
Abstract
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are inborn errors of heme biosynthesis with the same phenotype but resulting from autosomal recessive loss-of-function mutations in the ferrochelatase (FECH) gene and gain-of-function mutations in the X-linked erythroid-specific 5-aminolevulinate synthase (ALAS2) gene, respectively. The EPP phenotype is characterized by acute, painful, cutaneous photosensitivity and elevated erythrocyte protoporphyrin levels. We report the FECH and ALAS2 mutations in 155 unrelated North American patients with the EPP phenotype. FECH sequencing and dosage analyses identified 140 patients with EPP: 134 with one loss-of-function allele and the common IVS3-48T>C low expression allele, three with two loss-of-function mutations and three with one loss-of-function mutation and two low expression alleles. There were 48 previously reported and 23 novel FECH mutations. The remaining 15 probands had ALAS2 gain-of-function mutations causing XLP: 13 with the previously reported deletion, c.1706_1709delAGTG, and two with novel mutations, c.1734delG and c.1642C>T(p.Q548X). Notably, XLP represented ~10% of EPP phenotype patients in North America, two to five times more than in Western Europe. XLP males had twofold higher erythrocyte protoporphyrin levels than EPP patients, predisposing to more severe photosensitivity and liver disease. Identification of XLP patients permits accurate diagnosis and counseling of at-risk relatives and asymptomatic heterozygotes.
Similar content being viewed by others


Introduction
Erythropoietic protoporphyria (EPP) (OMIM 177000), the most common erythropoietic porphyria, is an autosomal recessive inborn error of heme biosynthesis because of the deficient activity (<35% of normal) of ferrochelatase (FECH) (E.C.4.99.1.1), the enzyme in the heme biosynthetic pathway that inserts ferrous iron into protoporphyrin to form heme. The enzyme deficiency results in the accumulation of protoporphyrin, primarily in marrow reticulocytes (1–3). Accumulated protoporphyrin is released from the marrow and circulating erythrocytes into the plasma, leading to its deposition in vascular cells as well as the liver, which is primarily responsible for its excretion.
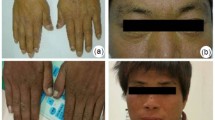
Clinically, EPP is characterized by the onset of acute cutaneous photosensitivity early in childhood (1,4). Exposure to sunlight or ultraviolet radiation for even a few minutes causes extreme pain that can be excruciating and is not relieved by narcotic analgesics. After prolonged sunlight exposure, the pain may persist for several days and full recovery may take a week. The exposed skin often becomes erythematous and edematous, but does not blister, although scarring may occur with multiple exposures.
Pathogenetically, sunlight photoactivates protoporphyrin in the superficial vessels, presumably triggering a singlet oxygen-mediated free radical reaction that results in cutaneous damage and painful symptoms. Protoporphyrin is insoluble in water and is not secreted into the urine, but is normally secreted into the bile. Excess biliary protoporphyrin is presumably responsible for the increased prevalence of gallstones in EPP patients. Accumulated hepatic protoporphyrin can precipitate in hepatocytes and bile canaliculi, causing hepatotoxicity, decreased bile formation and flow, and cholestatic liver failure in some patients (5).
The diagnosis of the EPP phenotype is based on the characteristically painful acute cutaneous photosensitivity and biochemical confirmation of markedly elevated levels of erythrocyte protoporphyrin. There is no specific treatment, so patients rigorously avoid sunlight exposure, which markedly limits their quality of life (6). Liver transplantation has been undertaken in patients with liver failure, and bone marrow transplantation has prevented recurrence (7,8). Recently, clinical trials of an α-melanocyte stimulating hormone analog (afamelanotide) to darken the skin have resulted in decreased photosensitivity, increased pain-free exposure to sunlight and improved quality-of-life for these patients (9,10).
FECH mutation analyses in Western Europeans with the EPP phenotype revealed that the phenotype is genetically heterogeneous (11,12). In Europe, about 92% of biochemically diagnosed patients with the EPP phenotype had autosomal recessive EPP with either two FECH loss-of-function mutations or, most often, one FECH loss-of-function mutation and the common FECH low expression mutation, IVS3-48T>C. The IVS3-48T>C allele creates a cryptic upstream acceptor site in intron 3 that modulates the alternative splicing of the normal FECH mRNA, generating about 25% of wild-type FECH mRNA and an mRNA that contains 63 intronic bases resulting in premature truncation and nonsensemediated decay (13). The IVS3-48T>C allele frequency varies widely with ethnicity and is ~10% among Western European Caucasians (14). Over 160 FECH loss-of-function mutations have been reported in the Human Gene Mutation Database (https://doi.org/www.hgmd.cf.ac.uk) (15).
