
Abstract
The operation of combined mass spectrometry and electrochemistry setups has recently become a powerful approach for the in situ analysis of gas evolution in batteries. It allows for real-time insights and mechanistic understanding into different processes, including battery formation, operation, degradation, and behavior under stress conditions. Important information is gained on the safety and stability window as well as on the effect of protecting strategies, such as surface coatings, dopings, and electrolyte additives. This review primarily aims at summarizing recent findings on the gassing behavior in different kinds of liquid- and solid-electrolyte-based batteries, with emphasis placed on novel cathode-active materials and isotope labeling experiments, to highlight the relevance of in situ gas analysis for elucidation of reaction mechanisms. Various instrumental and experimental approaches are presented to encourage and inspire both novices and experienced scientists in the field.
Graphical abstract

Similar content being viewed by others

Introduction
As the increasing societal and commercial demand for large-scale energy storage and electric vehicles continues to promote innovations in battery research, new or improved materials and concepts are in the focus of scientific interest. Main motives for these innovations are improvements in one or more of energy density, longevity, safety, sustainability, and ultimately costs.
For current generation lithium-ion batteries (LIBs) with graphite as anode material, the focus is on the tradeoff between larger energy density and lower costs by increased Ni content in the layered transition metal oxide cathode-active material (CAM) and reduced cycle life resulting from the change in composition and reactivity. While incremental improvements, mostly from suppressing degradation mechanisms, such as particle fracture (exposing additional [reactive] surface area to the electrolyte) or phase transitions/transformations, are made to these CAMs, their Ni content approaches 100%, thus limiting the possibilities for further energy density increases at the positive electrode side [1, 2].
For improvements beyond, Li-rich CAMs, both layered and with rock-salt-type structures, are currently being discussed. These materials gain additional capacity by involving lattice oxygen in the redox, but the often limited reversibility of this reaction remains a challenge [3,4,5].
A relatively large increase in energy density at the anode side can be achieved by substitution of the graphite as standard electrode material by Li metal. However, because of dendrite formation, cells with Li metal anodes are prone to failure. The solid-state battery (SSB) promises to solve this problem by replacing the liquid electrolyte with a solid electrolyte (SE), which also reduces the cell’s flammability [6]. For this reason, bulk SSBs are receiving increasing interest lately. Nevertheless, cell manufacturing and especially the development of the key component, an SE with favorable mechanical properties as well as high (electro)chemical stability and Li-ion conductivity, still require additional research in order to achieve SSBs with competitive energy and power densities [7].
Meanwhile, the much higher abundance of sodium compared to lithium makes sodium-ion batteries (SIB) a suitable candidate in the field of post-LIB technologies. While the CAMs introduced so far can rarely match their lithium equivalents in terms of specific capacity, the lower costs and environmental impact make SIBs promising for large-scale energy storage [8, 9].
For all battery materials and concepts mentioned so far, key to their continuous development and improvement is a thorough understanding of (side) reaction mechanisms at play during formation, operation, and degradation. In situ techniques allow for the characterization of materials within the relevant environment of a battery cell, while operando techniques go one step further and enable real-time monitoring during cycling operation (i.e., within a dynamic, operating system). Thus, it comes as no surprise that various review papers on in situ characterization studies are available in the literature [10,11,12,13]. However, most in situ or operando methods applied to battery materials, such as (electron) microscopy, spectroscopy and X-ray-based techniques, are restricted in their working principle to condensed matter or even crystalline phases only [13, 14].
As for the materials and concepts discussed above, reactions that include gas evolution are relevant to their evaluation, such as the surface (im)purity, structural (in)stability, interface formation, and electrolyte degradation. The in situ gas analysis is therefore a useful addition to routine electrochemical experiments and other analytical techniques for better understanding of reaction mechanisms. To this end, mass spectrometry (MS) is typically performed simultaneously with battery operation. While existing reviews focus mostly on instrumentation [15] and standard materials [16], in this work, an overview of recent developments in the field of in situ instrumentation and gas evolution studies is given, with emphasis placed on novel CAMs (with layered and rock-salt or related lattice structures) and cell concepts.
In situ gas evolution measurements
Instrumentation
The combination of electrochemical testing and MS allows for a variety of experiments, with in situ measurements usually referred to as differential electrochemical mass spectrometry (DEMS) [17, 18]. In the battery context, pioneering work has been reported by Imhof et al. as early as 1998, studying the solid-electrolyte interphase (SEI) formation on graphite by using a porous electrode and a hydrophobic membrane separating the cell from applied vacuum to selectively extract evolved gases from the working electrode [19]. The development history of in situ MS in the battery field has been described in more detail by Schiele et al. [15] and Lundström et al. [20]. As the hydrophobic membrane can be passed by organic solvents, especially those of low boiling point, a stream of carrier gas can be used instead to extract the evolving gasses from the cell’s headspace [21]. The use of carrier gas comes at the price of stressing the cell by electrolyte depletion, because the electrolyte solvents are carried out of the cell, albeit at a slower rate, and also detected by the mass spectrometer. Berkes et al. implemented both a bubbler to saturate the carrier gas with electrolyte solvents before entering the cell and a cold trap near the outlet of the cell to remove them from the carrier gas again [22]. While reducing noise and allowing for longer battery operation time, the introduction of carrier gas and cold trap affects the achievable time resolution. For this reason, Jusys et al. chose to keep working with a fluoropolymer membrane in their recent setup [23].
An alternative to carrier gas or membranes is the use of closed or semi-closed headspaces. In a semi-closed headspace, as introduced by McCloskey et al. [24], He et al. [25] and recently by Lundström et al. [20], see Fig. 1(a), the evolved gasses are purged at set time intervals, again sacrificing time resolution, but gaining an environment that closer resembles a standard battery, as well as a potentially higher detectability of trace gasses, as these have time to accumulate and are not constantly diluted in the carrier gas. An additional benefit of this approach compared to the continuous measurement of gas evolution is the possibility of monitoring the gassing of multiple cells with just one spectrometer by purging them subsequently. The choice of purging interval has to be reasonable compared to the timeframe of battery cycling, but with cells often cycled at a 0.1C rate, sampling every 10 min already generates 60 data points for a single charge or discharge cycle. The reader should keep in mind that even when “continuously monitoring,” a standard quadrupole mass spectrometer may only measure a single m/z ratio at a time, thus also affecting the possible time resolution, especially during broadband monitoring (e.g., all m/z from 1 to 100 are measured). Because of the discrete nature of the sampling times and the accumulation of gasses in the headspace, the gas analysis from a semi-closed headspace is typically referred to as online electrochemical mass spectrometry (OEMS) instead.

The closed headspace approach developed in the group of Gasteiger is also referred to as OEMS. In this case, a small capillary leak connects the headspace of the cell continuously and directly to the mass spectrometer, with only very small gas flow necessary, thereby eliminating the need for carrier gas, membranes, purge valves, and cold traps [26, 27]. Notably, as the headspace is not purged, the gas composition is measured and has to be differentiated to obtain gas evolution rates. As the headspace gas is not refilled, the pressure within the cell is decreasing over time, limiting the maximum measurement time. For this setup, a two-compartment solution via Li-ion conducting glass ceramic has been used to selectively measure the gas evolution at one electrode only [27].
We note that many of the experimental setups discussed herein are derived from setups for the study of metal–air batteries by in situ MS. Because of conceptual differences and the contrasting role of gas presence, this article excludes metal–air battery studies.
A further simplified OEMS design has been reported by Hahn et al., using an X-shaped Swagelok-type cell, yet with challenges in response time and quantification, and result dependency on the exact capillary position [28]. Recently, Geng et al. established OEMS measurements on pouch cells via the closed headspace principle [29] and Mattinen et al. even demonstrated an OEMS setup that is capable of monitoring the gassing of commercial 18650 cells, see Fig. 1(b) [30].
Irrespective of the chosen design approach, quantification of the gases evolved is achieved by flushing the cell with a calibration gas of known composition (e.g., in ppm for each gas of interest) after the measurement. If doing so in steps of different dilution with carrier gas, a calibration curve (e.g., ion current to ppm of gas) can be obtained. Using either the flow rate (open headspace) or cell volume (closed headspace), a conversion of concentration to evolution rate or amount of gas is possible. A detailed description for the calibration of a semi-closed headspace system is provided elsewhere [20].
Gas chromatography (GC) can also be applied to batteries in situ [31]. In this case, it is even possible to replace the mass spectrometer by a simpler detector, such as thermal conductivity [32] or barrier ionization discharge detectors [33]. However, it should be noted that the possible sampling rate is strongly decreased due to the chromatographic retention times of the gaseous species, so that measurements have to be performed very slowly or only at certain potentials [32, 33]. Horsthemke et al. used in situ GC–MS to examine the consumption of vinylene carbonate (VC) and the formation of electrolyte aging products, including their identification [34]. Because electrolyte and aging products are not gaseous and their detection relies on the extraction procedure and heating ramps applied during GC, such studies are outside the scope of this article. The same holds true for works limited to a specific gas or ion, such as the detection of evolved oxygen via reduction at a rotating ring disk electrode, as described by Yin et al. [35].
Readers keen on establishing own DEMS or OEMS setups are encouraged to take a look at the available literature, as well as to consider their own needs and interests, as the setups should be tailored to the system(s) of interest. This kind of tailoring is highlighted when comparing the OEMS setups of Mattinen et al. [30] and Lundström et al. [20], which, while published recently and almost at the same time, are fundamentally different in almost any aspect. Commercial turnkey DEMS solutions are available, but are in most cases not optimized for battery research. Some criteria to consider are the intended operation time and (dis)charge rate of the cell; the size and loading of electrodes; the use of standard (calendered) electrode tape versus the need to specifically coat membranes, separators, or mesh [15]; the ability to measure multiple cells at the same time; the expected amount of evolved gasses; the ability to do measurements in pouch cells, Swagelok-type cells, or in a customized cell setup; the ability to switch between electrode materials and electrolytes; the volume of electrolyte required; restrictions of electrolyte by vapor pressure or melting point, and the need for a cold trap.
State-of-the-art LIBs
For an introduction into and the history of gas evolution in batteries, the reader is referred to the existing literature [15, 16]. In this article, more recent findings relevant to the fundamental mechanistic understanding of gas evolving (side) reactions and Ni-rich CAMs will be reviewed.
