
Abstract
Psoriatic arthritis (PsA) is a chronic and erosive form of arthritis of unknown cause. We aimed to characterize the PsA phenotype using gene expression profiling and comparing it with healthy control subjects and patients rheumatoid arthritis (RA). Peripheral blood cells (PBCs) of 19 patients with active PsA and 19 age- and sex-matched control subjects were used in the analyses of PsA, with blood samples collected in PaxGene tubes. A significant alteration in the pattern of expression of 313 genes was noted in the PBCs of PsA patients on Affymetrix U133A arrays: 257 genes were expressed at reduced levels in PsA, and 56 genes were expressed at increased levels, compared with controls. Downregulated genes tended to cluster to certain chromosomal regions, including those containing the psoriasis susceptibility loci PSORS1 and PSORS2. Among the genes with the most significantly reduced expression were those involved in downregulation or suppression of innate and acquired immune responses, such as SIGIRR, STAT3, SHP1, IKBKB, IL-11RA, and TCF7, suggesting inappropriate control that favors proinflammatory responses. Several members of the MAPK signaling pathway and tumor suppressor genes showed reduced expression. Three proinflammatory genes—S100A8, S100A12, and thioredoxin—showed increased expression. Logistic regression and recursive partitioning analysis determined that one gene, nucleoporin 62 kDa, could correctly classify all controls and 94.7% of the PsA patients. Using a dataset of 48 RA samples for comparison, the combination of two genes, MAP3K3 followed by CACNA1S, was enough to correctly classify all RA and PsA patients. Thus, PBC gene expression profiling identified a gene expression signature that differentiated PsA from RA, and PsA from controls. Several novel genes were differentially expressed in PsA and may prove to be diagnostic biomarkers or serve as new targets for the development of therapies.
Similar content being viewed by others

Introduction
Psoriatic arthritis (PsA) is a chronic and erosive form of autoimmune arthritis of unknown cause that affects approximately 10% to 20% of patients with psoriasis, with an estimated prevalence of 0.3% to 1% (1). The synovial tissue of PsA is characterized by pronounced T- and B-cell infiltrates, marked angiogenesis, and synovial hyperplasia with an increased expression of cytokines and proteases (2,3). TNFα is a major mediator in the pathogenesis of PsA (2), and therapies that target the TNFα pathway induce a significant improvement (American College of Rheumatology 20, ACR20) in 73% of patients (4). However, the magnitude of the typical clinical improvement (20%) is still far from complete disease remission. Remission has been reported to occur in up to 17% of patients, but the disease in the majority of these patients flares up within a 2-year period (5). Therefore, better understanding of the pathogenesis of PsA is necessary to identify novel and better targets for the development of more effective therapies. Additionally, prognostic and diagnostic biomarkers are needed.
Genome-wide gene expression profiling has been used to better classify many cancers (6) and to understand the molecular pathways involved in several disease processes. Recently, peripheral blood cells have been used to obtain gene expression profiles of patients with systemic lupus erythematosus (SLE) (7), rheumatoid arthritis (RA) (8), and multiple sclerosis (MS) (9). In this study, we used a similar strategy to identify gene expression profiles that distinguish PsA patients from healthy control subjects and patients with RA.
Materials and Methods
Patients and Controls
PsA was diagnosed according to the criteria of Moll and Wright (10). The study included 19 Caucasian patients (10 men, 9 women), age 50.9 ± 13.9 years (mean ± SD) and disease duration 12.3 ± 10.4 years. All patients had active disease (Table 1) and were about to be enrolled in an anti-TNF agent study. None of the patients were on anti-TNF agents or disease-modifying antirheumatic drugs (DMARDs); all DMARDs had been discontinued at least 8 weeks before blood collection. Three patients were taking prednisone ≤ 10 mg/d. Blood was also obtained from a group of age- and sex-matched normal control individuals from Rochester and the New York City area. RA patients had been enrolled in an ongoing study of biomarkers for autoimmune diseases (ABCoN) and met the American College of Rheumatology classification criteria for RA (11). All RA patients had active disease, and blood was collected before starting therapy with anti-TNF agents. The study is part of Institutional Review Board (IRB)-approved protocols, and all patients and control subjects gave informed consent.