Whatley et al. (11) reported that 2–4% of unrelated UK patients with the EPP phenotype did not have FECH mutations but had a two- or four-base deletion in the last exon of the X-linked erythroid-specific 5-aminolevulinate synthase (ALAS2) gene, either c.1699-1700delAT (p.M567EfsX2; designated AAT) or c.1706-1709delAGTG (p.E569GfsX24; designated ΔAGTG) (16). Both deletions altered the carboxyl-terminal region of the enzyme such that its activity and/or stability were increased. These gain-of-function mutations markedly increased the erythroid synthesis of 5-aminolevulinic acid (ALA) resulting in the erythroid accumulation of protoporphyrin and the EPP phenotype (16). This X-linked disorder is termed X-linked protoporphyria (XLP). Of note, 6% of the UK patients with the EPP phenotype did not have FECH or ALAS2 mutations (11), indicating the occurrence of additional genetic heterogeneity.
In EPP, >90% of the elevated erythrocyte protoporphyrin is not zinc chelated. In contrast, affected XLP males have ~15–50% zinc-chelated protoporphyrin, despite normal FECH activity. The heterozygous females in the European XLP families had increased erythrocyte protoporphyrin and photosensitivity (16), although mutation-confirmed XLP heterozygotes can be asymptomatic, as expected for X-linked traits (16,17). In addition, about 17% of XLP patients had overt liver disease (16), higher than that reported for EPP (16,18). To date, in limited studies of unrelated North American patients with EPP (19), only two with XLP were reported (20). Here, we report the FECH and ALAS2 mutations in 155 unrelated North American patients with the EPP phenotype. Of these, ~90% (140 of 155) had FECH mutations causing EPP, including 23 novel loss-of-function lesions. All 15 remaining patients had ALAS2 exon 11 mutations causing XLP, including 13 with the ΔAGTG deletion and two with novel exon 11 lesions, a deletion and a nonsense mutation. Thus, about 10% of unrelated North American patients with the EPP phenotype had XLP, five times that reported for UK EPP phenotype patients (11), and at least twice that found in Western European patients with the EPP phenotype (12). Clearly, genotyping patients with the EPP phenotype is important to identify at-risk XLP family members, particularly the asymptomatic heterozygous females who can transmit the disease-causing genes to their offspring.
Materials and Methods
Patients with the EPP phenotype were referred for mutation analyses by the Porphyrias Consortium of the National Institutes of Health (NIH)-sponsored Rare Disease Clinical Research Network or by their physicians. Peripheral bloods or cheek swabs were obtained with informed consent. Erythrocyte protoporphyrin levels were determined by commercial laboratories or at the Porphyria Center (University of Texas Medical Branch, Galveston, TX, USA). Clinical information, including medical histories and family pedigrees, symptoms and results of biochemical testing were obtained. FECH and ALAS2 exon 11 mutation analyses were performed by the Porphyria Molecular Diagnostic Testing Laboratory at the Mount Sinai School of Medicine in New York City.
Genomic DNAs were extracted using the QIAGEN® DNA Isolation Kit (QIAGEN, Valencia, CA, USA). For FECH gene analyses, ~500 base pairs (bp) of the promoter, all exons and ~30–40 bp adjacent intronic or flanking sequences were amplified by using the GeneAmp PCR System 9700 (PE Applied Biosystems, Foster City, CA, USA) with previously obtained oligonucleotide primers (14) with the following modification. Primer 9R had a known intronic polymorphism (rs58628398 in the NCBI dbSNP [single nucleotide polymorphism database]), which deleted seven bases (AGGACAC) in 10% of European, 5% of African and 0% of East Asian chromosomes (21). Therefore, a new 9R primer was synthesized to avoid the polymorphism (Supplementary Table S1). The IVS3-48T>C region was also amplified with exon 4, and the T>C genotype was determined (22). For ALAS2, the entire exon 11 coding region and ~30–40 bp intronic and 3′-un-translated sequence were amplified by using the indicated polymerase chain reaction (PCR) primers (Supplementary Table S1). Sequences were analyzed by using Sequencer version 4.8 software (Gene Codes) (reference sequences: FECH transcript NM_000140.3; FECH gene: NG_008175; ALAS2 transcript GenBank NM_000032.4). Gene dosage analysis to detect large deletions that included entire exons was performed as described (23). Splicing mutations were analyzed by using SplicePort to predict effects on FECH function (https://doi.org/spliceport.cbcb.umd.edu/) (24).