Cathode gassing
For LiNixCoyMnzO2 (NCM) materials, CO2 is the main component of gasses released at the cathode side. Additionally, a concurrent evolution of CO is usually observed. Apart from surface carbonate impurities, which will be discussed separately in the next section, the CAM itself does not contain carbon, leaving conductive carbon black, polymer binder, and electrolyte as possible sources. Because O2 evolution from the CAM is often also detected, albeit at a lower level, the conclusion of oxidation of one of the aforementioned components is only logical. However, it should be noted that the organic carbonate electrolytes, such as the widely used ethylene carbonate (EC), can also release CO2 upon hydroxide-catalyzed hydrolysis.
The exact nature of the electrolyte oxidation reaction, with different onset potentials reported earlier, as well as of the O2 release, has been clarified by Jung et al. and Streich et al. They demonstrated that for NCM811 (80% Ni), NCM622 (60% Ni), and NCM111 (33% Ni), the onset of gas evolution varies, being at a lower potential with increasing Ni content (when the state of charge (SOC) reaches ≥ 80%) [36, 37]. From this observation, Jung et al. were able to assign the SOC-dependent CO2 evolution below 4.7 V vs. Li+/Li to the chemical oxidation of EC by O2 released from the NCM lattice. Above 4.7 V, also electrochemical oxidation was apparent, as demonstrated with an electrode containing no CAM. In a follow-up study, they further showed that the onset potential is not dependent on temperature, but only on SOC, which at a given potential only changes slightly with increasing temperature [38]. They also observed an increase in gas evolution at elevated temperature and, using 13C-labeled electrolyte, identified (and quantified) electrolyte hydrolysis and impurity oxidation as the reasons for gas evolution prior to the O2 release from the NCM.
As lattice oxygen is released, the layered oxide surface undergoes a reconstruction toward a redox-inactive rock-salt-like phase. The thickness of this layer can be calculated from the observed gas evolution (O2 and CO2 from chemical electrolyte oxidation) and the CAM’s specific surface area [37]. However, as Oswald et al. recently demonstrated in a comparison of polycrystalline and single-crystalline NCM CAMs, the increase in specific surface area due to particle fracture upon delithiation (charge) has to be taken into account [39]. In a follow-up study, they examined the role of particle morphology, i.e., primary particle size, finding both a lower total gas release, with no O2 but only CO2 evolution, for single-crystalline material [40].
Metzger et al. investigated the electrochemical oxidation of carbon black and EC containing different supporting lithium salts at various temperatures by using 13C-labeled carbon electrodes and common 12C-electrolyte in order to distinguish the reaction products (13CO2 and 12CO2) in OEMS [41]. They observed that the conductive carbon is oxidized in the presence of LiClO4, but not in the presence of LiPF6, lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) or LiBF4, whereas EC oxidation took place in the presence of LiPF6, thus concluding that LiBF4 is best suited for use at high potentials.
A major discovery in the field of battery gas evolution was the in situ observation of singlet oxygen (1O2) generation by both NCM and Li-rich CAMs, which will be discussed separately, by Wandt et al. [42]. In previous works, it has already been speculated that the released oxygen is highly reactive, because the electrolyte is not oxidized when handled in air. Using the 633 nm photon emission of 1O2 dimers upon return to ground state, Wandt et al. developed an operando photomultiplier setup that is capable of detecting the released photons, revealing the presence of 1O2 above ~ 80% SOC and correlation with the gas evolution from OEMS. This work has major implications, as it suggests that not stability against electrochemical oxidation, but instead against 1O2 is the foremost requirement for electrolytes when CAMs are operated at high SOC. Freiberg et al. also demonstrated the reaction of EC or dimethyl carbonate (DMC) with 1O2 using rose bengal dye to excite oxygen dissolved into the electrolyte upon irradiation while also monitoring the gas evolution [43]. In EC, they observed the formation of CO2 (but no CO) and consumption of O2, while in DMC no signal above a background experiment without dissolved oxygen was detected, highlighting the stability of DMC against 1O2. A reaction mechanism has been proposed from density functional theory (DFT) calculations, including (in the first step) the dehydrogenation of EC with the formation of VC and H2O2. The presence of H2O2 has been confirmed colorimetrically via the [Ti(O2)]2+ complex and its subsequent oxidation probed using OEMS, revealing the evolution of O2 at 4.4 V vs. Li+/Li, leaving H2O and acid protons behind. The formation of H2O and acid protons has multiple detrimental effects, such as the hydrolysis of LiPF6, producing HF and leading to transition metal leaching from the CAM [44].
The effect of increased Ni content in NCM CAMs on gas evolution has been studied by considering the endmember LiNiO2 (LNO). de Biasi et al. used DEMS to show that some (mostly minor) O2 evolution (and thus also CO2 evolution) already occurs in the H2 region (x(Li) ≈ 0.3), with the gassing being reduced during the H2–H3 phase transformation before a large increase in rate of gas evolution is observed (SOC > 80%). Surprisingly, they also detected O2 evolution in the H2 region during discharge [45]. A mechanistic insight on oxygen evolution has been given by Li et al. They combined DEMS with synchrotron X-ray absorption spectroscopy (XAS) and resonant inelastic X-ray scattering (RIXS) and showed that during charge, starting from 4.3 V vs. Li+/Li, O2 evolution is observed, with the Ni ions at and near the surface decreasing in oxidation state again. This result indicates the presence of oxidized oxygen, for which a RIXS feature remains present until discharge to 3.8 V [46]. Figure 2(a) shows the corresponding RIXS and DEMS results. The oxidation of oxygen anions to form molecular O2 thus goes in hand with the reduction of Ni4+. In a follow-up study, Li et al. demonstrated that doping with Al3+ leads to increased oxygen redox and O2 evolution, as the local concentration of redox-active cations is reduced [47].

Papp et al. compared the gas evolution of LiCoO2 (LCO) and LNO upon charging to 5 V vs. Li+/Li, focusing on the electrochemical electrolyte oxidation. They revealed that while LNO releases one order of magnitude more CO2 in the initial cycle, it shows much less gas evolution in the following cycles [48]. The authors inferred that LNO has a higher electrocatalytic activity for electrolyte degradation, but at the same time forms a passivation layer faster. A distinction between chemical and electrochemical oxidation was possible through the use of 18O-enriched CAM.
Surface modification is a well-established concept to mitigate CAM degradation, and it is not surprising that also the gas evolution can be affected by cathode electrolyte interphase (CEI)-forming additives [49, 50] or coatings [51,52,53]. For example, Zhu et al. demonstrated that by enclosing LCO in a shell of LiMn0.75Ni0.25O2, the oxygen redox can be utilized with no apparent O2 evolution, yielding a specific charge capacity of more than 250 mAh/g at 4.6 V vs. Li+/Li, see Fig. 2(b) [54]. Comparing the gassing behavior of coated and uncoated CAMs requires attention to the amount of surface impurities, as they will affect the gas evolution, especially in the case of carbonates, as discussed in the following.
The role of carbonates, peroxides, and surface treatments
Impurities are regularly found on the surface not only of NCM but of LIB CAMs in general, with Li2CO3 and LiOH being the most common. They are formed from excess reagents during synthesis and exposure to ambient air and moisture, thus being more or less unavoidable. Li2CO3 decomposes under release of CO2 and therefore is of great importance in gassing studies. In situ gas analysis setups can be modified to determine the amount of carbonates present by measuring the CO2 evolution upon addition of acid to the CAM, and the contribution of carbonate decomposition to the total CO2 evolution, which is significant in the initial cycle, can be quantified by isotope labeling [55, 56]. CO2 is released both by chemical decomposition in acidic environment (Eq. 1) or by electrochemical oxidation at potentials above 3.8 V vs. Li+/Li (Eq. 2).
While the rate of Li2CO3 decomposition has been found to increase with electrode potential and the reaction is known from metal–oxygen (air) batteries, no O2 evolution is usually detected. Using carbon/Li2CO3 electrodes, Mahne et al. demonstrated that the electrochemical oxidation leads to the formation of 1O2, similar to the release of lattice oxygen from NCM CAMs, which is typically not detected, as it reacts quickly with the electrolyte [57]. The authors achieved this in an experiment using 9,10-dimethylantracene as chemical probe in the electrolyte to trap the 1O2 and then detect the reaction product, and in another experiment by detecting O2 evolution after adding a quencher to the electrolyte. However, the slow and incomplete decomposition of carbonate species in SSB cells, i.e., in the absence of liquid electrolyte, raises the question of the rate at which the electrochemical decomposition occurs [58, 59].
Freiberg et al. have shown via OEMS that a carbon/Li213CO3 electrode releases the amount of 13CO2 equal to complete carbonate decomposition even when separated from the working electrode by a non-conducting polyester layer, making direct electrochemical oxidation impossible [60]. The source of acid protons necessary for the chemical decomposition of carbonates has been determined to be the oxidation of alcoholic impurities, which already occurs at 3.5 V vs. Li+/Li and helps explain the often observed early onset of carbonate decomposition. The presence of protons has a catalytic effect, as H2O formed by the carbonate decomposition hydrolyzes LiPF6, leading to the generation of additional HF and POF3. In contrast, Kaufman et al. observed no 13CO2 evolution when performing an experiment similar to that of Freiberg et al., in which they used a Li213CO3-containing separator instead of a disconnected carbon/Li213CO3 interlayer [61]. Figure 3(a) and (b) shows the results from both groups. Overall, the individual contributions of the chemical and electrochemical pathways to carbonate decomposition remain disputed.

Performing acid titration either on charged or discharged cathodes, Renfrew et al. found that charged cathodes have a larger carbonate content than the pristine ones and the content is only reduced below that of pristine cathodes upon discharge. This result indicates that the degradation of organic carbonates leads to the formation of a surface layer containing carbonate-type side products during charge, which are desorbed with discharge [55]. In the same study, the authors also observed O2 evolution in acid titration experiments using charged cathodes, which they explained by the formation of a peroxo-like surface layer, notably prior to the onset of lattice oxygen evolution. Upon acid titration, peroxides release oxygen according to (Eq. 3).
From a follow-up study, indicating that the thickness of the peroxo-like layer is not dependent on the electrolyte but the SOC [62], the authors suggested that organic carbonates are deposited onto the cathode, where they can react with lattice oxygen, explaining the origin of CO2 containing isotope-labeled lattice oxygen before the actual release of O2 from the lattice. The observation of electrolyte fragments attached to diatomic oxygen during acid titration, including both 16O from the lattice and 18O from Li2C18O3 by Kaufman et al. indicates a complex interplay of the carbonate species and the lattice reactivity [61]. Taking the peroxo-like surface layer and carbonate decomposition together, Houchins et al. proposed a mechanism for 1O2 generation based on superoxide formation and disproportionation [63].
