pathways, such as increased production of SMC-derived IFN-γ, also could be responsible for IL-18-associated perivascular fibrosis (44).
Diabetic patients are frequently at paucity of coronary collateral circulation, which suggests a limited angiogenesis (45,46). Altered characteristics of left ventricular capillaries in genetically obese diabetic rats have been reported (47). In a preliminary study, we also characterized capillary profiles in fructose-fed rats. Remodeling appeared to be constrained in fructose-fed rats (unpublished observations).
In the present study, IL-18 in serum and cardiac tissue in fructose-fed rats that received felodipine was reduced and hence the perivascular fibrosis was modulated. This appears to be a cardiac-specific effect of felodipine. Recently several suggest that some dihydropyridine-type calcium channel blockers, such as nifedipine (48), nilvadipine (49), and benidipine (50), have a direct beneficial effect by inhibiting the nuclear translocation and DNA-binding activity of nuclear factor-κ B, which plays a central role in this process by regulating the expression of several pro-inflammatory genes. Ding and Vaziri (51) demonstrated that felodipine increased nitric oxide (NO) production, endothelial nitric oxide synthase (eNOS) activity, and eNOS protein expression in cultured endothelial cells from the rat aorta. In their study, they suggest that the enhancement of endothelial NO production could account for the reported improvement in endothelial function associated with calcium channel blocker in the case of both renal and hypertensive disorders. These mechanisms may contribute to the favorable effects of calcium channel blocker seen in patients at high cardiovascular risk. The most important finding in our study is the role of felodipine in attenuating the metabolic syndrome-associated proinflammatory changes, hence the reduction in myocardial fibrosis. This direct effect may be independent of its blood pressure regulatory effects and suggests a therapeutic modality of the calcium channel blocker in treatment of cardiovascular complications in metabolic syndrome patients.
Our study is limited in several aspects. First, the present study did not have an additional antihypertensive treatment group using other calcium channel blockers or other blood pressure-controlling agents. Therefore, our data are not conclusive in ruling out the possibility that the blood pressure lowering may contribute to the attenuation of perivascular fibrosis. However, we speculate that the effect felodipine on cardiac perivascular fibrosis is less likely due to the blood pressure regulation. This hypothesis is supported by a recent finding in diabetic rats which were normotensive. Calcium channel blocker benidipine normalized the endothelin system and reduced cardiac perivascular fibrosis (52), indicating that this effect is independent of the blood pressure-lowering outcome. Second, it is not clarified in the current study whether IL-18 directly affects collagen synthesis or degradation, or both.
In summary, this study demonstrates that augmented IL-18 in serum and cardiac tissue in metabolic syndrome may contribute to the coronary perivascular fibrosis. Calcium channel blocker felodipine administration can attenuate the inflammatory and fibrosis process. Thus, felodipine could potentially be used in humans with multiple cardiovascular risk factors. Obviously, further prospective studies relating IL-18 to metabolic syndrome would be informative.
References
Bonora E et al. (2003) Carotid atherosclerosis and coronary heart disease in the metabolic syndrome: prospective data from the Bruneck study. Diabetes Care. 26:1251–7.
Reilly MP et al. (2004) Measures of insulin resistance add incremental value to the clinical diagnosis of metabolic syndrome in association with coronary atherosclerosis. Circulation. 110:803–9.
Reaven GM. (1988) Banting lecture 1988: role of insulin resistance in human disease. Diabetes. 37:1595–607.
Meigs JB. (2000) Invited commentary: insulin resistance syndrome? Syndrome X? Multiple metabolic syndrome? A syndrome at all? Factor analysis reveals patterns in the fabric of correlated metabolic risk factors. Am. J. Epidemiol. 152:908–12.
Sakkinen PA, Wahl P, Cushman M, Lewis MR, Tracy RP. (2000) Clustering of procoagulation, inflammation, and fibrinolysis variables with metabolic factors in insulin resistance syndrome. Am. J. Epidemiol 152:897–907.
Zaman AK et al. (2004) Salutary effects of attenuation of angiotensin II on coronary perivascular fibrosis associated with insulin resistance and obesity. J. Mol. Cell. Cardiol. 37:525–35.