Additional genotyping included FECH intragenic single nucleotide polymorphisms (SNPs). These SNPs were 5′-252A>G, tightly linked to IVS1-23C>T and IVS3-48T>C, as well as c.798C>G (P266P) linked to c.921A>G. The unlinked SNPs included c.287G>A (p.R96Q), IVS6-198C>T, IVS8+94T>A, IVS10-64G>A and 3′+248C>T.
Structural Characterization of FECH Missense Mutations
The potential impact of each novel missense mutation was evaluated by visual inspection of the environment of the altered residue in the FECH crystal structures of the catalytic structural states, that is, “open” (PDB #2QD3), “closed” with bound substrate (2QD1) and “release” with bound product (2QD2). Attention was directed to identify residues for which available structural data indicate a role in catalysis and/or macrocycle binding. The propensity for the observed mutations to be tolerated or not were assessed by the SIFT (https://doi.org/sift-dna.org) and PolyPhen programs (25,26). Low scores indicate deleterious mutations predicted by the SIFT program, whereas high scores (1.0 = max) indicate deleterious mutations predicted by the PolyPhen program. The PyMol program was used to produce a figure highlighting the locations of the novel FECH mutations (The PyMOL Molecular Graphics System, Version 1.2; Schrödinger LLC, New York, NY, USA).
All supplementary materials are available online at https://doi.org/www.molmed.org .
Results
Mutation Analysis of the FECH Gene in Patients with the EPP Phenotype
Genomic DNAs from 155 North American patients with the EPP phenotype (that is, photosensitivity and markedly elevated erythrocyte protoporphyrin levels) were analyzed for FECH loss-of-function mutations and the IVS3-48T>C allele by PCR amplification and sequencing. Of the 155 patients, 140 (~90%) had FECH loss-of-function mutations causing autosomal recessive EPP. All 15 remaining patients were found to have ALAS2 exon 11 gain-of-function mutations causing XLP (see below). Among the 140 patients with EPP, three had two loss-of-function FECH alleles: genotypes c.348A>C (p.S151F)/c.1001C>T (p.P334L), c.643C>T (p.R219W)/c.901_902delTG (p.W301Afs*22) and c.820G>A (p.D274N)/c.913G>T (a splicing mutation). Three patients were homozygous for the low-activity allele but also had a second mutation on one allele [genotypes: c.485G>A(G162E)+IVS3-48T>C/IVS3-48T>C, c.577C>T(Q193X)+IVS3-48T>C/IVS3-48T>C and c.794T>G(L265R)+IVS3-48T>C/IVS3-48T>C]. The remaining 132 patients had a loss-of-function mutation on one allele and the low-expression IVS3-48T>C lesion on the other. Altogether, 71 FECH mutations were detected, including 48 previously reported and 23 novel mutations (Figures 1 and 2, Table 1).

FECH loss-of-function mutations causing autosomal-recessive EPP. Mutations in black and blue type were previously reported and listed in the Human Gene Mutation Database, release date 29 June 2012. Previously published mutations in blue were those found in the patients of this study. Novel mutations are indicated in red. Asterisks indicate splicing mutations caused by exonic mutations. The gene depicted in the top line shows exonic mutations, and mutations in intronic sequences are shown on the bottom line.

Large FECH deletions and complex deletion/insertion rearrangements causing EPP. The Human Gene Mutation Database (HGMD) accession numbers are provided, and the extent of the deletion is listed to the right of each deleted region. In parentheses, the number of patients previously reported with the deletion is followed by the number of patients with the deletion in the present study. Novel mutations are those with only one incidence in this study. Question marks indicate the unknown positions of the breakpoints. Alu sequences and their orientations are indicated with open arrows. The published breakpoints have been mapped to the FECH sequence provided in GenBank accession number NG_008175 from the February 2009 HG-19 human genome assembly. HGMD accession numbers for mutations with identical breakpoints have been listed together with the respective deletion region.
Previously Reported FECH Mutations
The previously reported FECH mutations, identified in 111 patients, are indicated in Figures 1 and 2 and included 9 missense, 5 nonsense, 12 small insertions or deletions, 18 splicing mutations and 4 large deletions, with the latter 4 detected by SNP homozygosity and identified by breakpoint sequencing (Figure 2). Among the most common of the previously reported mutations were the following: IVS3+2T>G found in 13 patients, c.1232G>A (p.C411G) in 12 patients, the large deletion del10,379in-sTTCA (23) in 6 patients and splicing defects IVS1+1G>A, c.913G>T and c.1135A>T each in 6 patients. These mutations were all present in trans with the IVS3-48T>C allele. Of interest, one patient had two spicing mutations (IVS5+1G>T and IVS5-3C>T) on the same allele and the IVS3-48T>C mutation on the other, as defined by family studies. These two mutations were reported previously, but singly, in different probands (15).
