
Abstract
Inflammatory conditions characterized by excessive peripheral immune responses are associated with diverse alterations in brain function, and brain-derived neural pathways regulate peripheral inflammation. Important aspects of this bidirectional peripheral immune-brain communication, including the impact of peripheral inflammation on brain region-specific cytokine responses, and brain cholinergic signaling (which plays a role in controlling peripheral cytokine levels), remain unclear. To provide insight, we studied gene expression of cytokines, immune cell markers and brain cholinergic system components in the cortex, cerebellum, brainstem, hippocampus, hypothalamus, striatum and thalamus in mice after an intraperitoneal lipopolysaccharide injection. Endotoxemia was accompanied by elevated serum levels of interleukin (IL)-1β, IL-6 and other cytokines and brain region-specific increases in Il1b (the highest increase, relative to basal level, was in cortex; the lowest increase was in cerebellum) and Il6 (highest increase in cerebellum; lowest increase in striatum) mRNA expression. Gene expression of brain Gfap (astrocyte marker) was also differentially increased. However, Iba1 (microglia marker) mRNA expression was decreased in the cortex, hippocampus and other brain regions in parallel with morphological changes, indicating microglia activation. Brain choline acetyltransferase (Chat) mRNA expression was decreased in the striatum, acetylcholinesterase (Ache) mRNA expression was decreased in the cortex and increased in the hippocampus, and M1 muscarinic acetylcholine receptor (Chrm1) mRNA expression was decreased in the cortex and the brainstem. These results reveal a previously unrecognized regional specificity in brain immunoregulatory and cholinergic system gene expression in the context of peripheral inflammation and are of interest for designing future antiinflammatory approaches.
Similar content being viewed by others

Introduction
Dysregulated peripheral immune responses and cytokine release mediate pathogenesis in sepsis, type 2 diabetes, rheumatoid arthritis, lupus and other inflammatory and autoimmune conditions (1–4). Peripherally released cytokines and other inflammatory molecules have also been associated with brain immune activation and altered brain neuronal function in these disorders (5–13). Peripheral inflammation and elevated systemic cytokine levels have been proposed to contribute to brain inflammation and pathogenesis in Alzheimer’s disease (14,15), Parkinson’s disease (16) and schizophrenia (17). Furthermore, the brain plays an important role in controlling peripheral cytokine levels and inflammation via neural reflex circuits (1,2,18). We and others have shown that brain cholinergic signaling is an important component in this regulation during inflammatory conditions (19–23). This bidirectional peripheral immune-brain communication in inflammatory disorders is a topic of significant interest with potential therapeutic implications (18,24–29). Therefore, providing insight into specific aspects of this communication is necessary and important.
Peripheral endotoxin (lipopolysaccharide [LPS]) administration in mice has been used previously in studying peripheral immune-brain interactions. However, the impact of peripheral inflammation on brain cytokine gene expression within specific brain regions in mice has not been thoroughly examined. The predominant immune cells in the central nervous system are microglia and astrocytes; microglia activation and astrogliosis, resulting from immune challenge or neuronal damage, are associated with interleukin (IL)-1β, IL-6 and other inflammatory molecule production (30,31). Microglia and astrocytes also are active modulators of neuronal synaptic interaction and neuronal plasticity (32,33), and changes in microglia and astrocyte functional state can alter neuronal homeostasis and brain function (33–35). While a role for brain cholinergic signaling in the neural regulation of peripheral cytokine release during endotoxemia and other inflammatory conditions has been previously indicated (19–23), the intriguing question of whether acute peripheral inflammation affects the cholinergic system in various brain regions has not been adequately addressed.
Here we aimed at providing insight into the impact of peripheral inflammation on brain cytokine and cholinergic system constituent gene expression. We studied mRNA expression of cytokines (Il1b and Il6), immune cytokine-producing cell markers (Gfap and Iba1) and cholinergic system components (Chat, Ache and Chrm1) in seven brain regions after peripheral endotoxin administration in mice.
The results reveal a somewhat surprising brain region-specific profile of Il1b and Il6 gene expression in parallel with differential alterations in Gfap, Iba1, Chat, Ache, and Chrm1 gene expression during peripheral systemic inflammation, accompanied by high-serum IL1β, IL-6 and other cytokine levels. These results highlight new aspects of a peripheral immune-brain communication.
Materials and Methods
Animals and LPS Treatment
Male Balb/c mice (5–6 months old, Taconic) were used in the experiments. Animals were housed under standard conditions (room temperature 22°C with a 12-h light-dark cycle) and had free access to standard chow and water. Animals were allowed to acclimate for at least 20 d before experiments. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (36) under protocols approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research, North Shore-LIJ Health System (Manhasset, NY, USA).