Sample Processing and Microarray Hybridization
Peripheral blood was collected directly into PaxGene tubes (Qiagen, Valencia, CA, USA), which stabilize and protect RNA. PaxGene tubes were frozen at −80 °C until RNA extraction. Total RNA was extracted according to the manufacturer instructions using the RNeasy kit (Qiagen). Five µg of total RNA was used to synthesize cRNA using the Affymetrix expression protocol (expression analysis technical manual; Affymetrix, Santa Clara, CA, USA). Ten µg of labeled and fragmented cRNA was hybridized to a U133A chip, then stained and scanned.
Data Acquisition and Analysis
Affymetrix microarray suite (MAS) 5.0 software was used to obtain gene expression (signal) values for each gene. For accurate comparison between chips, and to correct for minor variations in the overall intensity of hybridization, each chip was scaled to an intensity of 1500. The U133A chip contains 22,215 probe sets and a total of 12,509 known genes.
A list of genes differentially expressed between PsA patients and normal controls was generated using all three of the following criteria: i) P < 0.001 using unpaired Student t test; ii) difference in expression of 100 signal units (SU) or greater when comparing the means of the two groups; and iii) ≥ 1.5-fold change in the mean gene expression between the two groups. The expression value for each gene was converted to a “fold difference” by dividing each signal value by the mean signal value of that gene in the control group. The ratios were then log2 transformed and hierarchically clustered using the program Cluster and visualized with Treeview software (12).
Identifying Differentially Expressed Genes by Logistic Regression
Single-gene logistic regression is a statistical model of how sample label information may depend on the expression level of one gene. In this model, the probability that a sample belongs to one group (for example, PsA) is a continuous function of the log expression level (x):
where a and b are 2-parameter values in the model to be fitted by the data. For a fitted logistic regression, a “deviance” measure is used to characterize how different the model is from the data; the smaller the deviance, the better the model and the more differentially expressed the gene is. A deviance value close to 0 implies that all samples in a group are expressed consistently high or consistently low. The logistic regression model fitting and deviance calculation was carried out with the R statistical package (https://doi.org/www.r-project.org/) (8,13). The statistical significance of a logistic regression result was obtained by comparing the deviance with the “null deviance.” This null deviance is the (−2)log-likelihood of a random model in which the probability for a sample to belong to a group (for example, PsA) is equal to the proportion of PsA samples in the dataset. The difference between the deviance and the null deviance follows the χ2 distribution with 1 degree of freedom by chance alone, and this χ2 distribution was used to determine the P value.
Analysis of Joint Action of Multiple Genes by Recursive Partitioning
Single-gene logistic regression, like any single-gene model or single-gene test, analyzes only one gene at a time. Use of recursive partitioning (RP) (or classification trees) (14,15) is one approach to analyze two or more genes jointly. RP first picks a gene (gene 1) that is most likely to separate sample labels based on the level of expression. Both the gene and the threshold value are determined by the data. Then, RP may pick another gene (gene 2) if further improvement on classification performance can be achieved.
This process can be visualized as a classification tree, in which the first branching at the top corresponds to gene 1, second-level branchings corresponds to gene 2, and so on. A node on the tree can be either a branching point or a terminal leaf. The “rpart” subroutine of the R statistical package was used to carry out the recursive partitioning (16). The rpart subroutine stops the branching process if the number of samples in a node is fewer than 20 or if the number of samples in a terminal node is fewer than 3.
Chromosomal Clustering Analysis of Differentially Expressed Genes
We examined whether the number of genes from a specific chromosomal band was enriched in the group of downregulated genes. Two proportions of genes from a chromosome band were calculated: one overall proportion of all genes on a chip, and the proportion of the top-ranking differentially expressed genes (reduced level in PsA compared with controls). If the first proportion is larger than the second proportion, that chromosome band is overrepresented in the selected differentially expressed genes. The comparison of two proportions was carried out by the standard χ2 test on the 2×2 table.