Removal of surface carbonates via washing of the CAM appears obvious. However, while indeed reducing their amount, the overall effect on gas release is complex. Depending on the exposure time to H2O and the applied drying procedure, the surface reactivity of the CAM varies, as discussed by Pritzl et al. [64] and Renfrew et al. [65]. Not only are washing steps directly decreasing the peroxo-like character, but also removing lithium from the CAM, thereby negatively affecting the capacity and forming a Li-deficient surface layer. Upon heating (drying), the latter may decompose to a rock-salt or spinel-type phase with increased impedance. For this reason, washing procedures have to be developed with care.
For Li-rich CAMs, similar surface treatments have been shown to suppress O2 evolution by the formation of a passivating surface film [66, 67]. For example, carbonates [68, 69] or ternary lithium metal oxide shells [70, 71] have been deliberately prepared to later be washed off, eventually showing lower gas evolution.
Anode/electrolyte gassing
The gassing behavior of electrolytes and anodes is interwoven, as the SEI formation is the most relevant process in terms of gas evolution. The SEI is a complex surface layer, with its formation and composition depending on many factors. Herein, only in situ studies on the SEI formation are reviewed.
Trace H2O and acid-derived protons are reduced to evolve H2 while leaving OH− ions and other anions behind. These ions lead to the hydrolysis of cyclic organic carbonates, such as EC or propylene carbonate (PC), resulting in the evolution of CO2 and generation of alkoxide anions, which in turn can react with the electrolyte solvent to produce glycol species [72]. The main gas evolution at the anode side is the electrolyte reduction, which below ~ 0.9 V vs. Li+/Li leads to the formation of lithium ethylene dicarbonate (LEDC) as an SEI component and C2H4 in the case of EC and lithium propylene dicarbonate and C3H6 in the case of PC [73]. Note that for the detection of C2H4, the (fragment) signal at m/z = 26 is suited best, because both N2 and CO are also detected at m/z = 28.
The role of H2O impurities present in the battery cell has been discussed by Bernhard et al. [74] and Kitz et al. [75], observing increased rates of H2 and CO2 evolution with increasing H2O level. With the closed headspace OEMS used by Bernhard et al., the subsequent consumption of CO2 has been seen. This consumption can be explained by the formation of Li2CO3 in a reaction of CO2 with OH− ions. Indeed, by combining OEMS with electrochemical quartz crystal microbalance (EQCM), Kitz et al. found that the addition of trace H2O leads to a thicker and more rigid SEI with increased Li2CO3 content. Using additionally surface-enhanced Raman spectroscopy, Mozhzhukhina et al. observed a carbonate band appearing at 1.8 V vs. Li+/Li, while LiF was found to deposit onto the electrode after the onset of H2 evolution [76]. The latter stems from HF, which results from the hydrolysis of LiPF6, as discussed previously.
Combining OEMS and EQCM, Melin et al. were also able to show that both EC and PC do form an SEI with accompanying gassing upon reduction. However, the gas evolution rate and mass deposition were much higher in the case of PC, forming a thicker layer that re-dissolves when current is no longer applied, thus explaining the lack of stable SEI formation in PC [73]. Figure 4 summarizes the discussed anode gas evolution processes and SEI formation in EC- and PC-based LIB electrolytes.

Gas evolution and SEI formation in (a) EC and (b) PC electrolytes. Reproduced with permission from [73].
The common electrolyte additives VC and fluoroethylene carbonate (FEC) have been investigated by Schwenke et al. [77] and Kitz et al. [78] regarding their effect on gas evolution during SEI formation and the resulting SEI properties. Both groups observed the evolution of CO2 upon reduction, as opposed to the evolution of C2H4 for EC-containing electrolyte. Because the additives are decomposed at higher potentials (1.3–1.1 V for VC and 1.45–0.95 V for FEC) [78] than EC, they mitigate the subsequent electrolyte reduction by passivating the anode, resulting in a thinner SEI (note that the evolved CO2 can lead to the formation of Li2CO3). While FEC also leads to the formation of LiF in the SEI, VC is capable of suppressing it. Based on these observations, Schwenke et al. demonstrated that a CO2 atmosphere in the cell can lead to the formation of a carbonate-containing SEI even for EC-free electrolytes. Specifically, they used OEMS to track the consumption of 13CO2 during cycling. Alternatively, Solchenbach et al. introduced lithium oxalate as an electrolyte additive, which is oxidized to CO2 in the first charge cycle, and demonstrated the effect on the SEI formation while using OEMS to verify a 1 e−/CO2 conversion [79].
Solchenbach et al. also studied the effect that cathode transition metal leaching has on the SEI by adding either Ni(TFSI)2 or Mn(TFSI)2 to the electrolyte and monitoring the C2H4 signal. They observed a larger evolution in the case of Ni and a larger and continuous evolution over multiple cycles in the case of Mn [80]. However, by preforming the SEI, the additional gassing could be strongly suppressed. By switching to DMC, which does not release C2H4, after preforming the anode and then still detecting C2H4 evolution in the presence of Mn, the authors were able to conclude that Mn0 species can catalytically reduce LEDC to Li2CO3 and C2H4, leading to a carbonate-rich SEI.
Regarding beneficial additives, Tezel et al. showed reduced CO2 and H2 evolution during SEI formation by tris(hexafluoroisopropyl)borate [81]. A systematic study of phosphate additives has been presented by Zhao et al. They found via DEMS that the unsaturated compounds, especially the alkyne-containing ones, greatly suppress gas evolution both at the cathode and anode, with thinner and more uniform CEI and SEI, respectively [49].
Although the reactivity of LiPF6, especially toward acid protons and hydrolysis, has already been mentioned earlier, some more observations shall be summarized here. As demonstrated by Solchenbach et al. and Guéguen et al., protic (electrolyte) oxidation products can already trigger decomposition, resulting in the formation of PF5, which is detected as POF3 owing to high reactivity with moisture [82, 83]. Bolli et al. demonstrated that tris(trimethylsilyl)phosphate (TMSPa) not only serves as chemical scavenger for HF and LiF, but that the product of this reaction, Me3SiF, can be detected by OEMS (m/z = 77) and therefore is suited as an operando probe for fluoride formation in batteries [84]. They were able to study the formation of LiF from FEC and the proton release by electrolyte oxidation at the cathode side and subsequent LiPF6 decomposition. They also demonstrated the presence of HF in a cell free of fluorinated compounds except for polyvinylidene fluoride (PVDF), proving that the binder is indeed dehydrofluorinated under operating conditions. Guéguen et al. added the similar component tris(trimethylsilyl)phosphite (TMSPi) in a follow-up study comparing TMSPa and TMSPi, demonstrating that both additives mainly work as acid scavengers [85]. Protons and protic side products lead to H2 evolution at the anode due to electrochemical cross-talk, which Metzger et al. revealed by employing a sealed diffusion barrier between cathode and anode [86].
Other anode materials for LIBs, such as Li3VO4 [87], TiNb2O7 [88], Si in carbon shells [89], and NbO2/carbon nanohybrids [90], to name a few, have also been investigated regarding their gassing behavior, typically to examine the stability of the SEI formed on these electrodes.
Lithium-rich cathode-active materials
A good introduction to the history and development of Li-rich CAMs can be found in the literature [4]. The same holds true for the recent progress and future perspectives [3]. Herein, we aim to discuss the role of gas evolution measurements in the characterization and design of these materials.
Layered cathodes
Substituting lithium for transition metals in the respective layer requires compensation for the lower charge of lithium ions compared to the transition metal ions. For this reason, the valence state of the remaining metals is increased, limiting the amount of lithium replacement to 1/3 of the atoms in the transition metal layer, where then all remaining ions are in oxidation state 4+. The resultant structure can be written as Li[Li1/3M2/3]O2 (M = Ni, Mn) or Li1.33M0.67O2 to express the similarity to NCM CAMs, or it can be summarized as Li2MO3. Intriguingly, lithium can be electrochemically de-intercalated from these materials, resulting in large specific charge capacities, even though all of the transition metals are in the highest (expected) valence state and cannot be oxidized further for charge compensation. This opens the possibility of anionic redox, i.e., the at best reversible oxidation of oxygen anions to either peroxide or even superoxide species or to molecular oxygen. Rana et al. have shown via DEMS that for Li2MnO3 almost all charge current can be attributed to oxidation of lattice oxygen to O2 (4 e−/O2 process) and reversible oxygen redox is negligible, as no corresponding RIXS feature was detected and MS titration experiments revealed minor amounts of peroxides (equivalent to 10 mAh/g) [91]. Because the O2 released from the lattice is 1O2 [42], CO2 evolution due to electrolyte oxidation also needs to be considered. Depending on the experimental procedures, either of these two gas species might be predominantly observed, as discussed by Guerrini et al., who also demonstrated that oxygen oxidation/loss is the main contributor to the charge capacity of Li2MnO3 [92]. For Li2NiO3, Bianchini et al. have shown via DEMS that almost all charge capacity of the CAM is due to O2 evolution, leaving a rock-salt-type structure behind, which after 100 cycles still delivered about 100 mAh/g [93].
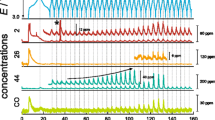
While detailed gas analysis indicates that reversible anion redox cannot be utilized in Li2MnO3, a reduced lithium content leads to materials of the form Li[M1−xLix/3Mn2x/3]O2 (M = Co, Ni, Mn), which can also be written as xLi2MnO3·(1 − x)LiMO2, representing a layered NCM-type CAM with nanoscale domains of Li2MnO3 [94]. For Li1.2Ni0.2Mn0.6O2, Armstrong et al. demonstrated already in 2006 that the charge plateau at 4.5 V vs. Li+/Li is associated with O2 evolution [95]. For the same material and also for Li1.2Ni0.13Co0.13Mn0.54O2, Luo et al. examined the anion redox in detail, observing only CO2 evolution at the 4.5 V plateau, with additional O2 evolution at higher potentials [96, 97]. By enriching the lattice oxygen with 18O, the authors were able to show that the CO2 (containing C16/18O2) in fact contains lattice oxygen, see Fig. 5(a). These CAMs revealed a stable cycling behavior from the second cycle onward, achieving specific discharge capacities of about 270 mAh/g. Using RIXS, the authors demonstrated the presence of oxidized oxygen in the charged cathodes. At the same time, the electrodes did not show Raman bands representing peroxide species, with the authors concluding that localized electron holes are formed on oxygen ions coordinated by Mn4+ and Li+. Based on the gas evolution in DEMS, they calculated an oxygen redox contribution of 0.5 e−/formula unit (~ 157 mAh/g) and of only 0.05 e−/formula unit (~ 16 mAh/g) by O2 evolution for Li1.2Ni0.13Co0.13Mn0.54O2.