Endotoxemia and Sample Preparation
Lethal inflammation in mice was induced by administering LPS (endotoxin, Escherichia coli 0111:B4; Sigma-Aldrich, St. Louis, MO, USA). LPS (in pyrogen-free saline) was sonicated for 30 min and injected intraperitoneally (IP) in mice in a dose of 8 mg/kg. Control mice were injected with saline (IP). Mice injected with LPS or saline (n = 15 per group) were euthanized 4 h after injection by carbon dioxide asphyxiation. Blood was collected immediately after euthanasia by cardiac puncture. Brains were isolated on ice and dissected through the midsagittal plane. The left hemisphere was transferred into 4% paraformaldehyde and subsequently processed for hematoxylin and eosin (H&E) staining and immunohistochemistry (see below for details). The cerebral cortex, cerebellum, brainstem, hippocampus, hypothalamus, striatum and thalamus were dissected on ice by using a binocular dissection microscope by a trained and highly experienced neuroscientist according to a modified method previously described by Glowinski and Iversen (37). Brain tissue was snap frozen on dry ice and transferred to storage at −80°C.
Serum Cytokine Determination
To obtain serum samples, blood was allowed to clot for 1.5 h and was centrifuged at 1,500g for 10 min. Supernatants (sera) were collected and kept at −20°C before cytokine analyses. IL-6, IL-1β, chemokine (C-X-C motif) ligand (CXCL1), IL-12p70, interferon (IFN)-γ, IL-10 and tumor necrosis factor (TNF) were determined by using the mouse proinflammatory 7-Plex electrochemiluminescent kit (Meso Scale Discovery, Gaithersburg, MD, USA), according to the manufacturer’s recommendations.
RNA Isolation and Quantitative Polymerase Chain Reaction
RNA from seven different brain regions (cortex, cerebellum, brain stem, hippocampus, hypothalamus, striatum and thalamus) was extracted using the RNeasy Plus Universal Mini-Kit (Qiagen, Germantown, MD, USA) after tissue homogenization with the Bullet Blender Homogenizer (Next Advance, Averill Park, NY, USA) and the recommended bead lysis kit. Because of limited RNA levels found in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples were combined from the same treatment groups before RNA extraction. RNA was quantified and analyzed for purity by using the Nanodrop 1000 (Thermo Scientific [Thermo Fisher Scientific Inc., Waltham, MA, USA]); 260:280 and 260:230 ratios were >2 and >1.8, respectively. Isolated RNA was frozen at −80°C until quantitative polymerase chain reaction (qPCR) assays. Specific primers for Il6, Il1b, acetylcholinesterase (Ache), choline acetyltransferase (Chat), glial fibrillary acidic protein (Gfap), ionized calcium-binding adapter molecule1 (Iba1), M1mAChR (Chrm1) and Hprt1 (internal reference gene) were designed by using the Roche Universal Probe Library Assay Design Center with the assigned probes: Il6, left, GCTAC CAAAC TGGATATAAT CAGGA, right, CCAGGTAGCTATGGT ACTCCAGAA, probe 6; Il1b, left, AGTTG ACGGACCCCA AAAG, right, AGCTG GATGCTCTCATCAGG, probe 38; Ache, left, TTAGGGCTGGGATATAATACGAC, right, GCCCCTAGTGGGAGGAAGT, probe 52; Chat, left, AAGCTTCCACGCCAC TTTC, right, AGAGCCTCCGACGAA GTTG, probe 3; Iba1, left, GGATTTGCAG GGAGGAAAAG, right, TGGGATCATC GAGGAATTG, probe 3; Chrm1, left, GGTCC CAGGA GACACTGC, right, TCAGAGTAAGGGCATCACCA, probe 73; and Hprt1, left, TCCTCCTCAGACCGC TTTT, right, CCTGGTTCATCATCG CTAATC, probe 95. RNA (100 ng/reaction) was amplified using a Eurogentec reverse-transcriptase qPCR master mix (AnaSpec, EGT Corporate Headquarters, Fremont, CA, USA) on a LightCycler 480 (F. Hoffmann-La Roche Ltd, Basel, Switzerland), and the data were analyzed by using the Roche LightCycler 480 SW 1.5 software. Relative changes in mRNA expression were calculated as fold-changes (versus Hprt) by using the comparative Ct (AACt method) (38).
H&E Staining and Microscopic Evaluation
Brains were immersed in 4% paraformaldehyde fixation fluid for at least 24 h. Postfixed brain tissue was processed by using standard techniques and was embedded in paraffin. Serial sections of fixed brain tissue were cut at 40 µm, mounted on glass slides and air-dried. The slides were stained and deparaffinized by using the Ventana symphony staining system for H&E staining. Images of brain sections were acquired at 20× and 40× magnification with the use of a Nikon Eclipse E400 microscope attached to a Nikon Digital DS-Fi1 camera (Nikon Inc., Melville, NY, USA) and Adobe Photoshop software (Adobe Systems Incorporated, San Jose, CA, USA). The morphology of the neurons in the cerebral cortex, hippocampus and cerebellum and Purkinje cells (in the cerebellum) were assessed and photographed (40× magnification) in at least three different high-power fields per brain region.