Reaven GM. (1988) Role of insulin resistance in human disease. Diabetes. 37:1595–606.
Haffner S, Taegtmeyer H. (2003) Epidemic obesity and the metabolic syndrome. Circulation. 108:1541–5.
Murakami H, Urabe K, Nishimura M. (1998) Inappropriate microvascular constriction produced transient ST-segment elevation in patients with syndrome X. J. Am. Coll. Cardiol. 32:1287–94.
Pickup JC, Mattock MB, Chusney GD, Burt D. (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and IL-6 with metabolic syndrome X. Diabetologia. 40:1286–92.
Pickup JC. (2004) Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 27:813–23.
Biondi-Zoccai GG, Abbate A, Liuzzo G, Biasucci LM. (2003) Atherothrombosis, inflammation, and diabetes. J. Am. Coll. Cardiol. 41:1071–7.
Okamura H, Tsutsui H, Kashiwamura S, Yoshimoto T, Nakanishi K. (1998) Interleukin-18: a novel cytokine that augments both acquired and innate immunity. Adv. Immunol. 70:281–312.
Gracie JA, Robertson SE, McInnes IB. (2003) Interleukin-18. J. Leukoc. Biol. 73:213–24.
Mallat Z et al. (2001) Expression of IL-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 104:1598–603.
Blankenberg S et al. (2002) Athero Gene Investigators. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 106:24–30.
Blankenberg S et al. (2003) PRIME Study Group. IL-18 and the risk of coronary heart disease in European men: the Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation. 108:2453–9.
Esposito K. et al. (2002) Weight loss reduces IL-18 levels in obese women. J. Clin. Endocrinol. Metab. 87:3864–6.
Escobar-Morreale HF, Botella-Carretero JI, Villuendas G, Sancho J, San Millan JL. (2004) Serum IL-18 concentrations are increased in the polycystic ovary syndrome: relationship to insulin resistance and to obesity. J. Clin. Endocrinol. Metab. 89:806–11.
Esposito K et al. (2002) Inflammatory cytokine concentrations are acutely increased by hyper-glycemia in humans: role of oxidative stress. Circulation. 106:2067–72.
Esposito K et al. (2003) Cytokine milieu tends toward inflammation in type 2 diabetes. Diabetes Care. 26:1647.
Aso Y, Okumura K, Takebayashi K, Wakabayashi S, Inukai T. (2003) Relationships of plasma IL-18 concentrations to hyperhomocysteinemia and carotid intimal-media wall thickness in patients with type 2 diabetes. Diabetes Care. 26:2622–7.
Stetler-Stevenson WG. (1999) Matrix metallopro-teinases in angiogenesis: a moving target for therapeutic intervention. J. Clin. Invest. 103:1237–41.
El Messal M et al. (2006) Elevated serum levels of proinflammatory cytokines and biomarkers of matrix remodeling in never-treated patients with familial hypercholesterolemia. Clin. Chim. Acta. 366:185–9.
Miatello R et al. (2005) Chronic administration of resveratrol prevents biochemical cardiovascular changes in fructose-fed rats. Am. J. Hypertens. 18:864–70.
Huang BW, Chiang MT, Yao HT, Chiang W. (2004) The effect of high-fat and high-fructose diets on glucose tolerance and plasma lipid and leptin levels in rats. Diabetes Obes. Metab. 6:120–6.
Hishikawa K, Lüscher TF. (1998) Felodipine inhibits free-radical production by cytokines and glucose in human smooth muscle cells. Hypertension. 32:1011–5.
Rödler S, Roth M, Nauck M, Tamm M, Block LH. (1995) Ca(2+)-channel blockers modulate the expression of interleukin-6 and interleukin-8 genes in human vascular smooth muscle cells. J. Mol. Cell. Cardiol. 27:2295–302.
Bunag RD. (1973) Validation in awake rats of a tail-cuff method for measuring systolic pressure. J. Appl. Physiol. 34:279–82.
Bunag RD. (1994) Blood pressure measurement in rats. In: Garten D, De Jong W. Experimental and genetic models of hypertension. Handbook of hypertension, vol 16. Amsterdam: Elsevier, pp:1–17.