Bulk transformation of the Li2MnO3 domains toward electrochemically active LiMnO2 under lithium extraction and O2 evolution, referred to as “activation,” is often used to explain the behavior of Li-rich CAMs in the first charge cycle. However, the amount of O2 detected in the aforementioned studies and by Strehle et al. for Li1.17[Ni0.22Co0.12Mn0.66]0.83O2 [98] is far too low to sustain the assumption of a bulk transformation. Instead, Strehle et al. calculated the thickness of a spinel surface layer based on the quantified gas evolution in OEMS, reaching 2–3 nm, in good agreement with transmission electron microscopy (TEM) observations. Teufl et al. analyzed the gas evolution of xLi2MnO3·(1 − x)LiMO2 compositions with varying x. They found significant increases in oxygen evolution and spinel surface layer thickness with increasing x, starting to also observe bulk spinel formation at x = 0.5 [99]. The authors emphasize that for a fair comparison between the different CAMs and also with the corresponding NCM (x = 0), the gas evolution has to be normalized to the specific surface area, which was ~ 10 times larger for the Li-rich materials. In contrast to the spinel surface layer formation, Yin et al. reported about a bulk phase transformation by adding a constant voltage charge at 4.8 V vs. Li+/Li for Li1.2Ni0.13Mn0.54Co0.13O2, upon which they observed strong O2 evolution and were able to detect the new bulk phase via in situ X-ray diffraction (XRD) [100].
The at least partially unavoidable gas evolution of Li-rich CAMs is a main challenge for commercial application. Recently, Schreiner et al. disclosed the production of multilayer pouch cells using Li1.14[Ni0.26Co0.14Mn0.60]0.86O2 on a pilot scale production line [101]. Via OEMS, they found that a formation step at 45 °C instead of 25 °C allows to concentrate most gas evolution into the initial cycle.
Multiple structural modifications and protection strategies have been reported to suppress the gas evolution of Li-rich CAMs. Cao et al. demonstrated that the preparation via chemical ion exchange from the Na-containing precursor results in less O2 loss compared to the preparation via electrochemical ion exchange [102]. Following a similar approach, Cao et al. obtained both a Li-deficient (in the lithium layer) and Li-rich (in the transition metal layer) CAM Li0.8[Li0.2Mn0.8]O2, for which they quantified the irreversible O2 loss via DEMS and observed an increasing peroxide character during charge [103].
The role of Ni and Co bulk doping in stabilizing Li-rich and Mn-rich CAMs has been examined by multiple groups. Shen et al. gradually replaced Ni by Co, starting from Li1.13Ni0.275Mn0.58O2. With increasing Co content, they observed a strong increase in O2 evolution via OEMS (also leading to increased spinel layer formation), which was barely detectable against the background signal in the Co-free material [104]. However, it should be noted that at the same time, the CO2 evolution rates decreased and the authors did not provide a comparative quantification of the total gas amounts released, thus leaving the contribution of chemical electrolyte oxidation unattributed. A similar observation of reduced gas evolution and spinel formation has been made by Huang et al. for Mn-rich NCM CAMs [105]. Boivin et al. compared the irreversible charge capacities resulting from gas evolution after doping Li2MnO3 with Ni and/or Co and observed reduced gassing for both dopants, yet with Ni having a more pronounced effect, see Fig. 5(b) [106]. The authors showed that unlike Co, Ni doping leads to a Ni-rich, Li-poor rock-salt-type shell, mitigating surface degradation due to gas evolution. Zhang et al. found that Zr doping of Li1.21Ni0.28Mn0.51O2 leads to the formation of Li2ZrO3 slabs in the structure, affecting the oxygen redox by reducing the formation of O2 versus electron holes localized on the oxygen anions [107]. Shin et al. performed a computational screening of dopants regarding their effectiveness in increasing oxygen retention, confirming experimentally that 2% Ta-doped Li1.3Nb0.3Mn0.4O2 shows surface enrichment effects and a substantially reduced O2 evolution [108]. Increased surface carbonate content, as confirmed by acid titration experiments, explained the larger CO2 evolution for the doped CAM, again highlighting the importance of surface carbonates for correct interpretation of in situ gas analysis results.
Wang et al. reported that the lattice oxygen release from Li1.2Ni0.27Mn0.53O2 was reduced by anion doping with chlorine (Li1.2Ni0.27Mn0.53O1.976Cl0.024), finding both lower O2 and CO2 evolution [109]. As early as 2008, Zheng et al. demonstrated via DEMS that by coating Li1.2Ni0.2Mn0.6O2 with AlF3, the ratio between the released gasses changes, with mostly O2 evolving from coated CAM and CO2 from pristine CAM [110]. A decade before the experimental detection of 1O2, the authors already proposed that the oxygen may become less reactive while passing through the coating. Li et al. presented a three-in-one strategy, consisting of a Na2SiO3 coating with concurrent Na and Si doping, for which they showed reduced O2 evolution via DEMS. However, they did not report the CO2 evolution profiles, thus hindering a quantitative comparison (including chemical oxidation of electrolyte) [111]. With a similar Na5AlO4 coating, Maiti et al. achieved suppressed gas evolution for Li-rich NCM up to 4.65 V vs. Li+/Li. A strong increase in O2 release was observed at higher potentials and explained by the decomposition of the coating, resulting in Na2O2 formation among others [112]. Gim et al. further reported the lack of O2 detection by coating of Li-rich CAM (40 nm thickness) using CoPO4 nanoparticles, albeit not discussing CO2 evolution [32]. Organometallic reagents like those used in atomic layer deposition (ALD) have been found by Evenstein et al. and Rosy et al. to alter the free surface of Li-rich CAMs by forming a layer of reduced transition metal oxide and metal species from the reagent, a process they refer to as atomic surface reduction [113, 114]. For both diethylzinc- and trimethylaluminum-treated Li-rich NCM, they observed reduced O2 and CO2 evolution rates. Sun et al. studied a thin lithium polyacrylate coating, for which reduced CO2 evolution, yet no change in O2 evolution, was observed at high potentials, thus indicating a beneficial effect mostly against electrochemical electrolyte oxidation, supported by the finding of reduced CO2 and POF3 evolution with glassy carbon electrodes [115].
With the inherent O2 release of Li-rich CAMs during the first charge cycle and the high working potential of these materials, the stability of electrolytes against oxidation is of great importance. By comparing the CO2 evolution for EC and FEC both on carbon black and NCM622 electrodes, Teufl et al. demonstrated that both electrolytes show a similar stability against electrochemical oxidation, while EC is more readily chemically oxidized by lattice oxygen [116]. Consequently, when using pre-activated CAM, the performance in EC is greatly improved.
Wu et al. were able to demonstrate that with the ionic liquid electrolyte N-butyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide a stable CEI is formed on Li1.2Ni0.2Mn0.6O2, leading to reduced gas evolution and rock-salt-type phase formation [117]. Han et al. investigated the working mechanism of lithium fluoromalonato(difluoro)borate as electrolyte additive, also observing the formation of a stable CEI. Using DEMS, they showed that after the initial decarboxylation of the additive, reduced O2 evolution is achieved [118].
Li-rich layered oxides are not necessarily based on Mn. Xu et al. found that the replacement of 3d Mn by 4d Ru in Li1.2Ni0.2M0.6O2 (M = Mn, Ru) leads to lower irreversible charge capacity and the absence of severe evolution of both O2 and CO2, even after increasing the potential to 5 V vs. Li+/Li [119]. A RIXS feature indicating anion redox was only present for Li1.2Ni0.2Mn0.6O2, while in situ XAS revealed reversible cation redox of Ru in Li1.2Ni0.2Ru0.6O2. Yu et al. probed the oxygen redox of Li2RuO3, demonstrating O–O coupling through extensive synchrotron characterizations, and used DEMS to quantify the oxygen loss during the first charge. The observed reduction of Ru at high potentials cannot be explained by oxygen loss alone, thus anion redox must be present [120]. In a follow-up study, Yu et al. examined the effect that Ru substitution in Li2RuO3 (by Ti, Cr, Mn, Fe, Ru, Sn, Ir, and Pt) has on the reversibility of oxygen redox, which they quantified from integration of dq/dV and DEMS curves, see Fig. 6 [121]. The irreversibility increased with the ionic character of the substituents, an observation explained by increased distortion of the M–O octahedra, enabling easier O–O dimer formation. Ning et al. demonstrated that O–O dimerization is suppressed in intralayer-disordered Li2RuO3, which they prepared via ion exchange from Na2RuO3 [122]. In contrast to common Li2RuO3, where cations in the intralayer show the so-called honeycomb arrangement, no O2 and less CO2 were detected for the disordered material.

The effect of transition metal substitution on gas evolution of Li2Ru0.75M0.25O3 (M = Ru, Ti, Mn, Fe). Adapted with permission from [121].
McCalla et al. synthesized the model compounds Li4FeTeO6 (specifically Li4.27Fe0.57TeO6) and Li4FeSbO6 for the detailed study of anion redox and O2 release [123, 124]. For the former, they found via DEMS that nearly all charge capacity is reflected in O2 evolution. For the latter, Fe oxidation and O oxidation upon charge (4.2 V plateau) have been observed, with incomplete reduction of the oxygenated species during discharge. However, when the material was charged beyond the plateau, strong CO2 and O2 evolution occurred. At 5 V, Fe was found to be reduced again, accompanied by O2 evolution. Ting et al. confirmed the existence of such a reductive couple (Fe and oxygenated species), with oxygen redox also for the Ni-containing equivalents Li4NiTeO6 and Li1.15Ni0.47Sb0.38O2, yet limited to the surface of the materials [125]. While observing O2 evolution via OEMS, the outgassing was much smaller for the latter materials than the Fe-containing ones.
Jaquet et al. studied the anion redox and gas evolution of layered CAMs of composition Li3RuyIr1−yO4 and reported reduced gassing for the Ir-free Li3RuO4. For Li3RuO4, they found a new degradation mechanism, namely, the dissolution of RuO4/RuO4−, oxidizing the electrolyte and forming CO2 and solid RuOx [126]. The presence of Ir can suppress the dissolution. However, in this case, increased gas evolution has been detected.