Reverse transcriptase (RT)-PCR studies of the previously suspected splicing mutation c.1077G>A (p.E359E) (27) demonstrated that the G>A transition in the last nucleotide of exon 9 altered the 5′ acceptor site consensus sequence from GAGgtaaat to GAAgtaaat caused 100% exon 9 skipping (data not shown), as predicted by the SplicePort program (24).
Novel FECH Mutations
The 23 novel FECH loss-of-function mutations (Table 1, Figures 1 and 2) included eight missense and nine nonsense mutations, three splice site lesions, one small deletion and two complex large deletion/insertions. Of the 17 novel missense and nonsense mutations, one (p.R215W) occurred at a cytosine guanine dinucleotide (CpG), a known hot spot for mutation (28), of which there are 38 in the FECH coding sequence.
Of the eight novel missense mutations, six occurred at residues having 96–100% amino acid conservation in 26 species from insects to humans (Supplementary Table S2). The novel FECH missense mutations were assessed on the FECH 3D crystal structure (29) (Figure 3) to determine their predicted affects on the structure and/or function of the enzyme (Table 2). Although none was predicted to be tolerated by both the SIFT and PolyPhen programs (Table 2), on the basis of the scores for both programs, the R215W mutation was relatively more tolerated, consistent with its low conservation (27%).

FECH three-dimensional structure and the location of novel deleterious FECH mutations. The crystal structure of one of the homodimeric FECH monomers is shown by using secondary structure cartoons in green and the surface in light gray. The view is from the side of the protein that is membrane-bound, looking into the active site region containing protoporphyrin IX and the iron-sulfur cluster. The residues changed by the novel mutations are shown as space-filling spheres and are the indicated native residues, not the mutations.
The novel small deletion c.663_665delGAG (p.W221_S222delinsC) (Figure 1) lost three bases across two codons, thereby substituting a cysteine for tryptophan in codon 221 and deleting the serine codon 222, after which it retained the normal amino acid sequence. Two “sequencing cryptic” deletions were identified by dosage analyses. These studies revealed two additional novel large rearrangements in three patients, involving exons 3 and 4 or exons 7, 8 and 9 (Figure 2).
FECH Single Nucleotide Polymorphisms
Of the 906 single nucleotide polymorphisms (SNPs) in the FECH gene recorded in the dbSNP database, about 125 were common (minor allele frequency [MAF] >5%). Of these, 11 were characterized in the EPP phenotype patients, since they were present in the promoter and exonic amplicons that were sequenced. These included rs17963905 (5′-252A>G) and rs7243988 (IVS1-23C>T), which are usually linked to the IVS3-48T>C mutation. Five other polymorphisms were relatively frequent (minor allele frequencies of 14–31% in Caucasians), including three exonic polymorphisms: rs1041951 (c.287G>A, p.R96Q), rs536765 (c.798C>G; P266P) and rs536506 (c.921A>G, p.P307P). Monitoring these SNPs for apparent homozygosity was used to detect possible “sequencing cryptic” FECH gene deletions that were further delineated by FECH gene dosage analyses (23).
ALAS2 Mutations Causing XLP
Of the 15 unrelated probands who did not have two FECH mutations causing EPP, 13 had the previously reported four-base ALAS2 deletion, ΔAGTG (16); one had a novel c.1734delG (p.Q581SfsX13) single-base deletion (designated AG); and one had a novel nonsense mutation, c1642C>T (p.Q548X). With the exception of an African-American and a Pacific Islander, the ΔAGTG XLP patients had ancestors of Western European origin, particularly from the United Kingdom. The novel AG mutation altered the seven carboxyl-terminal wild-type amino acids (QYVTTYA) to SMSPPMP and extended the enzyme polypeptide sequence by five residues (EKPAA) (Figure 4). Of note, the last 14 altered carboxyl-terminal residues of the elongated ΔAGTG and AG mutations were identical. The c.1642C>T (p.Q548X) nonsense mutation altered ALAS2 codon 548 from CAG encoding a glutamine to TAG, a chain termination signal that prematurely deleted the 40 wild-type carboxyl-terminal residues (548–587) of the enzyme polypeptide.