Results
Gene Expression Profiles of PBCs from PsA Patients Compared with Normal Controls
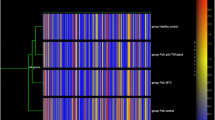
The profile of PBCs obtained from 19 PsA patients and 19 normal controls identified 313 genes meeting previously used criteria (8) for significant differential expression (P < 0.001, a difference in mean intensity of at least 100 and a fold-change ≥ 1.5). These data were clustered and visualized as shown in Figure 1. All 19 PsA patients clustered together. There was a predominance of genes with reduced expression in PsA: 257 (82%) had reduced expression, and 56 (18%) had increased expression, compared with controls.

Gene expression profiles of PBCs from 19 controls and 19 PsA patients. Unsupervised hierarchical clustering of 313 genes that distinguish PsA patients (red dendrogram) from healthy controls (blue dendrogram). Each row represents a gene; each column shows the expression for 313 genes expressed by each individual. Red indicates genes that are expressed at higher levels compared with the control mean. Green indicates genes that are expressed at lower levels relative to the control mean. PsA patients cluster to the left, and control samples cluster to the right of the figure (see supplemental Tables I and II for complete list of genes and individual expression data).
Genes Expressed in Increased Levels in PBCs from PsA Patients
Fifty-six genes were expressed at increased levels in PsA patients compared with controls (Table 2, and Supplemental Table I). These genes included caspase 5 and apoptotic protease activating factor, genes known to regulate apoptosis, and CD36, a gene involved on the removal of apoptotic cells.
Increased expression of proinflammatory mediators such as S100 calcium binding protein A12 (S100A12; calgranulin C), S100 calcium binding protein A8 (S100A8; calgranulin A), and thioredoxin was also detected in PsA.
Genes Expressed in Reduced Levels in PBCs from PsA Patients
Two hundred fifty-seven genes had reduced expression in PsA patients compared with controls (Table 2 and Supplemental Table II). There was an overrepresentation of members of the MAP kinase (MAPK) signaling pathway among the genes with reduced levels of expression, including GNAI2, TCF7, TAK1, MAP4K1, MAP3K3, MAP2K5 (MEK5), CD81, PKCB1, PKCZ, PKCBP1, Dynactin 1 (DCTN1), and TNFRSF7 (CD27), a receptor capable of activating the MAPK pathway.
B cell-specific genes involved in maturation, activation, and signaling (B lymphoid tyrosine kinase, immunoglobulin heavy constant µ, immunoglobulin κ constant, immunoglobulin heavy constant δ, B-cell associated protein, CD72, CD79B, CD81, and SHP1) and T-cell genes (LCK, SHP1, SCAP2, TCF7, and CABIN1) were expressed in lower levels in PsA. Genes involved in antigen presentation such as HLA-E, HLA-B, HLA-DQA, and HLA-DMA were also expressed in reduced levels in PsA. Several genes implicated in downregulating immune responses, such as STAT3, SIGIRR (TOLL/IL1R 8, or TIR8), NOD1 (CARD4), IKBKB, and IL-11RA, also had reduced expression in PsA compared with controls.
Additional genes expressed in reduced levels in PsA included genes involved in ubiquitination (ubiquitin-specific protease 11, ubiquitin-conjugating enzyme E2G2, and ubiquitin-activating enzyme E1-like), genes involved in apoptosis homeostasis (TNFRSF25, PDCD6, CARD4/NOD1, REQ, TRADD), and genes involved in trafficking of RNA, proteins, and organelles such as nucleoporin 62 kDa (NUP62), TIM22, and KIF5B. Additional pathway/functional classification of the differentially expressed genes is available in the supplementary materials (webtables-networks 1–12).
Logistic Regression Analysis Identifies the Best Discriminators between PsA-Controls and PsA-RA
Log-transformed data were used for the logistic regression analyses, and genes were ranked based on classification performance. The best discriminator between PsA and control was the level of expression of nucleoporin 62 kDa (NUP62) (PsA when < 817 SU and control when ≥ 817 SU), followed in order by MAP3K3 and ASXL2 (Figure 2). NUP62 expression correctly classified all controls and 94.7% of the PsA patients (18 of 19). MAP3K3 correctly classified all PsA patients and 89.4% of the controls (17 of 19).