Li3RuO4 has a rock-salt structure and can either possess Li-only layers alternating with Li/Ru layers or show a random arrangement of Li and Ru, with the former structure being referred to as ordered and the latter as disordered rock-salt (DRX). Li et al. reported similar gas evolution in the initial charge cycle for both polymorphs [127]. In a recent study, Li et al. demonstrated that Li1.2Ni0.4Ru0.4O2 can be prepared with intergrown layered and DRX phases, exhibiting anion redox with minimal O2 evolution, thus combining the benefits of both layered and DRX materials [128].
Disordered rock-salt cathodes
While the formation of a rock-salt-type surface layer in NCM CAMs is detrimental, in DRX materials, the percolation of the so-called 0-TM diffusion channels, in which the tetrahedrally coordinated (activated) lithium site is not neighbored by a transition metal ion, can lead to high and SOC-independent Li mobility, as demonstrated by Lee et al. [129]. A main benefit of cation disorder is the much lower and isotropic volume change of the CAM upon battery operation. For a detailed review of mechanisms, possibilities and constraints of DRX materials, the reader is again referred to the literature [5]. DRX materials require a certain amount of Li excess to enable 0-TM percolation, and like layered Li-rich CAMs therefore show limited capacity by transition metal redox. They rely on anion redox to achieve high specific capacities, with the risk of showing substantial O2 evolution. As an example, Cambaz et al. observed via DEMS that O2 evolution only occurs in the Li-rich DRX Li1.2Ni1/3Ti1/3W2/15O2, but not in stoichiometric LiNi0.5Ti0.5O2 [130]. No longer constrained by the requirements of a layered structure, the partial replacement of oxygen by fluorine has been shown to suppress the evolution of O2, while the lower valence of fluoride ions allows for an increase in the redox-active transition metal content, as demonstrated by Lee et al. in a comparison of Li1.15Ni0.375Ti0.375Mo0.1O2 and Li1.15Ni0.45Ti0.3Mo0.1O1.85F0.15 [131]. Not only Ni, but also both Mn redox couples (Mn2+/Mn4+ and Mn3+/Mn4+) can be utilized in DRX cathodes, where again fluorination reduced the O2 evolution [132,133,134,135]. Combining DEMS and the titration of peroxide-like species and carbonates, Crafton et al. examined the effect of fluorination by comparing Li1.2Mn0.6Nb0.2O2 (LMNO) and Li1.2Mn0.625Nb0.175O1.95F0.05 (LMNOF) [136]. They found reduced and delayed O2 evolution, see Fig. 7(a), and were able to show that the contribution of anion redox as a whole is reduced by fluorination, as the fraction of redox-active cations with lower initial oxidation number is increased. Using TMSPa as a probe (evolution of gaseous Me3SiF), the authors also showed that fluorinated cathodes suffer from fluoride dissolution near the end of charge over multiple cycles. Sathish et al. demonstrated that soaking the same CAM in electrolyte leads to the removal of Li and F, thereby increasing the material’s capacity retention and suppressing O2 evolution, similar to the already discussed acid washing steps [137].

Huang et al. recently showed that DEMS and related acid titration methods in combination with 18O enrichment can be used to deconvolute the redox processes in Li1.15Ni0.45Ti0.3Mo0.1O1.85F0.15. During acid titration, oxidized lattice oxygen (the contribution of anion redox) is released as (18O-enriched) O2 according to Eq. (3), while at the same time the CAM is dissolved and both Ni3+ and Ni4+ (the contribution of cation redox) oxidize H2O, leading to O2 evolution without isotope enrichment. This way, the relative contributions can be calculated from the observed isotope ratio, see Fig. 7(b) [138]. Consequences of a large reliance on anion redox have been demonstrated by Kan et al. for single-crystalline Li1.3Nb0.3Mn0.4O2, where the authors reported not only gas evolution, but also volume changes leading to particle fracture [139].
The choice of the redox-active transition metal and its effect on the anion redox and electrochemical properties have been analyzed by Jaquet et al. by comparing Li1.3Ni0.27Ta0.43O2 and Li1.3Mn0.4Ta0.3O2 [140]. They found that the Ni-containing CAM exhibits a large voltage hysteresis and increased gas evolution coinciding with partial Ni reduction, which the authors both attributed to a smaller charge-transfer band gap (supported by DFT calculations).
To stabilize DRX structures, redox-inactive (in the given voltage range) d0 metal ions, such as Ti4+ and Nb5+, are often required [141]. While being redox-inactive, they still affect the anion redox, as demonstrated for Li1.3M0.3Mn0.4O2 (M = Nb, Ti) by Chen et al. [142]. For Nb, the authors found increased O2 evolution and a larger capacity contribution of anion redox in the initial cycles, while Ti stabilized oxidized oxygen species, thereby increasing the reversibility of anion redox and leading to a lower O2 evolution and reduced CAM degradation. By comparing RIXS data of cycled electrodes, the authors also demonstrated that anion redox features decrease stronger for Nb than Ti. Yue et al. found that the introduction of Mo into Li1.15Ni0.35Ti0.5O1.85F0.15 (yielding Li1.15Ni0.45Ti0.3Mo0.1O1.85F0.15) results in a lower voltage hysteresis after migration of Mo into the tetrahedral sites, yet with increased O2 evolution [143]. Cambaz et al. observed via DEMS and X-ray photoelectron spectroscopy (XPS) that concurrent with O2 evolution from Li1.2Ni1/3Ti1/3Mo2/15O2, partial reduction of Mo6+ to Mo4+ is occurring at the particle surface, indicating a reductive couple and formation of a densified surface layer [144].
Finally, the introduction of configurational entropy (high-entropy concept) into DRX materials and their gas evolution behavior have been reported by Breitung et al. and Lun et al. [145, 146]. Lun et al. found improved energy density and better rate capability for high-entropy Li1.3Co0.1Cr0.1Mn0.2Nb0.2Ti0.1O1.7F0.3 compared to low-entropy Li1.3Mn0.4Ti0.3O1.7F0.3. However, the former material showed stronger CO2 evolution, which the authors attributed to increased interfacial side reactions with the electrolyte.
In summary, in situ gas analysis has proven indispensable in the study of Li-rich CAMs both with layered and DRX structures, as it can provide quantitative information on the irreversibility and extent of anion redox, a key feature of these electrode materials.
Solid-state batteries
On the one hand, the gas analysis of SSBs is simplified by the lack of continuous liquid electrolyte degradation and accompanied electrolyte fragment detection. On the other hand, the assembly of a test cell is complicated by the fact that most SSBs are cycled under external pressure to assure proper contacting and conductivity. Bartsch et al. first reported about DEMS measurements on SSBs with Li3PS4 as SE in 2018, utilizing pellets of NCM622 CAM, SE separator and In anode, as well as a rather robust cell housing [58]. They attributed the H2 evolution to the initial reduction of trace H2O, while both CO2 and O2 were detected clearly in the charge cycle. The only possible source for CO2 in this configuration were residual carbonates, as proven by 13C-labeling experiments. The authors observed minor amounts of CO2 compared to the overall carbonate content, presumably due to the lack of acid protons. O2 evolved from the NCM lattice at high SOC and from the electrochemical carbonate decomposition, as discussed previously. Oxidation of the SE by the reactive oxygen has been observed, leading to traces of SO2 being detected. In later studies, the same group examined the effect of Li2CO3, Li2CO3/LiNbO3 (with Li3PS4 as SE) [147], Li2CO3/Li2ZrO3 (with argyrodite Li6PS5Cl as SE) [148], and more complex nanoparticle coatings [149] on NCM CAMs on the gas evolution, observing that a Li2CO3 coating alone leads to increased CO2 release as expected, but in a dual coating, the CO2 evolution is greatly reduced and minor or no SO2 is detected. From this observation, DEMS allowed for the conclusion of a uniform (hybrid) coating structure, as opposed to areas in which only the carbonate is present. Li2ZrO3 prevented the formation of SO2, even when the coated CAM was annealed in air (note that in this case the carbonate content and CO2 evolution were larger than for the uncoated material). Extending the scope of CAMs to LNO, CO2 has only been detected in the initial cycle, while the release of lattice O2 continued at high potentials during the second cycle [150]. In all aforementioned studies, H2 and CO2 have been detected at the onset of charge (first cycle). In a liquid cell, such gassing behavior would be attributed to organic carbonates reacting at the anode side, which does not apply to SSBs. Because the H2 evolution is explained by the reduction of trace H2O in the cell, the authors assume a correlation between H2 and CO2 evolution.
Major challenges of SSBs are the variation in performance and lack of scalability for pellet-stack cells. Tape-cast electrodes can alleviate these issues while requiring the development of slurry recipes and the use of polymer binders. Teo et al. studied the binder choice for SSB cathodes containing NCM622 CAM, Li3PS4 SE, conductive carbon, and one of three binders using a design of experiments (DOE)-guided approach [151]. The binders tested were polyisobutene (OPN), poly(styrene-co-butadiene) rubber (SBR) and hydrogenated nitrile butadiene rubber (hNBR) having different functional groups on the polymer chain. Using DEMS, the influence of the binder choice has been investigated, initially revealing a distinct double peak in CO2 evolution, not observed in previous SSB studies, indicating that in addition to carbonate gassing also binder oxidation occurs. OPN-based cathodes showing the best performance overall revealed the most pronounced gas evolution (O2, CO2, and SO2), which seems counterintuitive, but was explained by the higher SOC achieved with this binder. Using LiNbO3-coated NCM622 instead, a comparison of OPN- and SBR-based cathodes at similar SOC was possible, see Fig. 8(a). In this case, a larger O2 evolution was found for OPN, but a more distinct double peak and cumulative amount of CO2 for SBR, demonstrating that SBR is more readily oxidized than OPN.

For an SSB, the properties of the SE have a profound effect on the cell characteristics. For this reason, the influence of SEs on the gas evolution has been studied by various groups. In an OEMS comparison of a solid polymer electrolyte (SPE) based on trimethylene carbonate units with liquid organic carbonates containing LiTFSI as supporting salt, Sångeland et al. noted that during reduction, the SPE releases CO2, while the liquid electrolyte releases C2H4 [152]. The formation of CO2 has been explained by the presence of trace H2O, forming H2 and OH− ions upon reduction, the latter leading to hydrolysis with subsequent decarboxylation of the electrolyte. At the cathode side, both electrolytes degrade under evolution of CO2 and H2 (formed at the anode from protic degradation species) and, interestingly, SO2 originating from the decomposition of TFSI upon radical attack, due to the weak N–S bond.