XLP mutations and gene structure. (A) ALAS2 loss-of-function and gain-of-function mutations causing X-linked sideroblastic anemia and EPP. Mutations in black and blue are those reported in the Human Gene Mutation Database, release date 29 June 2012. The mutations in bold are published mutations also found in patients of this study. The mutations in bold italics are the novel mutations. (B) Variations in the C-terminal sequences of the XLP gain-of-function mutations. The partial wild-type exon 11 ALAS2 sequence is boxed. The hybrid sequences of wild-type ALAS2and the sequence following the mutation sites are aligned below the wild-type sequence. The termination codons are denoted by asterisks. Note that for the pE569GfsX24 and pQ581SfsX13 mutations, the last 12 mutated residues are identical to each other and different from the wild-type.
EPP and XLP Genotype and Protoporphyrin Levels
Assuming that the erythrocyte and plasma protoporphyrin concentrations can be correlated with (or predict) disease severity, the available erythrocyte and plasma levels in EPP and XLP patients were stratified by mutation type (Table 3). For 120 EPP patients, the mean and median erythrocyte protoporphyrin levels were 2,220 and 1,740 µg/dL, respectively, and ranged from 407 to 8,240 µg/dL, with the highest levels mostly in patients with liver disease or liver transplants (Table 3, Supplementary Figure S1). The mean erythrocyte protoporphyrin levels in males and females were 2,113 and 2,344 µg/dL, respectively, which were similar. There were no effects of age on erythrocyte protoporphyrin levels (data not shown), but there was a significant increase in risk of liver disease above a threshold of 2,500–3,000 µg/dL. The mean percentage of erythrocyte metal-free protoporphyrin for the different FECH genotypes in Table 3 ranged from 92 to 97% of the total erythrocyte protoporphyrin with a range of 84 to 98%. The 25 patients with missense mutations (including unrelated patients with the same mutation), which may have residual activity, had the lowest mean erythrocyte protoporphyrin concentration (1,430 ± 660 µg/dL), whereas patients with a splicing defect, nonsense mutation or small or large deletion had higher mean concentrations ranging from 2,170 to 3,580 µg/dL. Similarly, the mean plasma porphyrin levels were lowest in patients with missense mutations (5.7 µg/dL) and higher in the other FECH genotype groups (8.7–14.0 µg/dL). Of note, the patient with two missense alleles (S151F and P334L) and the patient with two low-activity alleles (one having a missense mutation [G162E]) had concentrations of 3,210 and 7,750 µg/dL, respectively.
For eight XLP male patients with the ΔAGTG mutation, the mean erythrocyte protoporphyrin level was 5,153 µg/dL and the range was from 2,293 to 10,650 µg/dL, about twofold higher than that in the 120 EPP patients (Supplementary Figure S1). Notably, the mean level of free erythrocyte protoporphyrin for patients with the ΔAGTG mutation was 67%, indicating an average ratio of 67/34 for free versus zinc-chelated protoporphyrin. Of note, the Q548X proband had a ratio of 92/8, which was similar to those of EPP patients.
Discussion
Fifty years ago, Magnus et al. (30) reported the first patients with EPP, described the photo-induced cutaneous manifestation as “solar urticaria” and identified the presence of accumulated protoporphyrin in the patient’s erythrocytes and stools. Their description of the photo-induced cutaneous manifestations and the protoporphyrin accumulation not only characterized the clinical and biochemical phenotype of the disease, but remains today the basis for its diagnosis (1). Subsequently, the partially deficient FECH activity (<35% of normal in cultured cells and < 25% of normal in bone marrow) was shown in EPP patients (31,32).
Early studies of the inheritance of EPP suggested that the disease was either an autosomal-dominant trait with reduced penetrance (33) or an autosomal-recessive disorder (34). Initially, EPP was thought to be an autosomal-dominant disorder when the FECH cDNA and genomic sequences were isolated and characterized (35,36) and the causative mutations were identified (15,23,37,38) (https://doi.org/www.hgmd.cf.ac.uk). Over 90% of symptomatic patients had only one loss-of-function FECH mutation supporting autosomal-dominant inheritance. The small percentage of EPP patients with two FECH mutations was classified as having “recessive” EPP. The fact that symptomatic patients had 15–35% of normal activity was a dilemma that led to various hypotheses concerning the molecular genetics of the disease (1).
The inheritance was clarified by Gouya et al. (13,14,22,39), who discovered the common intronic FECH low expression allele. With the identification of this allele, it was quickly realized that over 90% of EPP patients had a loss-of-function FECH mutation on one allele and the IVS3-48T>C mutation on the other (14), consistent with autosomal-recessive inheritance.