Iba1 Staining and Microscopic Evaluation
Fixed brains were cryoprotected in 30% sucrose, frozen and cut using cryostat. Coronal sections (30 µm) were collected in phosphate-buffered saline (PBS). The immunohistochemistry was carried out on free-floating sections. The sections were incubated for 30 min in 0.3% H2O2-PBS to inactivate endogenous peroxidase activity. The sections were blocked and incubated with primary antibodies (anti-rabbit IBA1 1:100; Wako, Cambridge, MA, USA) in PBS containing 0.3% (v/v) Triton-100, 2% (v/v) normal goat serum in PBS. The incubations were performed overnight at 4°C with gentle shaking. After several washes in PBS, the sections were incubated with biotinylated secondary antibodies (1:200; Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. The staining was visualized by using the biotin-avidin-peroxidase method (Elite Kit; Vector Laboratories) with diaminobenzidine as the chromogen/substrate and counterstained with hematoxylin (Sk-4100 and HR-3401; Vector Laboratories). Images of Iba1-immunostained brain sections were acquired at 40× magnification with the use of a Nikon Eclipse E400 microscope attached to a Nikon Digital DS-Fi1 camera and Adobe Photoshop software. Microglial morphology in the cerebral cortex and hippocampus were assessed and photographed (40× magnification) in at least three different high-power fields per brain region.
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM). Differences between experimental groups were assessed by using a Student t test (GraphPad Prism 6; GraphPad Software Inc., La Jolla, CA, USA). Differences with P < 0.05 were considered statistically significant.
All supplementary materials are available online at https://doi.org/www.molmed.org .
Results
Peripheral Inflammation Is Associated with Brain Region-Specific Increases in Proinflammatory Cytokine Gene Expression
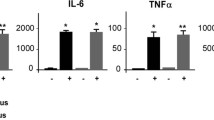
After the administration of LPS (8 mg/kg, IP), increased serum IL-6, IL-1β, CXCL1, IFN-γ, IL-12p70, IL-10 and TNF levels were determined at 4 h compared with saline-injected control mice (Figure 1). These elevated serum cytokine levels indicated peripheral systemic inflammatory responses, which can ultimately lead to 80% mortality within 4 d, as demonstrated in a parallel set of animals (Supplementary Figure S1). IL-1β and IL-6 are major cytokine mediators of brain inflammatory conditions (39). Peripheral LPS administration dramatically increased brain Il1b and Il6 gene expression compared with saline-injected controls, and these (relative to basal level in saline-treated mice) increases were region-specific (Figures 2, 3). The highest upregulation in Il1b mRNA expression was found in the cortex (144-fold) and the lowest in the cerebellum (29-fold) (Figure 2). The highest Il6 mRNA expression elevation was observed in the cerebellum (173-fold) and the lowest in the striatum (29-fold) (Figure 3). These levels of peripheral and brain inflammation were not associated with increased brain cell damage, as evaluated by the number of eosin-positive neurons identified in the cortex, cerebellum and hippocampus (Figure 4).

Peripheral LPS administration increases serum cytokine and chemokine levels. Mice were injected with LPS (8 mg/kg, IP) or saline (S) and euthanized 4 h later. Serum IL-6, IL-1β, CXCL1, IFN-γ, IL-12p70, IL-10 and TNF were analyzed by using a multiplex platform, as described in Materials and Methods. Data are shown as mean ± SEM (n = 15 mice per group); ****P < 0.0001.

Relative Il1b mRNA expression is differentially increased in various brain regions after peripheral LPS administration. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain regions were isolated and processed for Il1b mRNA expression analysis by qPCR, as described in Materials and Methods. Data are shown as mean ± SEM and fold-change (n = 15 mice per group); ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. Because of limited RNA levels in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples (from the same region and treatment group) were combined before RNA extraction; therefore, n = 5 was used for statistical analyses in these regions.

Relative Il6 mRNA expression is differentially increased in various brain regions after peripheral LPS administration. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain regions were isolated and processed for Il6 mRNA expression analysis by qPCR, as described in Materials and Methods. Data are shown as mean ± SEM and fold-change (n = 15 mice per group); ****P < 0.0001, ***P < 0.0005, **P < 0.001, *P < 0.005. Because of limited RNA levels in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples (from the same region and treatment group) were combined before RNA extraction; therefore, n = 5 was used for statistical analysis in these regions.

Lethal inflammation is not associated with early brain neuronal cell death and morphology alterations. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain neuronal cell morphology was evaluated (using H&E-stained sections) in the cortex, cerebellum and hippocampus by assessing three visual fields per brain region per mouse. Representative photomicrographs (40×) are shown of H&E-stained sections of cortex-saline (A), cortex-LPS (B), cerebellum-saline (C), cerebellum-LPS (D), hippocampus-saline (E) and hippocampus-LPS (F). Scale bar, 50 µm.