Logistic regression analysis showing the three best discriminators between PsA and controls. Nucleoporin 62 kDa was expressed in reduced levels and had the best classifying performance (P = 1.2 × 10−10), followed by MAP3K3 (P = 4 × 10−10), which was also expressed in reduced levels, and by ASXL2 (P = 1.4 × 10−10), which was expressed at increased levels in PsA compared with controls.
The best individual discriminators between PsA and RA were MAP3K3, KIF5B (involved in the transport of organelles), and SFRS2IP (a splicing factor) (Figure 3). MAP3K3, the best discriminator, correctly classified 42 of 48 RA patients (87.5%) and 17 of 19 PsA patients (89.5%).

Logistic regression analysis showing the three best discriminators between PsA and RA. MAP3K3 was expressed in reduced levels in PsA and had the best discriminating performance between PsA and RA (P = 2.3 × 10−10), whereas KIF5B (P = 1.4 × 10−11) and SFRS2IP (P = 1.4 × 10−8) were expressed in increased levels and were the second- and third-ranked genes.
Recursive Partitioning (RP) Analyses of PsA Compared with Controls and PsA Compared with RA
RP analysis of PsA-controls determined that no other gene further contributed to the discriminatory property of NUP62 expression, as identified in the logistic regression (817 SU as the cutoff) (Figure 4A).

Recursive partitioning analysis tree. (A) PsA versus controls: levels of expression of nucleoporin 62 kDa (NUP62) expression correctly classified all controls (≥ 817 SU) and 18 of the 19 (94.7%) PsA patients (< 817 SU). (B) PsA versus RA: levels of MAP3K3 expression correctly classified all PsA patients (< 4,042 SU) and 42 of the 48 RA patients (87.5%). The stepwise addition of CACNA1S expression data correctly classified the remaining 6 RA patients.
In the PsA-RA analysis, MAP3K3 correctly classified 42 of 48 RA and 17 of 19 PsA, as described above. Whereas CACNA1S (a component of the L-type voltage-dependent calcium channel) was not a good independent discriminator/classifier in the logistic regression analysis, its use in stepwise RP was informative and helped to correctly classify the remaining 6 RA patients (Figure 4B).
Chromosomal Clustering of Differentially Expressed Genes
Genes expressed in reduced levels in PsA were predominantly located in specific chromosomal bands (Table 3), and this clustering was statistically significant. Interestingly, two of these chromosomal regions contain the psoriasis susceptibility loci PSORS1 (6p21) (17) and PSORS2 (17q25) (18), raising the possibility that a genetic variant in one of the differentially expressed genes could account for the detected differences in expression.
No obvious clustering was detected in the group of genes expressed in increased levels in PsA patients. Two genes, however, calgranulins A and C (S110A8 and S100A12), were expressed in increased levels and are located within PSORS4 on chromosome 1q21 (19).
Please note that supplementary information is available on the Molecular Medicine website (https://doi.org/www.molmed.org).
Discussion
Gene expression studies have been able to better diagnose and prognosticate cancers (6), as well as identify novel pathways implicated in the pathogenesis of autoimmune diseases (7–9). The present study is the first to comprehensively characterize the gene expression signature of PsA. Freshly obtained peripheral blood samples were used, and RNA was immediately stabilized in Pax-Gene tubes. This approach is critical, because it has been previously shown that even short-term ex vivo incubation of blood cells can alter expression profiles (20).
The analysis of the gene expression in PsA identified an overrepresentation of downregulated genes not previously described in autoimmune diseases such as SLE, RA, or MS (7–9). We identified a gene, nucleoporin 62 kDa (NUP62), which alone could differentiate 94.75% of the PsA patients from controls. RP analysis determined that MAP3K3 in combination with CACNA1S could correctly classify all RA and all PsA patients. Thus, the expression analysis of NUP62, MAP3K3, and CACNA1S could become useful for the diagnosis and/or differential diagnosis of PsA.