To further complicate the inheritance, it was noted that loss-of-function FECH mutations were absent in about 5–10% of symptomatic patients with elevated erythrocyte protoporphyrin. The inheritance pattern in some of these patients suggested X-linkage. While loss-of-function mutations in the ALAS2 gene cause X-linked sideroblastic anemia (Figure 4) (40), Whatley et al. in 2008 (16) reported two deletions in exon 11 of the ALAS2 gene that resulted in frameshift mutations and increased ALAS2 activity. Notably, both affected and asymptomatic heterozygotes for the same ALAS2 gain-of-function mutation were reported, demonstrating that it is not a dominant disorder (17).
Here, we report mutation analyses of 155 unrelated North American patients with the EPP phenotype that identified 140 with two FECH mutations causing EPP and 15 with ALAS2 mutations causing XLP. These studies identified 23 novel FECH loss-of-function mutations and two novel ALAS2 exon 11 mutations: a small deletion (AG) and a nonsense mutation (p.Q548X). These novel mutations add to the previously reported 164 FECH (Figures 1 and 2, Table 1) and two ALAS2 exon 11 mutations causing XLP (and over 60 loss-of-function ALAS2 mutations that cause X-linked sideroblastic anemia) (Figure 4). Of the 140 patients with EPP, most (96%) had a loss-of-function FECH allele and the IVS3-48T>C allele, similar to the findings in Western Europe, where ~95% of EPP patients had a loss-of-function mutation and the IVS3-48T>C allele, and only ~5% had two loss-of-function alleles (11,16).
Of the North American EPP patients with the 23 novel FECH mutations, eight had missense mutations that mostly replaced highly conserved amino acids. Structural modeling predicted that these mutations would likely alter FECH function and/or stability (Table 2, Figure 3). The FECH coding sequence contains 38 CpGs, which are mutational hot spots for C→T or G→A transitions (29). However, of the 60 previously published missense and nonsense mutations, only eight occurred at CpGs, whereas only one of the 17 novel base substitutions reported here was at a CpG. Thus, EPP mutations do not appear to favor these hot spots. Of the 44 previously reported and three novel splicing mutations, 1–12 occurred in each of the 10 introns (Figure 1).
Gene dosage analyses were required to identify four additional large FECH deletions involving entire exons and not detectable by sequence analysis, thereby confirming the EPP diagnoses in these patients. In total, we found about 8% of the EPP phenotype patients had large deletions, similar to the frequency of large deletions (10%) reported in European patients (11,23).
Of the 15 patients with the EPP phenotype who did not have FECH mutations, all had ALAS2 exon 11 gain-of-function mutations. Thirteen had the ΔAGTG deletion and two had novel mutations: a single-base deletion (c.1734delG), which altered the nine carboxyl-terminal amino acids and elongated the enzyme polypeptide by five residues, and a nonsense mutation (p.Q548X), which deleted the terminal 40 amino acids (Figure 4). For these mutations, the mechanism(s) by which the structural changes are responsible for their increased enzymatic activity and/or stability remain to be determined. It has been hypothesized that the carboxy-terminal amino acids may normally restrict release of the product, since it was demonstrated that numerous hyperactive variants created by mutations in this region resulted in increased rates of conformational change leading to increased ALA synthesis (41). It is notable that the human ALAS2 exon 11 encodes 54 terminal amino acids, of which the terminal 30 amino acids are only present in vertebrates and that this region was recently shown to be required for binding of the precursor enzyme succinyl-CoA synthetase (42). In addition, other mutations in this region can cause X-linked sideroblastic anemia (Figure 4) by alterations in enzyme stability in vivo (43). Thus, these terminal residues play a critical role in the regulation of erythroid heme biosynthesis in humans.
Notably, the three exon 11 deletions (ΔAGTG, AAT and AG) all occur within a span of 36 nucleotides. The ΔAGTG deletion was at a direct repeat (AGTG AGTG), which only occurred at this location in the coding region of the human ALAS2 gene. The AAT mutation occurs three bases upstream from ΔAGTG, and the ΔG mutation occurs downstream in a region containing four guanine residues followed by four cytosines. The mutational mechanism responsible for the ΔAGTG and ΔG mutations may be homologous, but unequal, with crossing-over of their repeats during recombination. Comparison of the distribution of the erythrocyte protoporphyrin levels in the available EPP and XLP patients (Supplementary Figure S1) indicated a higher concentration in the XLP compared with EPP patients. The finding of higher protoporphyrin levels may correlate with disease severity (particularly increased cutaneous photosensitivity and risk for liver damage, with the latter due to delivery of excess hepatotoxic protoporphyrin to the liver for biliary excretion). Thus, physicians should closely monitor liver function in EPP and especially in XLP patients. Notably, the erythrocyte zinc protoporphyrin in XLP males and most XLP females averaged >30% of the total protoporphyrin, compared with ~5% for EPP patients, a previously noted finding of diagnostic value (16).