Peripheral Inflammation Alters Microglia and Astrocyte Marker Gene Expression
Microglia and astrocytes are the main immune cell subtypes, associated with innate immune responses and cytokine production in the brain (30,31). qPCR quantification of Gfap (astrocyte marker) and Iba1 (microglia marker) mRNA expression have been used previously for studying astrogliosis and microglia activation, respectively (13,40,41). Here, we examined whether peripheral lethal inflammation affects astrocyte and/or microglia functional status by determining Gfap and Iba1 mRNA expression in the various brain regions. As shown in Figure 5, brain Gfap mRNA expression was upregulated after peripheral LPS administration. Although the magnitude of this upregulation was markedly lower when compared with increases in Il1b and Il6 gene expression, it was significant in the cortex (P < 0.0001), cerebellum (P < 0.0002), brainstem (P < 0.05), hippocampus (P < 0.05) and thalamus (P < 0.05). This upregulation was consistent with the proposed role of astrocytes in the innate immune response within the central nervous system and as cytokine producers (30). In contrast to the elevated astrocyte marker gene expression, Iba1 gene expression was slightly but significantly decreased in the cortex (P < 0.0001), brainstem (P < 0.0001), hippocampus (P < 0.05), striatum (P < 0.05) and thalamus (P < 0.05) (Figure 6) during peripheral inflammation. This somewhat unexpected observation prompted us to additionally perform Iba1 protein staining in two brain regions with significantly decreased Iba1 gene expression (the cortex and hippocampus), to highlight the activation state of microglia. As shown in Figure 7, Iba1 staining revealed a microglial morphological reorganization, as evidenced by increased density of the Iba1-stained material, thickening of ramifications and processes and increased cytoplasm. These alterations were predominantly observed in the hippocampus of the LPS-treated mice (Figure 7).

Gfap mRNA expression is increased in specific brain regions during lethal inflammation. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain regions were isolated and processed for Gfap mRNA expression analysis by qPCR, as described in Materials and Methods. Data are shown as mean ± SEM (n = 15 mice per group); ***P < 0.0001, **P < 0.0002, *P < 0.05. Because of limited RNA levels in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples (from the same region and treatment group) were combined before RNA extraction; therefore, n = 5 was used for statistical analysis in these regions.

Iba1 mRNA expression is decreased in specific brain regions during lethal inflammation. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain regions were isolated and processed for Iba1 mRNA expression analysis by qPCR, as described in Materials and Methods. Data are shown as mean ± SEM (n = 15 mice per group); **P < 0.0001, *P < 0.05. Because of limited RNA levels in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples (from the same region and treatment group) were combined before RNA extraction; therefore, n = 5 was used for statistical analysis in these regions.

Lethal inflammation is associated with morphological alterations in microglia indicative of activation. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain tissues were stained for Iba1 immunohistochemistry, as described in Materials and Methods. Iba1-stained microglia were evaluated in the cortex and hippocampus by assessing three visual fields per brain region, per mouse. Microglia were faintly stained with anti-Iba1 in the cortex and the hippocampus of saline-injected mice, while morphological changes of activated microglia, including cell body enlargement, shortening and thickening of processes, were observed in these brain areas of LPS-injected mice. Representative photomicrographs (40×) of Iba1-stained sections are shown: cortex-saline (A), cortex-LPS (B), hippocampus-saline (C) and hippocampus-LPS (D). Enlarged single cell images are shown on the upper right corners. Scale bar, 50 µm.
Peripheral Inflammation Results in Specific Alterations in Cholinergic System Component Gene Expression
Next, we examined the impact of lethal inflammation on the expression of major genes associated with the brain cholinergic system. The gene expression of choline acetyltransferase (ChAT) (the enzyme implicated in acetylcholine biosynthesis), acetylcholinesterase (AChE) (the major enzyme in acetylcholine degradation) and the M1 subtype of muscarinic acetylcholine receptor (M1 mAChR) (an important receptor involved in processing neurotransmission in the brain cholinergic synapse) were studied. As shown in Figure 8, lethal inflammation did not alter Chat gene expression, with the exception of the striatum, where it was decreased (P < 0.01).

Lethal inflammation results in decreased Chat mRNA expression in the striatum. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain regions were isolated and processed for Chat mRNA expression analysis by qPCR, as described in Materials and Methods. Data are shown as mean ± SEM (n = 15 mice per group); *P < 0.01. Because of limited RNA levels in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples (from the same region and treatment group) were combined before RNA extraction; therefore, n = 5 was used for statistical analysis in these regions. No Chat mRNA was detected in the cerebellum and hippocampus.
No Chat mRNA was detected in the cerebellum or hippocampus. Ache mRNA expression was significantly decreased in the cortex (P < 0.004) and increased in the hippocampus (P < 0.01) (Figure 9). In addition, lethal LPS-induced inflammation decreased Chrm1 (M1mAChR) mRNA expression in the cortex (P < 0.002) and reduced gene expression to undetectable levels in the brainstem (Figure 10). No Chrm1 mRNA expression was detected in the cerebellum of control or LPS-treated mice.

Lethal inflammation results in region-specific alterations in brain Ache mRNA expression. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain regions were isolated and processed for Ache mRNA expression analysis by qPCR, as described in Materials and Methods. Data are shown as mean ± SEM (n = 15 mice per group); ***P < 0.004, **P < 0.01. Because of limited RNA levels in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples (from the same region and treatment group) were combined before RNA extraction; therefore, n = 5 was used for statistical analysis in these regions.