Strauss et al. reported a quantitative comparison of the gas evolution of 13C-carbonate-labeled NCM622 CAM in combination with two SEs (Li3PS4 and Li6PS5Cl) and a liquid electrolyte (LP57), noting minor differences in the cumulative amounts of O2 and CO2 (virtually only 13CO2) between the SEs. However, SO2 evolution has been observed when using Li3PS4 [153]. Because SO2 was absent for Li6PS5Cl, a higher stability of this electrolyte against reactive oxygen and/or formation of only solid degradation products was suggested. In comparison, in liquid electrolyte cells, much larger CO2 evolution, with more 12CO2 from electrolyte oxidation than 13CO2, and lower O2 evolution have been detected, indicating almost complete consumption of the released reactive oxygen. By acid titration after cycling, it was shown that in the SSB cathodes, a larger fraction of Li213CO2 remained, presumably due to the lack of acid protons and the known issue of inactive (isolated) CAM in SSBs. To ensure comparability of the gas evolution, a similar SOC was targeted by restricting the specific charge capacity of the liquid electrolyte cells to 240 mAh/g, resulting in a lower upper cutoff potential.
Teo et al. compared the crystalline SE Li6PS5Cl with the glassy SE 1.5Li2S-0.5P2S5-LiI via DEMS among others (in slurry-cast cathodes with LiNbO3-coated NCM622). They observed relatively more O2, but no SO2 evolution in the former and less O2, but also SO2 evolution in the latter, see Fig. 8(b) [154]. This finding is well explainable by the consumption of oxygen for the formation of SO2 and supported by the observation of oxygenated sulfur and phosphorus species via XPS and time-of-flight secondary ion mass spectrometry (ToF–SIMS). However, although more SE degradation has been observed, the electrochemical performance of SSB cells using the glassy SE was better, indicating that a stable and ion-conducting layer of degradation products with good contact to the CAM particles forms and the increased SO2 evolution is a side effect of tight contact between CAM and SE.
The decomposition of SE, in this case based on polyethylene oxide (PEO), has also been studied by Nie et al. [155]. They found that while PEO itself only starts to decompose via dehydrogenation and formation of protonated TFSI (HTFSI) at 4.5 V vs. Li+/Li, in the presence of LCO, this reaction can occur at potentials as low as 4.2 V vs. Li+/Li, due to undercoordinated surface oxygen of LCO, a problem solved via coating the cathode with Li1.4Al0.4Ti1.6(PO4)3. Seidl et al. identified methanol and 2-methoxyethanol as degradation products of HTFSI formation, but observed limited capability for the determination of onset voltages due to low sampling rate [156]. Li et al. prepared a sandwich composite polymer SE, consisting of reduction-resistant PEO and oxidation-resistant polyacrylonitrile with a PVDF layer in between, each containing Li3xLa2/3−xTiO3 fibers. Using DEMS, they showed that while the individual layers alone degrade under gas generation in an NCM811|SE|Li cell, no gas evolution is detected for the sandwich composite [157]. However, the authors reported only the evolution rates of C2H2, C2H4, C2H6, and H2 and did not include CO2.
Lastly, the gassing behavior of garnet SEs has also been investigated. Delluva et al. demonstrated that Li7La3Zr2O12 (LLZO) will release CO2 and O2 at potentials above 3.8 V vs. Li+/Li, as expected for the electrochemical oxidation of Li2CO3 impurities, not only in an LiMn2O4|LLZO|Li cell, but also in an Au|LLZO|Li cell, ruling out CAM contributions to the gas evolution [158]. The authors concluded that at the cathode|SE interface, Li2CO3 impurities of the SE are oxidized, with the resulting gas release leading to delamination, thus highlighting the need for fast and carbonate-free processing of LLZO. For the related Li6.4La3Zr1.4Ta0.6O12, Yang et al. have shown that by coating the garnet SE with a thin layer of LCO, surface contamination is suppressed and stability against Li2CO3 formation in air is achieved [159].
In summary, the (out)gassing tendency of CAMs is similar for liquid- and solid-electrolyte-based cells. However, because many follow-up reactions are occurring at the interface to the electrolyte, SSBs have unique features that allow for an advanced characterization of SE and said interface.
Sodium-ion batteries
Driven by the scarcity of lithium and relative abundance of sodium, many efforts are made to introduce SIBs as a cheaper and more sustainable or complementary alternative to LIBs [160], mostly in the field of stationary energy storage [9]. Among the existing CAMs, layered oxides of either P2 or O3 structure, differing in the coordination and amount of Na per formula unit, show the largest resemblance to LIB cathodes. Apart from that, polyanionic cathodes, foremost Na3V2(PO4)2F3 (NVPF) and Prussian blue analogs (PBAs), also hold promise for application in SIBs [8]. Herein, the outgassing of all three material families is reviewed as well the application of gas evolution measurements to study the SEI formation on different anode materials.
A systematic OEMS study of SIBs has been presented recently by Zhang et al., including screening of common electrolyte solvents (EC, DMC, PC) and a comparison of the CAMs NVPF (polyanionic and showing biphasic behavior), NaNi0.45Zn0.05Mn0.4Ti0.1O2 (NNZMTO, layered and showing purely cation redox and solid-solution behavior), and NaLi1/3Mn2/3O2 (NLMO, involving anion redox) [161]. Versus Na metal anodes, they observed recurring strong H2 evolution at higher potentials with NNZMTO and NVPF cathodes and explained this by the reduction of protic electrolyte degradation products at the anode. A direct comparison of gas evolution rates may be misleading, as the two cathodes were charged to different potentials. Using hard carbon as negative electrode, the H2 evolution was reduced, probably due to better passivation of the anode. Instead, C2H4 was observed as a result of SEI formation. NNZMTO also released CO2 at high potentials, yet only during the first cycle, thus suggesting surface carbonates as the cause. For NLMO, significant O2 release at high potentials in the initial cycle has been detected, with evolution of CO2 also in the subsequent cycles, proving the partial irreversibility of anion redox. It is worth to point out that the authors attempted isotope labeling experiments, but ran into purity issues with the labeled solvents, highlighting another challenge that comes with such experiments.
Starting with P2 cathodes, Maitra et al. demonstrated that Na2/3Mg0.28Mn0.72O2 (NMMO) not only shows oxygen redox without alkali ions in the transition metal layer, but also that no O2 loss occurs during charge, with the only gas evolution contributions coming from surface carbonates and electrolyte decomposition [162]. In a follow-up study, House et al. compared NMMO with the Li-containing Na0.78Li0.25Mn0.75O2 (NLMO) and found that both materials do not show O2 loss at 4.5 V vs. Na+/Na. Utilizing 18O-labeled CAMs under CV charge at 5.0 V, a cell with NMMO only released C16O2 stemming from electrolyte decomposition, while a cell with NLMO also released 18O2 as well as C16O18O and C18O2, clearly indicating the irreversible oxygen loss and chemical oxidation of electrolyte, see Fig. 9(a) [163]. However, shortly after, the same group demonstrated via high-resolution RIXS that molecular oxygen formed both in NMMO and NLMO at 4.5 V, but has no way of escaping the solid phase and is reduced again during discharge. Note that a slight change in stoichiometry of NLMO to Na0.6Li0.2Mn0.8O2 led to a ribbon structure instead of a honeycomb structure, thereby preventing the formation of O2 [164, 165]. This observation emphasizes the rare case that even with good signal-to-noise ratio and low detection limit, the lack of gas detection is necessary but not sufficient to exclude the underlying reaction mechanism.

Zhao et al. observed O2 evolution during charge to 4.5 V vs. Na+/Na for NLMO of again slightly different stoichiometry (Na0.66Li0.22Mn0.78O2) and found that partial bulk substitution of O by F, yielding NLMOF (Na0.65Li0.22Mn0.78O1.99F0.01), suppresses this O2 release [166].
Kulka et al. used OEMS to rule out O2 evolution as a reason for capacity loss in Na0.6MnO2, albeit only at potentials up to 4.0 V vs. Na+/Na [167]. It is in the nature of the P2 phase that as-synthesized materials are Na-deficient, and a higher degree of sodium intercalation is only achieved during operation. While not a problem in half-cells with near infinite Na supply, the capacity of full cells is thus reduced. Adding sacrificial sodium salts that decompose in the initial cycle to the cathode composite can alleviate this issue. Marelli et al. demonstrated that the addition of sodium rhodizonate (Na2C6O6) increases the full cell performance of Na0.67Mn0.5Fe0.5O2 and used OEMS to verify the decomposition to CO2 (the onset of oxidation was as low as 3.8 V vs. Na+/Na) [168].
Only recently, CAMs with O3-type structure have been characterized via in situ gas analysis. Wang et al. used OEMS to quantify the first cycle O2 loss due to anion redox for NaLi1/3Mn2/3O2. From that, they calculated the composition of the charged cathode to be Na0.09Li1/3Mn2/3O1.86 while demonstrating the presence of peroxo-like species within the structure via hard XPS (HAXPES) and RIXS [169]. Two other studies have shown that both in NaMn1/3Fe1/3Ni1/3O2 [170] (charged up to 4.6 V vs. Na+/Na) and in NaNi2/3Ru1/3O2 [171] (charged up to 4.1 V vs. Na+/Na) no O2 evolution occurs, although the formation of peroxo-like species has been observed via X-ray absorption near edge spectroscopy (XANES) and XPS.
The DEMS investigation of the high-entropy PBA Nax(Fe0.2Mn0.2Ni0.2Cu0.2Co0.2)[Fe(CN)6]1−y for SIBs revealed that next to H2 and CO2 as expected gasses from crystal water, surface carbonates and electrolyte decomposition, ethanedinitrile [(CN)2, cyanogen] evolves at high potentials, see Fig. 9(b) [172]. Overall, it has been shown clearly that the oxidative dimerization of anions is not limited to oxide CAMs.
In their extended study [161], Zhang et al. observed continuous gassing of linear carbonates in contact with Na metal, suggesting their inability to form a stable SEI. On hard carbon as the standard anode material, more and diffuse gas evolution has been observed for linear carbonates, compared to sharp gas evolution peaks with cyclic carbonates, indicating again a better SEI formation with the latter. H2 evolution, due to cross-talk via the formation of soluble protic species, is also observed in the SIB systems. Equivalent findings to those in LIBs have been made when discussing the role of electrolyte additives. Both VC and FEC lead to suppressed C2H4 evolution, but increased CO2 evolution during SEI formation, while TMSPi reacts with NaPF6 resulting in constant POF3 release. Sodium difluoro(oxalate)borate was able to suppress most of the gas evolution during SEI formation and cycling.