Complete blood counts and lymphocyte subpopulations were not determined in the present study. However, to our knowledge no abnormalities in peripheral blood cell counts have been described in PsA patients (21). In a single study, peripheral blood B-cell numbers were increased in PsA; however, in vitro responses to mitogens were reduced (22). We cannot completely exclude the possibility that some of the identified differences in gene expression between PsA and controls are accounted for by differences in peripheral blood cell counts. However, the reduced expression of B cell-specific genes, including those involved in cell activation and BCR signaling such as SYK, B lymphoid tyrosine kinase (BLK), B-cell associated protein (PHB2), CD72, CD79B, and CD81, provide a possible explanation for the reduced in vitro B-cell responsiveness. Although B cells and plasma cells are present in increased numbers in the synovial tissues of PsA patients (3), their role in PsA has not been established. Increased activation of B cells and plasma cells is generally thought to favor autoimmune and inflammatory processes. However, a recent study demonstrated that antigen-specific B cells also have a critical role in the recovery of autoimmunity and in the control of Th1 responses (23). Therefore, an alternative possibility is that an inefficient B-cell “regulatory” function, as suggested by the reduced expression of B-cell genes, could be contributing to the pathogen-esis of PsA. Finally, it is possible that chronic inflammation in the setting of PsA might selectively downregulate expression of these B-cell signaling genes.
T-cell activation and signaling genes such as LCK and CD3Z were expressed in reduced levels in PsA, similarly to what has been described in tumor-infiltrating T cells (24), in autoimmune diabetes (25), and following bacterial superantigen stimulation of T cells (26). Reduced levels of LCK correlate with reduced responsiveness to stimulation through the TCR (25) and may affect the survival of autoreactive T cells. Bacterial infections and superantigens have been previously suggested to have a role in PsA exacerbations (27), and perhaps the reduced expression of LCK and CD3Z provides a footprint for that process.
Several genes involved in the downregulation of acquired and innate immune responses were expressed in reduced levels in PsA compared with controls, including STAT3, IKBKB, SIGIRR, TCF7, CABIN1, SHP1, NOD1, and IL-11RA. Several studies have described that changes in expression of some of these genes, including STAT3 (28), SHP1 (29), TCF7 (30), and SIGIRR (31), affect the expression of rodent autoimmune diseases, suggesting that their reduced expression in PsA could also be influencing disease susceptibility or severity.
The downregulation of NOD1/CARD4 in PsA is also of interest. A deletion and loss of function of this gene has been associated with susceptibility to inflammatory bowel disease (IBD), particularly with the presence of extra-articular manifestations such as peripheral arthritis and spondylitis (32), two common clinical features of PsA. PsA and IBD often cluster together in families with spondyloarthropathies, and therefore could have common genetic susceptibility factors. Our data in conjunction with the observations by McGovern et al. (32) raise the possibility that genetic variation in the NOD1/CARD4 genes could account for the reduced expression of this gene in PsA.
Among the genes expressed in reduced levels in PsA were several members of the MAPK pathway. Reduced levels of these genes and proteins could interfere with the integrity of these pathways. MAPK pathways are involved in cell proliferation and proinflammatory processes, and therefore increased expression and/or activation, and not the opposite, was expected in PsA. However, specific MAPK pathways have distinct roles in cellular differentiation (33,34). Therefore, one interpretation of our data could be that an imbalance in MAPK pathways, as represented by reduced levels of mRNA of certain members, could be affecting the differentiation or maturation of immune cells in PsA, favoring the development of proinflammatory cells versus regulatory cells. These observations have to be carefully considered because the activation status and regulation of MAPK pathways are typically regulated at the post-translational level, and very little is actually known about the importance of transcriptional regulation for pathway integrity.
Although 12,000 genes were probed in this study, only 56 were expressed in increased levels in PsA PBCs compared with controls, according to criteria that we have used previously. Similar to other autoimmune diseases, including psoriasis (35), RA (8), juvenile RA (36), and diabetes (37), the proinflammatory S100A8 and S100A12 proteins (calgranulins) were upregulated in PsA compared with controls. Increased protein expression of S100A8 has been reported in PsA synovium and serum, and levels correlated with PsA activity (38). S100A8 and S100A12 are part of a 13-gene cluster located within PSORS4, a psoriasis susceptibility locus mapped to chromosome 1q21 (19). S100 proteins are expressed by neutrophils and macrophages and induce endothelial cell activation, chemotaxis, and increased production of TNFα and IL-1β via their binding to RAGE (39) (for review see Foell and Roth [40]). Blockade of S100-RAGE interaction ameliorated inflammatory colitis (39) and autoimmune arthritis (41) in mice. A recently described rodent model of psoriasis and PsA was also associated with very early increased expression of S100A8 (42), further implicating this gene in disease pathogenesis. Psoriasis pedigrees with linkage to PSORS4 had increased expression of S100 proteins (35), raising the possibility that the increased expression detected in PsA could be genetically determined, perhaps by a polymorphism that affects the transcription or message stability of these genes.