Lethal inflammation results in brain region-specific decreases in Chrm1 mRNA expression. Mice were injected with LPS (8 mg/kg, IP) or saline and euthanized 4 h later. Brain regions were isolated and processed for Chrm1 mRNA expression analysis by qPCR, as described in Materials and Methods. Data are shown as mean ± SEM (n = 15 mice per group); ***P < 0.002. Because of limited RNA levels in the hippocampus, hypothalamus, striatum and thalamus, three tissue samples (from the same region and treatment group) were combined before RNA extraction; therefore, n = 5 was used for statistical analysis in these regions. No Chrm1 mRNA was detected in the cerebellum of LPS-treated mice.
Discussion
Herein, we show that peripheral administration of LPS, accompanied by significant elevations of IL-1β and IL-6 and other inflammatory mediators in the circulation, results in significant increases in Il1 and Il6 gene expression in the mouse brain within 4 h and reveal a previously unrecognized pattern of region specificity, characteristic for each of these two cytokines. These increases were associated with increased Gfap mRNA expression and downregulation of Iba1 mRNA expression in most of the brain areas studied, along with specific alterations in the gene expression of main constituents of the brain cholinergic system.
Administration of a lethal dose of LPS, a constituent of the outer membrane of gram-negative bacteria, has been extensively used to model gram-negative microorganism-induced septic shock, a lethal condition in humans with no treatment available. Accordingly, evaluation of the survival rate has been an important outcome in numerous studies using the murine endotoxemia model. However, the effect of a lethal dose of LPS on brain cytokine gene expression has not been clearly elucidated. Moreover, we and others have demonstrated that stimulation of brain cholinergic signaling by M1 mAChR agonists or centrally acting acetylcholinesterase inhibitors suppresses peripheral proinflammatory cytokine release and improves survival in endotoxemia and other inflammatory conditions (20–23,42). Likewise, the impact of lethal LPS-induced peripheral inflammation on the expression of genes involved in the brain cholinergic system has not been examined.
Previous studies have demonstrated significant upregulation in brain Il1b and Il6 gene expression within 1–6 h after peripheral LPS administration. For instance, induction of Il1b mRNA expression has been reported in the rat brain as early as 1 h after LPS (2.5 mg/kg, intravenously [IV]) administration, peaking at 6 h, and microglia have been identified as the main producers of IL-1β (43). In a nonlethal model of endotoxemia, significant increases in Il1b and Il6 gene expression were reported within 1–4.5 h after LPS (5 mg/kg, IV) injection in rats (44). Likewise, a sublethal dose of LPS administered systemically to rats has been shown to induce significantly increased hypothalamic Il6 mRNA expression as early as 2 h after LPS administration (45). On the basis of these previous findings, we considered the 4-h time point after LPS administration as an appropriate time for evaluating inflammatory gene expression in the brain of endotoxemic versus control mice.
Peripheral administration of LPS significantly increased both Il1b and Il6 mRNA expression in all brain regions studied, with the highest magnitude in the cortex and hippocampus for Il1b and in the cerebellum and brainstem for Il6. These increases were associated with upregulation in Gfap mRNA expression in the cortex, hippocampus, cerebellum and other brain regions. These observations are consistent with the important role of astrocytes in IL-1β and IL-6 production. The correlation between upregulated cytokine and astrocyte marker mRNA expression are in accordance with data characterizing brain inflammation in the context of diet-induced obesity in mice. Gfap mRNA expression has been shown to be significantly elevated in hypothalamus of mice with diet-induced obesity simultaneously with increased expression of mRNA encoding myeloid cell-specific markers Cd68 and Emr1 (13). These changes, indicating active gliosis, occur in parallel with increased hypothalamic Il1b and Il6 mRNA expression (13).
In contrast to increased Gfap, Iba1 mRNA expression was decreased in the cortex, hippocampus and other brain regions 4 h after LPS administration. These observations suggest a different effect of peripheral inflammation, at this early time point, on mRNA expression of markers of astrocyte and microglia activation. Furthermore, the decrease in Iba1 gene expression in the cortex and hippocampus was accompanied by increased Iba1 protein staining in these regions, indicating morphological transformations pointing toward microglia activation. These transformations in microglia morphology were consistent with increased Il1b and Il6 mRNA expression in these two brain regions. Our observations suggest intriguing microglia morphofunctional fluctuations at the time point studied (4 h), during which the decreased Iba1 mRNA expression could be a stop signal preventing further microglia activation.
Microglial activation has been traditionally associated with morphological transformations from a ramified “resting state” to an amoeboid, macrophage-like cell “activation state.” However, this concept has now evolved, and microglia have been characterized at multiple morphofunctional stages, depending on the condition studied, activating stimulus and the time of study (32–34,46). We should note that the morphological changes observed in Iba1-positive microglia (increased processes density and enlarged cytoplasm) do not entirely represent the notion of the amoeboid microglia activation state. Most likely, they indicate a stage of microglia alteration (33) that, in this case, occurs simultaneously with decreased Iba1 mRNA levels and increased Il1b and Il6 mRNA expression. These microglia alterations at relatively early time points are in accordance with previous studies demonstrating increased staining of microglia in the hippocampus and other brain regions as early as 3 h after LPS (5 mg/kg, IV) injection in rats (47), or in the cortex and hippocampus after LPS (5 mg/kg, IP) injection in mice (48). A recent study using intraperitoneal administration of a sublethal dose of LPS revealed alterations in microglial morphological plasticity as early as 2 h after LPS administration manifested by reductions in the length of microglial processes in the hippocampus (46).