While graphite cannot reversibly intercalate large amounts of sodium when using standard electrolyte solvents and therefore hard carbon is applied as anode material instead, the use of ether-based electrolytes allows for the highly reversible co-intercalation of solvated sodium [173]. This raises questions about the nature of the SEI involved. Goktas et al. used OEMS to probe the SEI formation on graphite in diglyme containing 1 M NaPF6 as electrolyte, with the surprising finding that no SEI formation is observed via TEM, and yet gas evolution is restricted to the first cycle only [174]. The gas evolution is explained by the not recurring reaction of graphite surface groups, forming soluble reaction products (rather than an SEI). Hence, graphite in diglyme has been reported as the first SEI-free anode. In a following study, again utilizing OEMS, the authors showed that this observation depends on the conductive salt used. While diglyme containing NaOTf and NaPF6 releases the least gas and forms no SEI on graphite, the use of sodium bis(fluorosulfonyl)imide (NaFSI) or NaTFSI leads to significant gas evolution, SEI formation, and capacity fading, as these salts have been found not to be stable against the anode [175].
Tin (Sn) is an alternative anode candidate in SIBs. However, the low initial Coulombic efficiency and poor cycle life are challenging. Both effects have been shown via DEMS to be caused by excessive gas evolution and poor SEI formation, due to electrolyte decomposition at the relatively higher electrode potential of tin than hard carbon. As found by Liu et al., lowering the potential by mechanically pre-alloying Sn and Na leads to the formation of a stable SEI with less concurrent gas evolution and a drastically improved Coulombic efficiency [176]. Qin et al. demonstrated that the use of glyme electrolytes leads to largely suppressed gas evolution and a more stable, inorganic SEI [177].
In summary, the (out)gassing of SIB electrode materials often shows analogies to their respective LIB counterparts, yet the broader chemistry range considered for application leads to a larger variety of possible reaction mechanisms and evolving gasses.
Challenges and future perspectives
As the search for solutions to improve or replace today’s LIB technologies continues, novel and creative ideas are presented and for these, the in situ gas analysis plays an important role in understanding stability, redox activity, and degradation—mostly related to side reactions occurring at electrode interfaces. Thus, it comes as no surprise that more and more research groups develop their own, customized capabilities for in situ gas evolution measurements. After first being applied to understand reaction mechanisms in LIBs in detail, a second spring for gas analysis investigations is now imminent as part of the characterization and evaluation of new battery materials and concepts.
Gas evolution studies on various novel cell chemistries will certainly increase in the near future. First DEMS experiments on potassium-ion batteries have already been reported, showing that the KxCrO2 cathode does not undergo O2 loss during potassium extraction at high potentials, while the evolved CO2 could be attributed to electrolyte (EC/DEC) decomposition (after proving the absence of carbonates via acid titration) [178]. Similarly, DEMS has been used to probe FxReO3 as a host material for fluoride ions, demonstrating that it does not release oxygen upon fluorination and oxidative current is in fact due to operation of the cathode rather than electrolyte decomposition [179]. Beneficial O2 evolution in the first charge cycle has been reported for the zinc-ion battery cathode material Ca2Mn3O8 by Wang et al., allowing for the controlled introduction of performance-enhancing vacancies [180].
In situ gas evolution measurements can, after a comparatively low initial investment [181], be performed on a daily basis in a regular laboratory. However, establishing an experimental setup tailored to the needs not only of today’s, but also tomorrow’s research remains a challenging task (because of many choices and necessary work in design, manufacture, and assembly of custom-built parts). Apart from that, correct operation, data acquisition, and assignment, also of unusual gassing phenomena or m/z signals, require mental and resource commitment, even more so when involving isotope labeling experiments. It is to hope that academic cooperation and exchange are fostered by the DEMS/OEMS community, working on making gas evolution measurements available to a broader range of researchers.
While the study of gas evolution has been shown to allow for insights into many reaction mechanisms, some conclusions can be drawn too fast, especially when comparing materials, due to the complex interplay of SOC/potential, composition, and surface impurities. Moreover, even mechanisms that seem established in the community, such as the 1O2 evolution during battery operation, can be challenged based on the possibility of side reactions and theoretical considerations [182].
Conclusions
In this review article, recent developments in the field of in situ gas analysis of batteries have been discussed, spanning from instrumentation and state-of-the-art LIBs over Li-rich cathode materials and SIBs to SSBs, emphasizing the versatility of the method and its role in the evaluation of current and next-generation electrode materials. The unique gas detection and quantification capabilities of DEMS/OEMS allow elucidating formation, operation and degradation mechanisms in batteries that cannot be assessed by other in situ techniques. Isotope enrichment has been shown to be a powerful and versatile tool, with which multiple processes evolving the same gas species can be distinguished from one another. Still, reaction mechanisms can be more complex than they initially seem, highlighted by the development in understanding the decomposition of surface carbonate impurities among others. At the same time, the combination of gas analysis with complementary techniques, such as EQCM in the study of SEI formation or RIXS in the study of anion redox, can help to provide detailed insights into complex phenomena and becomes more relevant as DEMS/OEMS matures.
The increasing number of research groups performing in situ gas evolution measurements demonstrates the method’s appeal, and we hope that this article will inspire and encourage readers to include gas analysis in their future work.
Data availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski, J. Janek, Angew. Chem. Int. Ed. 58, 10434–10458 (2019)
H.J. Noh, S. Youn, C.S. Yoon, Y.K. Sun, J. Power Sources 233, 121–130 (2013)
X. Ji, Q. Xia, Y. Xu, H. Feng, P. Wang, Q. Tan, J. Power Sources 487, 229362 (2021)
P. Rozier, J.M. Tarascon, J. Electrochem. Soc. 162, A2490–A2499 (2015)
R.J. Clément, Z. Lun, G. Ceder, Energy Environ. Sci. 13, 345–373 (2020)
J. Janek, W.G. Zeier, Nat. Energy 1, 16141 (2016)
C. Li, Z. Wang, Z. He, Y. Li, J. Mao, K. Dai, C. Yan, J. Zheng, Sustain. Mater. Technol. 29, e00297 (2021)
N. Tapia-Ruiz, A.R. Armstrong, H. Alptekin, M.A. Amores, H. Au, J. Barker, R. Boston, W.R. Brant, J.M. Brittain, Y. Chen, M. Chhowalla, Y.S. Choi, S.I.R. Costa, M.C. Ribadeneyra, S.A.M. Dickson, E.I. Eweka, J.D. Forero-Saboya, C.P. Grey, Z. Li, S.F.L. Mertens, R. Mogensen, L. Monconduit, D.M.C. Ould, R.G. Palgrave, P. Poizot, A. Ponrouch, S. Renault, E.M. Reynolds, A. Rudola, R. Sayers, D.O. Scanlon, S. Sen, V.R. Seymour, B. Silv, G.S. Stone, C.I. Thomas, M.M. Titirici, J. Tong, T.J. Wood, D.S. Wright, R. Younesi, J. Phys. Energy 3, 031503 (2021)
K. Chayambuka, G. Mulder, D.L. Danilov, P.H.L. Notten, Adv. Energy Mater. 8, 1800079 (2018)
F. Strauss, D. Kitsche, Y. Ma, J.H. Teo, D. Goonetilleke, J. Janek, M. Bianchini, T. Brezesinski, Adv. Energy Sustain. Res. 2, 2100004 (2021)
Z. Shadike, E. Zhao, Y.N. Zhou, X. Yu, Y. Yang, E. Hu, S. Bak, L. Gu, X.Q. Yang, Adv. Energy Mater. 8, 1702588 (2018)
D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q.H. Yang, M. Wagemaker, F. Kang, X.Q. Yang, B. Li, Adv. Mater. 31, 1806620 (2019)
D. Goonetilleke, J.H. Stansby, N. Sharma, Curr. Opin. Electrochem. 15, 18–26 (2019)