Another gene expressed in increased levels in PsA was thioredoxin (TRX). TRX is a proinflammatory mediator that also regulates cell proliferation and apoptosis (43). Increased levels of TRX were previously described in RA serum and synovial fluid, and TRX increased the production of TNFα by RA fibroblast-like synoviocytes (44). The increased expression of TRX in PsA suggests that it could mediate disease pathogenesis directly, or indirectly via the regulation of the expression of TNFα.
Previous studies involved genome-wide gene expression analyses using skin biopsies from psoriasis patients (45–48); to our knowledge only one study used peripheral blood mononuclear cells (PBMCs) (49), and one used PBCs in a comprehensive analysis (50). The studies of skin biopsies identified several upregulated genes, including the proinflammatory IL-1, IL-8, and S100 (45,46), as well as others such as IGFBP3, SLURP-2, hRDH-E2, and CD68 (45,47,48). The study conducted with PBMCs identified increased expression of IL-8, COX-2, annexin-3, pre-B cell enhancing factor, and S100P; however, it did not describe the list of downregulated genes (49). The increased expression of S100 family genes is a consistent finding in psoriasis and now in PsA studies. The study of PBCs (50) focused on gene expression before and after therapy with a macrolactam derivative and did not have a non-psoriasis control group for comparison. Methodological differences, including the specimens used (PBMCs versus PBCs, which include neutrophils), processing methodology, analytical strategy, study population (disease, age, and sex), and characteristics of the controls likely contributed to differences between studies. One previous study analyzed PBMCs from a small number of patients with PsA (n = 6) using a limited 588-gene microarray (51).
Interestingly, genes expressed in reduced levels in PsA tended to cluster to a few (11) chromosomal bands. This clustering is highly significant, as shown in Table 3, and is in marked contrast to what we have observed previously for dysregulated genes in RA, which showed no evidence of such clustering (8). Some of these clusters are known to contain psoriasis susceptibility genes, such as PSORS1 (6p21) (17) and PSORS2 (17q25) (18), raising the possibility that a genetic variant in one of these genes could account for the detected differences in expression. The other clusters could conceivably point to a PsA-specific locus, such as that containing 15 genes, including NUP62, the best discriminator between PsA and controls, on chromosome 19q13.
Finally, by employing logistic regression and recursive partitioning, we have identified candidate genes whose expression may be useful diagnostically in discriminating PsA, RA, and control subjects, as shown in Figures 3 and 4. However, we wish to emphasize that these observations must be viewed as preliminary and hypothesis generating, and replication in an independent dataset and expansion of the overall sample size is required. Overall, these new differentially expressed genes may generate novel candidates for genetic analysis in PsA case-control association studies, and have the potential to become novel targets for the development of therapies and diagnostic or prognostic tests.
References
Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. (2005) Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann. Rheum. Dis. 64 Suppl 2:ii14–7.
Veale DJ, Ritchlin C, FitzGerald O. (2005) Immunopathology of psoriasis and psoriatic arthritis. Ann. Rheum. Dis. 64 Suppl 2:ii26–9.
Kruithof E, Baeten D, De Rycke L, et al. (2005) Synovial histopathology of psoriatic arthritis, both oligo- and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res. Ther. 7:R569–80.
Mease PJ, Goffe BS, Metz J, VanderStoep A, Finck B, Burge DJ. (2000) Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet 356:385–90.
Gladman DD, Hing EN, Schentag CT, Cook RJ. (2001) Remission in psoriatic arthritis. J. Rheumatol. 28:1045–8.
Staudt LM. (2002) Gene expression profiling of lymphoid malignancies. Annu. Rev. Med. 53:303–18.