Mechanisms underlying the impact of LPS-induced peripheral inflammation on the brain have been previously highlighted (48–51). Direct activation of immune responses in the brain by peripherally administered LPS does not seem to play a primary role because of poor LPS penetration across the blood-brain barrier (48). Instead, cytokines and other inflammatory molecules, released during endotoxemia, have been shown to signal the brain and trigger immune responses. The functional anatomy of this cytokine-mediated transfer of peripheral inflammation to the brain involves several mechanisms: crossing the blood-brain barrier by a saturable carrier-mediated mechanism; binding receptors at the surface of the endothelium of brain capillaries resulting in the release of soluble mediators, such as prostaglandins and nitric oxide; and acting via circumventricular organs that lack blood-brain barrier function (28,51). Accordingly, in our study, alterations in gene expression of inflammatory molecules in the brain were detected on the background of significantly increased circulatory IL-1β, IL-6 and other cytokine levels, with a previously indicated mediating role in immune cell activation in the brain (49). The expression of TLR4 and cytokine receptors on microglia and astrocytes provide a triggering mechanism for the activation of NF-κB and other intracellular signal transduction molecular components attributed to IL-6, IL-1β and other cytokine production.
The transmission of peripheral cytokine signals to the brain can also occur through vagus nerve sensory fibers, and the role of this neural pathway has been shown to depend on the magnitude of the immune challenge (50,51). This neural signaling constitutes the afferent arm of the inflammatory reflex, which controls peripheral cytokine release and inflammation through efferent vagus nerve signaling, termed the cholinergic antiinflammatory pathway (1,52–57). This efferent vagus nerve-based antiinflammatory mechanism can be modulated by brain cholinergic signaling. Centrally acting acetylcholinesterase inhibitors and M1 mAChR agonists have been shown to suppress peripheral cytokine release, to improve survival in endotoxemia (20,21) and to have antiinflammatory effects in other inflammatory conditions (22,23,42). Despite the important role of brain cholinergic signaling in controlling peripheral inflammation, the intriguing question of whether lethal systemic inflammatory responses affect the brain cholinergic system has not been evaluated. Here we show that peripheral inflammation leads to brain region-specific alterations in the expression of genes, encoding important molecules linked to cholinergic signaling. These alterations occur in parallel with a lack of increased brain neuronal damage in LPS-treated mice, an observation consistent with a previous study reporting no necrotic tissue damage or other visible abnormalities in the brain after LPS (5 mg/kg, IV) injection in rats (47). We detected no major changes in brain Chat mRNA expression during peripheral inflammation, with the exception of a decrease in the striatum—an area with large cholinergic neurons (intraneurones), which do not project to other brain areas, and in which ACh is locally released (58). Apart from the striatum, brain cholinergic neurons are predominantly localized in basal forebrain cholinergic nuclei and in the mesopontine/upper brainstem pedunculopontine and laterodorsal tegmental nuclei in proximity or giving projections to the cortex, hippocampus, hypothalamus and thalamus (58–60). Extremely low Chat mRNA expression in projections to cerebellum and hippocampus (61) could explain the lack of measurable Chat gene expression in these brain regions. We also show here that Ache mRNA expression is decreased in the cortex and increased in the hippocampus. In addition to revealing a differential early impact of peripheral inflammation on Ache gene expression, these observations are interesting from the perspective of suggesting brain regions that can be targeted by centrally acting AChE inhibitors with previously indicated anti-inflammatory function (21,22,42). In addition, our data show the downregulation of Chrm1 (M1 mAChR) mRNA expression in the cortex and a dramatic decline of Chrm1 mRNA in the brainstem. We have previously shown that central M1 mAChR agonist treatments suppress proinflammatory cytokine release in the periphery (20,22). Together, these observations suggest that the antiinflammatory role of the brain M1 mAChR might, at least at the mRNA level, be mitigated during inflammation. Interestingly, decreased Chrm1 gene expression has been previously determined in cortex of patients with schizophrenia (62–64), a disease associated with central and peripheral inflammation with a proposed pathogenic role (17). The lack of measurable Chrm1 mRNA expression in the cerebellum is in line with the extremely low (<5%) contribution of this subtype to the total M1 mAChR number (65,66). The brain regions studied here have specialized physiological functions and most of them are involved in the central control of the autonomic nervous system regulation, including peripheral immune modulation (67,68). Future investigation of the impact of alterations in brain cholinergic and cytokine gene expression on specialized brain functions, attributed to a specific region, would contribute to further assessment of the importance of these alterations in inflammatory conditions.
Conclusion
These results reveal a previously unrecognized regional specificity of brain Il1b and Il6 mRNA expression in acute systemic inflammatory settings. This result is associated with differential increases in brain Gfap mRNA expression, consistent with the role of astrocytes in cytokine production.