Although all 155 North American patients with the EPP phenotype were found to have a causative FECH or ALAS2 mutation, that was not the case in the Western European patients (16), where FECH or ALAS2 mutations were not detected in ~5% of patients with the EPP phenotype. This finding suggested that other gene defects may cause the EPP phenotype (11,12). Notably, the percentage of patients with the EPP phenotype who had XLP in our North American cohort was ~10%, which was two- to fivefold greater than that in the Western European studies (11,16).
Conclusion
ALAS2 mutation analysis should be undertaken in patients with the EPP phenotype who have no loss-of-function FECH mutations. Identification of the XLP patients will permit accurate diagnosis of at-risk family members, particularly asymptomatic heterozygotes who have a 50% risk for affected male offspring. Moreover, the finding of higher mean erythrocyte protoporphyrin levels in XLP probands compared with EPP probands, suggests that XLP probands may have a more severe disease course (particularly, an increased risk of liver damage).
Disclosure
KE Anderson received funding for educational programs from Lundbeck. HL Bonkovsky served as a consultant to Boehringer-Ingelheim, Clinuvel, Lund-beck and Novartis and has received research support from Clinuvel, Novartis and Vertex. RJ Desnick, KE Anderson, DM Bissell, J Bloomer, HL Bonkovsky and JD Phillips are principal investigators on the Phase 2 and 3 clinical trials for Clinuvel.
References
Anderson KE, Sassa S, Bishop DF, Desnick RJ. (2001) Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: The Metabolic and Molecular Bases of Inherited Disease. 8th edition. Scriver CR, et al. (eds.) New York, McGraw-Hill, pp. 2991–3062.
Lecha M, Puy H, Deybach JC. (2009) Erythropoietic protoporphyria. Orphanet. J. Rare. Dis. 4:19.
Todd DJ. (1994) Erythropoietic protoporphyria. Br. J. Dermatol. 131:751–766.
Anderson KE, et al. (2005) Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 142:439–50.
Gross U, Frank M, Doss MO. (1998) Hepatic complications of erythropoietic protoporphyria. Photodermatol. Photoimmunol. Photomed. 14:52–7.
Frank J, Poblete-Gutierrez P. (2011) Delayed diagnosis and diminished quality of life in erythropoietic protoporphyria: results of a cross-sectional study in Sweden. J. Intern. Med. 269:270–4.
McGuire BM, et al. (2005) Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 11:1590–6.
Rand EB, et al. (2006) Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 118:e1896–9.
Harms J, Lautenschlager S, Minder CE, Minder EI. (2009) An alpha-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. N. Engl. J. Med. 360:306–7.
Minder EI. (2010) Afamelanotide, an agonistic analog of alpha-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert Opin. Investig. Drugs. 19:1591–602.
Whatley SD, et al. (2010) Molecular epidemiology of erythropoietic protoporphyria in the U.K. Br. J. Dermatol. 162:642–6.
Schmitt C, Ducamp S, Gouya L, Deybach JC, Puy H. (2010) Erythropoietic protoporphyria: one disease, two genes and three molecular mechanisms. Pathol. Biol. (Paris). 58:372–80.
Gouya L, et al. (1996) Modulation of the phenotype in dominant erythropoietic protoporphyria by a low expression of the normal ferrochelatase allele. Am. J. Hum. Genet. 58:292–9.
Gouya L, et al. (2006) Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. Am. J. Hum. Genet. 78:2–14.
Stenson PD, et al. (2009) The Human Gene Mutation Database: 2008 update. Genome Med. 1:13.
Whatley SD, et al. (2008) C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 83:408–14.
Di Pierro E, Brancaleoni V, Tavazzi D, Cappellini MD. (2009) C-terminal deletion in the ALAS2 gene and X-linked dominant protoporphyria. Haematologica. 94:315.
Bloomer JR. (1997) Hepatic protoporphyrin metabolism in patients with advanced protoporphyric liver disease. Yale J. Biol. Med. 70:323–30.
Risheg H, Chen FP, Bloomer JR. (2003) Genotypic determinants of phenotype in North American patients with erythropoietic protoporphyria. Mol. Genet. Metab. 80:196–206.
Wang Y, et al. (2011) Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria. Exp. Hematol. 39:784–94.