Baechler EC, Batliwalla FM, Karypis G, et al. (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U. S. A. 100:2610–5.
Batliwalla FM, Baechler EC, Xiao X, et al. (2005) Peripheral blood gene expression profiling in rheumatoid arthritis. Genes Immunol. 6:388–97.
Bomprezzi R, Ringner M, Kim S, et al. (2003) Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum. Mol. Genet. 12:2191–9.
Moll JM, Wright V. (1973) Psoriatic arthritis. Semin. Arthritis Rheum. 3:55–78.
Arnett FC, Edworthy SM, Bloch DA, et al. (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 31:315–24.
Eisen MB, Spellman PT, Brown PO, Botstein D. (1998) Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95:14863–8.
Li W, Yang Y. (2002) Zipf’s law in importance of genes for cancer classification using microarray data. J. Theor. Biol. 219:539–51.
Breiman L, Friedman J, Olshen R, Stone C. (1984) Classification and Regression Trees. CRC Press.
Zhang H, Yu CY, Singer B, Xiong M. (2001) Recursive partitioning for tumor classification with gene expression microarray data. Proc. Natl. Acad. Sci. U. S. A. 98: 6730–5.
Therneau T, Atlinson E. (1997) An Introduction to Recursive Partitioning Using the RPART Routines. Mayo Foundation.
Jenisch S, Henseler T, Nair RP, et al. (1998) Linkage analysis of human leukocyte antigen (HLA) markers in familial psoriasis: strong disequilibrium effects provide evidence for a major determinant in the HLA-B/-C region. Am. J. Hum. Genet. 63:191–9.
Tomfohrde J, Silverman A, Barnes R, et al. (1994) Gene for familial psoriasis susceptibility mapped to the distal end of human chromosome 17q. Science 264: 1141–5.
Capon F, Novelli G, Semprini S, et al. (1999) Searching for psoriasis susceptibility genes in Italy: genome scan and evidence for a new locus on chromosome 1. J. Invest. Dermatol. 112:32–5.
Baechler EC, Batliwalla FM, Karypis G, et al. (2004) Expression levels for many genes in human peripheral blood cells are highly sensitive to ex vivo incubation. Genes Immunol. 5:347–53.
Bennett RM. (1997) Psoriatic arthritis. In: Koopman WJ (ed.) Arthritis and Allied Conditions: A Textbook of Rheumatology. Williams & Wilkins, Baltimore, pp. 1229–44.
Ventura M, Colizzi M, Ottolenghi A, et al. (1989) Cell-mediated immune response in psoriasis and psoriatic arthritis. Rec. Prog. Med. 80:449–54.
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. (2002) B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 3:944–50.
Riccobon A, Gunelli R, Ridolfi R, et al. (2004) Immunosuppression in renal cancer: differential expression of signal transduction molecules in tumor-infiltrating, near-tumor tissue, and peripheral blood lymphocytes. Cancer Invest. 22:871–7.
Nervi S, Atlan-Gepner C, Kahn-Perles B, et al. (2000) Specific deficiency of p56lck expression in T lymphocytes from type 1 diabetic patients. J. Immunol. 165:5874–83.
Criado G, Madrenas J. (2004) Superantigen stimulation reveals the contribution of Lck to negative regulation of T cell activation. J. Immunol. 172:222–30.
Yamamoto T, Katayama I, Nishioka K. (1999) Peripheral blood mononuclear cell proliferative response against staphylococcal superantigens in patients with psoriasis arthropathy. Eur. J. Dermatol. 9:17–21.
Welte T, Zhang SS, Wang T, et al. (2003) STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc. Natl. Acad. Sci. U. S. A. 100:1879–84.
Zhang J, Somani AK, Siminovitch KA. (2000) Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signaling. Semin. Immunol. 12:361–78.
Smith SS, Patterson T, Pauza ME. (2005) Transgenic Ly-49A inhibits antigen-driven T cell activation and delays diabetes. J. Immunol. 174:3897–905.
Garlanda C, Riva F, Polentarutti N, et al. (2004) Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. U. S. A. 101:3522–6.
McGovern DP, Hysi P, Ahmad T, et al. (2005) Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 14:1245–50.