In contrast, under these conditions, brain Iba1 mRNA expression is differentially decreased in parallel with morphological changes, indicative of microglial activation, thus highlighting an intriguing complexity of brain immunoregulatory events. In addition, our results show that brain cholinergic signaling known to play a role in the regulation of peripheral inflammation can in turn be specifically altered by a peripheral inflammatory insult. Inflammation mediates pathogenesis in a variety of human diseases, and murine and human endotoxemia have been widely used to investigate aspects of adverse inflammatory effects. One of these aspects is the impact of peripheral inflammation on brain immunoregulatory and neuronal function, with particular relevance to sepsis and septic shock, type 2 diabetes, rheumatoid arthritis and brain neurodegenerative diseases in which cholinergic signaling also is dysregulated. Therefore, our findings provide a platform for future studies of the brain immunoregulatory and neuromodulatory systems in inflammatory conditions, which may lead to developing new therapeutic approaches.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Tracey KJ. (2002) The inflammatory reflex. Nature. 420:853–9.
Pavlov VA, Tracey KJ. (2012) The vagus nerve and the inflammatory reflex: linking immunity and metabolism. Nat. Rev. Endocrinol. 8:743–54.
McInnes IB, Schett G. (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 7:429–42.
Diamond B, Volpe BT. (2012) A model for lupus brain disease. Immunol. Rev. 248:56–67.
Chavan SS, et al. (2012) HMGB1 mediates cognitive impairment in sepsis survivors. Mol. Med. 18:930–7.
Sonneville R, et al. (2013) Understanding brain dysfunction in sepsis. Ann. Intensive Care. 3:15.
Strachan MW, Reynolds RM, Marioni RE, Price JF. (2011) Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat. Rev. Endocrinol. 7:108–14.
Samaras K, Sachdev PS. (2012) Diabetes and the elderly brain: sweet memories? Ther. Adv. Endocrinol. Metab. 3:189–96.
Lampa J, et al. (2012) Peripheral inflammatory disease associated with centrally activated IL-1 system in humans and mice. Proc. Natl. Acad. Sci. U. S. A. 109:12728–33.
Hess A, et al. (2011) Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc. Natl. Acad. Sci. U. S. A. 108:3731–6.
Diamond B, Tracey KJ. (2011) Mapping the immunological homunculus. Proc. Natl. Acad. Sci. U. S. A. 108:3461–2.
Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT. (2009) Losing your nerves? Maybe it’s the antibodies. Nat. Rev. Immunol. 9:449–56.
Thaler JP, et al. (2012) Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 122:153–62.
Holmes C, et al. (2009) Systemic inflammation and disease progression in Alzheimer disease. Neurology. 73:768–74.
Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. (2011) Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 77:212–8.
Ferrari CC, Tarelli R. (2011) Parkinson’s disease and systemic inflammation. Parkinsons Dis. 2011:436813.
Meyer U, Schwarz MJ, Muller N. (2011) Inflammatory processes in schizophrenia: a promising neuroimmunological target for the treatment of negative/cognitive symptoms and beyond. Pharmacol. Ther. 132:96–110.
Andersson U, Tracey KJ. (2012) Reflex principles of immunological homeostasis. Annu. Rev. Immunol. 30:313–35.
Guarini S, et al. (2004) Adrenocorticotropin reverses hemorrhagic shock in anesthetized rats through the rapid activation of a vagal anti-inflammatory pathway. Cardiovasc. Res. 63:357–65.
Pavlov VA, et al. (2006) Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc. Natl. Acad. Sci. U. S. A. 103:5219–23.
Pavlov VA, et al. (2009) Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav. Immun. 23:41–5.
Ji H, et al. (2014) Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal. Immunol. 7:335–47.
Lee ST, et al. (2010) Cholinergic anti-inflammatory pathway in intracerebral hemorrhage. Brain Res. 1309:164–71.
Kelley KW, McCusker RH. (2014) Getting nervous about immunity. Semin. Immunol. 26:389–93.
Steinman L. (2012) Lessons learned at the intersection of immunology and neuroscience. J. Clin. Invest. 122:1146–8.
Capuron L, Miller AH. (2011) Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol. Ther. 130:226–38.
Bonaz B. (2013) Inflammatory bowel diseases: a dysfunction of brain-gut interactions? Minerva Gastroenterol. Dietol. 59:241–59.
Maier SF, Watkins LR. (2003) Immune-to-central nervous system communication and its role in modulating pain and cognition: implications for cancer and cancer treatment. Brain Behav. Immun. 17 (Suppl. 1):S125–31.
Doeuvre L, Plawinski L, Toti F, Angles-Cano E. (2009) Cell-derived microparticles: a new challenge in neuroscience. J. Neurochem. 110:457–68.
Farina C, Aloisi F, Meinl E. (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28:138–45.
Lampron A, Elali A, Rivest S. (2013) Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron. 78:214–32.
Garden GA, Moller T. (2006) Microglia biology in health and disease. J. Neuroimmune Pharmacol. 1:127–37.
Hanisch UK, Kettenmann H. (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10:1387–94.