(2010) A map of human genome variation from population-scale sequencing. Nature. 467:1061–73.
Gouya L, et al. (2002) The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat. Genet. 30:27–8.
Whatley SD, et al. (2007) Gene dosage analysis identifies large deletions of the FECH gene in 10% of families with erythropoietic protoporphyria. J. Invest. Dermatol. 127:2790–4.
Dogan RI, Getoor L, Wilbur WJ, Mount SM. (2007) SplicePort: an interactive splice-site analysis tool. Nucleic Acids Res. 35:W285–91.
Sim NL, et al. (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40:W452–7.
Adzhubei IA, et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods. 7:248–9.
Wang X, et al. (1999) Haplotype analysis of families with erythropoietic protoporphyria and novel mutations of the ferrochelatase gene. J. Invest. Dermatol. 113:87–92.
Cooper DN, Youssoufian H. (1988) The CpG dinucleotide and human genetic disease. Hum. Genet. 78:151–5.
Wu CK, et al. (2001) The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 8:156–60.
Magnus IA, Jarrett A, Prankerd TA, Rimington C. (1961) Erythropoietic protoporphyria: a new porphyria syndrome with solar urticaria due to protoporphyrinaemia. Lancet. 2:448–51.
Bonkowsky HL, Bloomer JR, Ebert PS, Mahoney MJ. (1975) Heme synthetase deficiency in human protoporphyria: demonstration of the defect in liver and cultured skin fibroblasts. J. Clin. Invest. 56:1139–48.
Bottomley SS, Tanaka M, Everett MA. (1975) Diminished erythroid ferrochelatase activity in protoporphyria. J. Lab. Clin. Med. 86:126–31.
Reed WB, et al. (1970) Erythropoietic protoporphyria: a clinical and genetic study. JAMA. 214:1060–6.
Went LN, Klasen EC. (1984) Genetic aspects of erythropoietic protoporphyria. Ann. Hum. Genet. 48:105–17.
Nakahashi Y, Taketani S, Okuda M, Inoue K, Tokunaga R. (1990) Molecular cloning and sequence analysis of cDNA encoding human ferrochelatase. Biochem. Biophys. Res. Commun. 173:748–55.
Taketani S, Inazawa J, Nakahashi Y, Abe T, Tokunaga R. (1992) Structure of the human ferrochelatase gene: exon/intron gene organization and location of the gene to chromosome 18. Eur. J. Biochem. 205:217–22.
Lamoril J, et al. (1991) Human erythropoietic protoporphyria: two point mutations in the ferrochelatase gene. Biochem. Biophys. Res. Commun. 181:594–9.
Elder GH, et al. (2009) The molecular genetics of erythropoietic protoporphyria. Cell. Mol. Biol. (Noisy-le-grand). 55:118–26.
Gouya L, et al. (1999) Inheritance in erythropoietic protoporphyria: a common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Blood. 93:2105–10.
Cotter PD, Baumann M, Bishop DF. (1992) Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proc. Natl. Acad. Sci. U. S. A. 89:4028–32.
Hunter GA, Ferreira GC. (2011) Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta. 1814:1467–73.
Bishop DF, Tchaikovskii V, Hoffbrand AV, Fraser ME, Margolis S. (2012) X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the beta-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 287:28943–55.
Kadirvel S, et al. (2012) The carboxyl-terminal region of erythroid-specific 5-aminolevulinate synthase acts as an intrinsic modifier for its catalytic activity and protein stability. Exp. Hematol. 40:477–6, e471.
Acknowledgments
The authors thank the Porphyrias Consortium coordinators for their assistance, Nicole Kelly for manuscript preparation and Jungmin Kim for technical assistance. This research was supported in part by grants from the American Porphyria Foundation and grants from the NIH, including a research grant (5 R01 DK026824) and a grant (1 U54 DK083909) for the Porphyrias Consortium of the NIH Rare Diseases Clinical Research Network. The views expressed in written materials or publications do not necessarily reflect the official policies of the Department of Health and Human Services.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
MB, DD, and DFB contributed equally to this work.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Balwani, M., Doheny, D., Bishop, D.F. et al. Loss-of-Function Ferrochelatase and Gain-of-Function Erythroid-Specific 5-Aminolevulinate Synthase Mutations Causing Erythropoietic Protoporphyria and X-Linked Protoporphyria in North American Patients Reveal Novel Mutations and a High Prevalence of X-Linked Protoporphyria. Mol Med 19, 26–29 (2013). https://doi.org/10.2119/molmed.2012.00340
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2012.00340