Arbour N, Naniche D, Homann D, Davis RJ, Flavell RA, Oldstone MB. (2002) c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8(+) T cell-mediated antiviral immunity. J. Exp. Med. 195:801–10.
Constant SL, Dong C, Yang DD, Wysk M, Davis RJ, Flavell RA. (2000) JNK1 is required for T cell-mediated immunity against Leishmania major infection. J. Immunol. 165:2671–6.
Semprini S, Capon F, Tacconelli A, et al. (2002) Evidence for differential S100 gene over-expression in psoriatic patients from genetically heterogeneous pedigrees. Hum. Genet. 111:310–3.
Schulze zur Wiesch A, Foell D, Frosch M, Vogl T, Sorg C, Roth J. (2004) Myeloid related proteins MRP8/MRP14 may predict disease flares in juvenile idiopathic arthritis. Clin. Exp. Rheumatol. 22:368–73.
Bouma G, Lam-Tse WK, Wierenga-Wolf AF, Drexhage HA, Versnel MA. (2004) Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes 53:1979–86.
Kane D, Roth J, Frosch M, Vogl T, Bresnihan B, FitzGerald O. (2003) Increased perivascular synovial membrane expression of myeloid-related proteins in psoriatic arthritis. Arthritis Rheum. 48:1676–85.
Hofmann MA, Drury S, Fu C, et al. (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901.
Foell D, Roth J. (2004) Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum. 50:3762–71.
Hofmann MA, Drury S, Hudson Bl, et al. (2002) RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immunol. 3:123–35.
Zenz R, Eferl R, Kenner L, et al. (2005) Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 437:369–75.
Burke-Gaffney A, Callister ME, Nakamura H. (2005) Thioredoxin: friend or foe in human disease? Trends Pharmacol. Sci. 26:398–404.
Yoshida S, Katoh T, Tetsuka T, Uno K, Matsui N, Okamoto T. (1999) Involvement of thioredoxin in rheumatoid arthritis: its costimulatory roles in the TNF-alpha-induced production of IL-6 and IL-8 from cultured synovial fibroblasts. J. Immunol. 163:351–8.
Bowcock AM, Shannon W, Du F, et al. (2001) Insights into psoriasis and other inflammatory diseases from large-scale gene expression studies. Hum. Mol. Genet. 10:1793–805.
Quekenborn-Trinquet V, Fogel P, Aldana-Jammayrac O, et al. (2005) Gene expression profiles in psoriasis: analysis of impact of body site location and clinical severity. Br. J. Dermatol. 152:489–504.
Tsuji H, Okamoto K, Matsuzaka Y, Iizuka H, Tamiya G, Inoko H. (2003) SLURP-2, a novel member of the human Ly-6 superfamily that is up-regulated in psoriasis vulgaris. Genomics 81:26–33.
Matsuzaka Y, Okamoto K, Tsuji H, et al. (2002) Identification of the hRDH-E2 gene, a novel member of the SDR family, and its increased expression in psoriatic lesion. Biochem. Biophys. Res. Commun. 297:1171–80.
Koczan D, Guthke R, Thiesen HJ, et al. (2005) Gene expression profiling of peripheral blood mononuclear leukocytes from psoriasis patients identifies new immune regulatory molecules. Eur. J. Dermatol. 15:251–7.
Rappersberger K, Komar M, Ebelin ME, et al. (2002) Pimecrolimus identifies a common genomic anti-inflammatory profile, is clinically highly effective in psoriasis and is well tolerated. J. Invest. Dermatol. 119:876–87.
Gu J, Marker-Hermann E, Baeten D, et al. (2002) A 588-gene microarray analysis of the peripheral blood mononuclear cells of spondyloarthropathy patients. Rheumatology (Oxford) 41:759–66.
Acknowledgements
This work was supported by the National Institutes of Health Autoimmune Biomarkers Collaborative Network contract NO1-AR-1-2256.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Batliwalla, F.M., Li, W., Ritchlin, C.T. et al. Microarray Analyses of Peripheral Blood Cells Identifies Unique Gene Expression Signature in Psoriatic Arthritis. Mol Med 11, 21–29 (2005). https://doi.org/10.2119/2006-00003.Gulko
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2006-00003.Gulko