Ransohoff RM, Perry VH. (2009) Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27:119–45.
Yirmiya R, Goshen I. (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25:181–213.
Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Studies, National Research Council of the National Academies. (2011) Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press.
Glowinski J, Iversen LL. (1966) Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J. Neurochem. 13:655–69.
Cikos S, Bukovska A, Koppel J. (2007) Relative quantification of mRNA: comparison of methods currently used for real-time PCR data analysis. BMC Mol. Biol. 8:113.
Steinman L. (2013) Inflammatory cytokines at the summits of pathological signal cascades in brain diseases. Sci. Signal. 6:e3.
Chen PC, et al. (2009) Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc. Natl. Acad. Sci. U. S. A. 106:2933–8.
Moss A, et al. (2007) Spinal microglia and neuropathic pain in young rats. Pain. 128:215–24.
Satapathy SK, et al. (2011) Galantamine alleviates inflammation and other obesity-associated complications in high-fat diet-fed mice. Mol. Med. 17:599–606.
Buttini M, Boddeke H. (1995) Peripheral lipopolysaccharide stimulation induces interleukin-1 beta messenger RNA in rat brain microglial cells. Neuroscience. 65:523–30.
Wolff S, et al. (2009) Endotoxin-induced gene expression differences in the brain and effects of iNOS inhibition and norepinephrine. Intensive Care Med. 35:730–9.
Thaler JP, et al. (2009) Atypical protein kinase C activity in the hypothalamus is required for lipopolysaccharide-mediated sickness responses. Endocrinology. 150:5362–72.
Madore C, et al. (2013) Early morphofunctional plasticity of microglia in response to acute lipopolysaccharide. Brain Behav. Immun. 34:151–8.
Buttini M, Limonta S, Boddeke HW. (1996) Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem. Int. 29:25–35.
Qin L, et al. (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 55:453–62.
Nadeau S, Rivest S. (1999) Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 93:1449–64.
Goehler LE, et al. (2000) Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton. Neurosci. 85:49–59.
Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. (2003) The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol. Med. 9:125–34.
Tracey KJ. (2009) Reflex control of immunity. Nat. Rev. Immunol. 9:418–28.
Pavlov VA, Tracey KJ. (2005) The cholinergic anti-inflammatory pathway. Brain Behav. Immun. 19:493–9.
Pavlov VA. (2008) Cholinergic modulation of inflammation. Int. J. Clin. Exp. Med. 1:203–12.
Huston JM, et al. (2009) Cholinergic neural signals to the spleen down-regulate leukocyte trafficking via CD11b. J. Immunol. 183:552–9.
Metz CN, Tracey KJ. (2005) It takes nerve to dampen inflammation. Nat. Immunol. 6:756–7.
Olofsson PS, et al. (2012) Alpha7 nicotinic acetylcholine receptor (alpha7nAChR) expression in bone marrow-derived non-T cells is required for the inflammatory reflex. Mol. Med. 18:539–43.
Woolf NJ. (1991) Cholinergic systems in mammalian brain and spinal cord. Prog. Neurobiol. 37:475–524.
Mesulam MM, Mufson EJ, Wainer BH, Levey AI. (1983) Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6). Neuroscience. 10:1185–201.
Hallanger AE, Wainer BH. (1988) Ascending projections from the pedunculopontine tegmental nucleus and the adjacent mesopontine tegmentum in the rat. J. Comp. Neurol. 274:483–515.
Ibanez CF, Ernfors P, Persson H. (1991) Developmental and regional expression of choline acetyl-transferase mRNA in the rat central nervous system. J. Neurosci. Res 29:163–71.
Dean B, McLeod M, Keriakous D, McKenzie J, Scarr E. (2002) Decreased muscarinic1 receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry. 7:1083–91.
Mancama D, Arranz MJ, Landau S, Kerwin R. (2003) Reduced expression of the muscarinic 1 receptor cortical subtype in schizophrenia. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 119B:2–6.
Scarr E, et al. (2013) Decreased cortical muscarinic M1 receptors in schizophrenia are associated with changes in gene promoter methylation, mRNA and gene targeting microRNA. Transl. Psychiatry. 3:e230.
Caulfield MP. (1993) Muscarinic receptors: characterization, coupling and function. Pharmacol. Ther. 58:319–79.
Hamilton SE, et al. (1997) Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. U. S. A. 94:13311–6.
Ulrich-Lai YM, Herman JP. (2009) Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 10:397–409.
Wrona D. (2006) Neural-immune interactions: an integrative view of the bidirectional relationship between the brain and immune systems. J. Neuroimmunol. 172:38–58.
Acknowledgments
This work was supported by the following grants from the National Institute of General Medical Sciences, National Institutes of Health: R01GM057226 (to KJ Tracey) and R01GM089807 (to VA Pavlov).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Silverman, H.A., Dancho, M., Regnier-Golanov, A. et al. Brain Region-Specific Alterations in the Gene Expression of Cytokines, Immune Cell Markers and Cholinergic System Components during Peripheral Endotoxin-Induced Inflammation. Mol Med 20, 601–611 (2014). https://doi.org/10.2119/molmed.2014.00147
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00147