Matthews DR et al. (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 28:412–9.
Masson P. (1992) Trichrome stainings and their preliminary technique. J. Tech. Methods. 2:75–90.
Livak KJ, Schmittgen TD. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods. 25:402–8.
Basciano H, Federico L, Adeli K. (2005) Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2:5.
Frohlich M et al. (2000) Association between C-reactive protein and features of the metabolic syndrome: a population-based study. Diabetes Care. 23:1835–9.
Festa A et al. (2000) Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation. 102:42–7.
Sakkinen PA, Wahl P, Cushman M, Lewis MR, Tracy RP. (2000) Clustering of procoagulation, inflammation, and fibrinolysis variables with metabolic factors in insulin resistance syndrome. Am. J. Epidemiol. 152:897–907.
Hak AE et al. (2001) Markers of inflammation and cellular adhesion molecules in relation to insulin resistance in nondiabetic elderly: the Rotterdam study. J. Clin. Endocrinol. Metabol. 86:4398–405.
Hung J, McQuillan BM, Chapman CM, Thompson PL, Beilby JP. (2005) Elevated interleukin-18 levels are associated with the metabolic syndrome independent of obesity and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 25:1268–73.
Zirlik A et al. (2007) Interleukin-18, the metabolic syndrome, and subclinical atherosclerosis: results from the Dallas Heart Study. Arterioscler. Thromb. Vasc. Biol. 27:2043–9.
Greenberg AS, McDaniel ML. (2002) Identifying the links between obesity, insulin resistance and beta-cell function: potential role of adipocyte adipocyte-derived cytokines in the pathogenesis of type 2 diabetes. Eur. J. Clin. Invest. 32:24–34.
McLaughlin T et al. (2002) Differentiation between obesity and insulin resistance in the association with C-reactive protein. Circulation. 106:2908–12.
Reddy VS et al. (2008) Interleukin-18 stimulates fibronectin expression in primary human cardiac fibroblasts via PI3K-Akt-dependent NF-kappaB activation. J. Cell. Physiol. 215:697–707.
Gerdes N et al. (2002) Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J. Exp. Med. 195:245–57.
Abaci A et al. (1999) Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 99:2239–42.
Schaper W, Buschmann I. (1999) Collateral circulation and diabetes. Circulation. 99:2224–6.
Zaman AK et al. (2001) Angiotensin-converting enzyme inhibition attenuates hypofibrinolysis and reduces cardiac perivascular fibrosis in genetically obese diabetic mice. Circulation. 103: 3123–8.
Matsumori A, Nunokawa Y, Sasayama S. (2000) Nifedipine inhibits activation of transcription factor NF-kappaB. Life Sci. 67:2655–61.
Iwasaki Y et al. (2004) Nilvadipine inhibits nuclear factor-kappaB-dependent transcription in hepatic cells. Clin. Chim. Acta. 350:151–7.
Matsubara M, Hasegawa K. (2004) Effects of benidipine, a dihydropyridine-Ca2+ channel blocker, on expression of cytokine-induced adhesion molecules and chemoattractants in human aortic endothelial cells. Eur. J. Pharmacol. 498:303–14.
Ding Y, Vaziri ND. (1998) Calcium channel blockade enhances nitric oxide synthase expression by cultured endothelial cells. Hypertension. 32:718–23.
Jesmin S et al. (2006) Subdepressor dose of benidipine ameliorates diabetic cardiac remodeling accompanied by normalization of upregulated endothelin system in rats. Am. J. Physiol. Heart Circ. Physiol. 290:2146–54.
Acknowledgments
This study was supported by the National Natural Science Foundation of P. R. China (No. 30670874 to W Zhang) and by the research funding of Astra-Zeneca Pharmaceutical Co. Ltd., China (QD02 to W Zhang).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Xing, SS., Tan, HW., Bi, XP. et al. Felodipine Reduces Cardiac Expression of IL-18 and Perivascular Fibrosis in Fructose-Fed Rats. Mol Med 14, 395–402 (2008). https://doi.org/10.2119/2008-00024.Xing
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2008-00024.Xing