S. Lou, Z. Yu, Q. Liu, H. Wang, M. Chen, J. Wang, Chem 6, 2199–2218 (2020)
A. Schiele, H. Sommer, T. Brezesinski, J. Janek, B.B. Berkes, Differential Electrochemical Mass Spectrometry in Lithium Battery Research (Elsevier, Amsterdam, 2018)
B. Rowden, N. Garcia-Araez, Energy Rep. 6, 10–18 (2020)
T. Herl, F.M. Matysik, ChemElectroChem 7, 2498–2512 (2020)
H. Baltruschat, J. Am. Soc. Mass Spectrom. 15, 1693–1706 (2004)
R. Imhof, P. Novák, J. Electrochem. Soc. 145, 1081–1087 (1998)
R. Lundström, E.J. Berg, J. Power Sources 485, 229347 (2021)
P. Novák, D. Goers, L. Hardwick, M. Holzapfel, W. Scheifele, J. Ufheil, A. Würsig, J. Power Sources 146, 15–20 (2005)
B.B. Berkes, A. Jozwiuk, H. Sommer, T. Brezesinski, J. Janek, Electrochem. Commun. 60, 64–69 (2015)
Z. Jusys, M. Binder, J. Schnaidt, R.J. Behm, Electrochim. Acta 314, 188–201 (2019)
B.D. McCloskey, D.S. Bethune, R.M. Shelby, G. Girishkumar, A.C. Luntz, J. Phys. Chem. Lett. 2, 1161–1166 (2011)
M. He, L. Boulet-Roblin, P. Borel, C. Tessier, P. Novák, C. Villevieille, E.J. Berg, J. Electrochem. Soc. 163, A83–A89 (2016)
N. Tsiouvaras, S. Meini, I. Buchberger, H.A. Gasteiger, J. Electrochem. Soc. 160, A471–A477 (2013)
M. Metzger, C. Marino, J. Sicklinger, D. Haering, H.A. Gasteiger, J. Electrochem. Soc. 162, A1123–A1134 (2015)
H. Hahn, R. Wagner, F. Schappacher, M. Winter, S. Nowak, J. Electroanal. Chem. 772, 52–57 (2016)
L. Geng, D.L. Wood, S.A. Lewis, R.M. Connatser, M. Li, C.J. Jafta, I. Belharouak, J. Power Sources 466, 228211 (2020)
U. Mattinen, M. Klett, G. Lindbergh, R. Wreland Lindström, J. Power Sources 477, 228968 (2020)
K.N. Shitaw, S.C. Yang, S.K. Jiang, C.J. Huang, N.A. Sahalie, Y. Nikodimos, H.H. Weldeyohannes, C.H. Wang, S.H. Wu, W.N. Su, B.J. Hwang, Adv. Funct. Mater. 31, 2006951 (2021)
J. Gim, J. Song, S. Kim, J. Jo, S. Kim, J. Yoon, D. Kim, S.G. Hong, J.H. Park, V. Mathew, J. Han, S.J. Song, J. Kim, Sci. Rep. 6, 23394 (2016)
J.-P. Schmiegel, M. Leißing, F. Weddeling, F. Horsthemke, J. Reiter, Q. Fan, S. Nowak, M. Winter, T. Placke, J. Electrochem. Soc. 167, 060516 (2020)
F. Horsthemke, M. Leißing, V. Winkler, A. Friesen, L. Ibing, M. Winter, S. Nowak, Electrochim. Acta 338, 135894 (2020)
W. Yin, S. Mariyappan, A. Grimaud, J.M. Tarascon, J. Electrochem. Soc. 165, A3326–A3333 (2018)
R. Jung, M. Metzger, F. Maglia, C. Stinner, H.A. Gasteiger, J. Phys. Chem. Lett. 8, 4820–4825 (2017)
D. Streich, C. Erk, A. Guéguen, P. Müller, F.F. Chesneau, E.J. Berg, J. Phys. Chem. C 121, 13481–13486 (2017)
R. Jung, P. Strobl, F. Maglia, C. Stinner, H.A. Gasteiger, J. Electrochem. Soc. 165, A2869–A2879 (2018)
S. Oswald, D. Pritzl, M. Wetjen, H.A. Gasteiger, J. Electrochem. Soc. 168, 120501 (2021)
S. Oswald, M. Bock, H. Gasteiger, J. Electrochem. Soc. (2022). https://doi.org/10.1149/1945-7111/ac5f7f
M. Metzger, P. Walke, S. Solchenbach, G. Salitra, D. Aurbach, H.A. Gasteiger, J. Electrochem. Soc. 167, 160522 (2020)
J. Wandt, A.T.S. Freiberg, A. Ogrodnik, H.A. Gasteiger, Mater. Today 21, 825–833 (2018)
A.T.S. Freiberg, M.K. Roos, J. Wandt, R. de Vivie-Riedle, H.A. Gasteiger, J. Phys. Chem. A 122, 8828–8839 (2018)
N. Laszczynski, S. Solchenbach, H.A. Gasteiger, B.L. Lucht, J. Electrochem. Soc. 166, A1853–A1859 (2019)
L. de Biasi, A. Schiele, M. Roca-Ayats, G. Garcia, T. Brezesinski, P. Hartmann, J. Janek, ChemSusChem 12, 2240–2250 (2019)
N. Li, S. Sallis, J.K. Papp, J. Wei, B.D. McCloskey, W. Yang, W. Tong, ACS Energy Lett. 4, 2836–2842 (2019)
N. Li, S. Sallis, J.K. Papp, B.D. McCloskey, W. Yang, W. Tong, Nano Energy 78, 105365 (2020)
J.K. Papp, N. Li, L.A. Kaufman, A.J. Naylor, R. Younesi, W. Tong, B.D. McCloskey, Electrochim. Acta 368, 137505 (2021)
H. Zhao, Y. Qian, S. Hu, G. Luo, C. Nie, P. Qiu, Y. Kang, H. Wang, Y. Chu, Q. Wang, J. Wang, H. Shao, K. Xu, Y. Deng, ACS Appl. Mater. Interfaces 13, 29676–29690 (2021)
M.S. Milien, H. Beyer, W. Beichel, P. Klose, H.A. Gasteiger, B.L. Lucht, I. Krossing, J. Electrochem. Soc. 165, A2569–A2576 (2018)
D. Weber, Đ. Tripković, K. Kretschmer, M. Bianchini, T. Brezesinski, Eur. J. Inorg. Chem. 2020, 3117–3130 (2020)
S.L. Dreyer, K.R. Kretschmer, Đ. Tripković, A. Mazilkin, R. Chukwu, R. Azmi, P. Hartmann, M. Bianchini, T. Brezesinski, J. Janek, Adv. Mater. Interfaces 9, 2101100 (2022)
L. Eisele, J. Skrotzki, M. Schneider, C. Bolli, C. Erk, T. Ludwig, A. Schaub, P. Novák, I. Krossing, J. Electrochem. Soc. 167, 120505 (2020)
Z. Zhu, D. Yu, Z. Shi, R. Gao, X. Xiao, I. Waluyo, M. Ge, Y. Dong, W. Xue, G. Xu, W.K. Lee, A. Hunt, J. Li, Energy Environ. Sci. 13, 1865–1878 (2020)
S.E. Renfrew, B.D. McCloskey, ACS Appl. Energy Mater. 2, 3762–3772 (2019)
T. Hatsukade, A. Schiele, P. Hartmann, T. Brezesinski, J. Janek, ACS Appl. Mater. Interfaces 10, 38892–38899 (2018)
N. Mahne, S.E. Renfrew, B.D. McCloskey, S.A. Freunberger, Angew. Chem. Int. Ed. 57, 5529–5533 (2018)
T. Bartsch, F. Strauss, T. Hatsukade, A. Schiele, A.-Y. Kim, P. Hartmann, J. Janek, T. Brezesinski, ACS Energy Lett. 3, 2539–2543 (2018)
F. Strauss, S. Payandeh, A. Kondrakov, T. Brezesinski, Mater. Futures 1, 023501 (2022)
A.T.S. Freiberg, J. Sicklinger, S. Solchenbach, H.A. Gasteiger, Electrochim. Acta 346, 136271 (2020)
L.A. Kaufman, B.D. McCloskey, Chem. Mater. 33, 4170–4176 (2021)
S.E. Renfrew, B.D. McCloskey, J. Electrochem. Soc. 166, A2762–A2768 (2019)
G. Houchins, V. Pande, V. Viswanathan, ACS Energy Lett. 5, 1893–1899 (2020)
D. Pritzl, T. Teufl, A.T.S. Freiberg, B. Strehle, J. Sicklinger, H. Sommer, P. Hartmann, H.A. Gasteiger, J. Electrochem. Soc. 166, A4056–A4066 (2019)
S.E. Renfrew, L.A. Kaufman, B.D. McCloskey, ACS Appl. Mater. Interfaces 11, 34913–34921 (2019)
S. Ramakrishnan, B. Park, J. Wu, W. Yang, B.D. McCloskey, J. Am. Chem. Soc. 142, 8522–8531 (2020)
S. Maiti, H. Sclar, Rosy, J. Grinblat, M. Talianker, L. Burstein, M. Noked, B. Markovsky, D. Aurbach, ACS Appl. Mater. Interfaces 12, 32698–32711 (2020)
Q. Li, D. Ning, D. Zhou, K. An, D. Wong, L. Zhang, Z. Chen, G. Schuck, C. Schulz, Z. Xu, G. Schumacher, X. Liu, J. Mater. Chem. A 8, 7733–7745 (2020)
B. Qiu, M. Zhang, L. Wu, J. Wang, Y. Xia, D. Qian, H. Liu, S. Hy, Y. Chen, K. An, Y. Zhu, Z. Liu, Y.S. Meng, Nat. Commun. 7, 12108 (2016)
Q. Luo, Y. Xie, Z. Wu, Q. Xie, D. Yan, H. Zou, W. Yang, S. Chen, ACS Appl. Energy Mater. 4, 4867–4878 (2021)
Z. Zhu, D. Yu, Y. Yang, C. Su, Y. Huang, Y. Dong, I. Waluyo, B. Wang, A. Hunt, X. Yao, J. Lee, W. Xue, J. Li, Nat. Energy 4, 1049–1058 (2019)
M. Metzger, B. Strehle, S. Solchenbach, H.A. Gasteiger, J. Electrochem. Soc. 163, A1219–A1225 (2016)
T. Melin, R. Lundström, E.J. Berg, Adv. Mater. Interfaces 9, 2101258 (2022)
R. Bernhard, M. Metzger, H.A. Gasteiger, J. Electrochem. Soc. 162, A1984–A1989 (2015)
P.G. Kitz, P. Novák, E.J. Berg, ACS Appl. Mater. Interfaces 12, 15934–15942 (2020)
N. Mozhzhukhina, E. Flores, R. Lundström, V. Nyström, P.G. Kitz, K. Edström, E.J. Berg, J. Phys. Chem. Lett. 11, 4119–4123 (2020)
K.U. Schwenke, S. Solchenbach, J. Demeaux, B.L. Lucht, H.A. Gasteiger, J. Electrochem. Soc. 166, A2035–A2047 (2019)
P.G. Kitz, M.J. Lacey, P. Novák, E.J. Berg, J. Power Sources 477, 228567 (2020)
S. Solchenbach, M. Wetjen, D. Pritzl, K.U. Schwenke, H.A. Gasteiger, J. Electrochem. Soc. 165, A512–A524 (2018)
S. Solchenbach, G. Hong, A.T.S. Freiberg, R. Jung, H.A. Gasteiger, J. Electrochem. Soc. 165, A3304–A3312 (2018)
A.O. Tezel, D.K. Streich, A. Guéguen, M. Hahlin, S. Sunde, K. Edström, P. Novák, A.M. Svensson, J. Electrochem. Soc. 167, 130504 (2020)
S. Solchenbach, M. Metzger, M. Egawa, H. Beyer, H.A. Gasteiger, J. Electrochem. Soc. 165, A3022–A3028 (2018)
A. Guéguen, D. Streich, M. He, M. Mendez, F.F. Chesneau, P. Novák, E.J. Berg, J. Electrochem. Soc. 163, A1095–A1100 (2016)
C. Bolli, A. Guéguen, M.A. Mendez, E.J. Berg, Chem. Mater. 31, 1258–1267 (2019)
A. Guéguen, C. Bolli, M.A. Mendez, E.J. Berg, ACS Appl. Energy Mater. 3, 290–299 (2020)
M. Metzger, B. Strehle, S. Solchenbach, H.A. Gasteiger, J. Electrochem. Soc. 163, A798–A809 (2016)
R. Asakura, C. Bolli, P. Novák, R. Robert, ChemElectroChem 7, 2033–2041 (2020)
D. Parikh, L. Geng, H. Lyu, C.J. Jafta, H. Liu, H.M. Meyer, J. Chen, X.G. Sun, S. Dai, J. Li, ACS Appl. Mater. Interfaces 13, 55145–55155 (2021)
A. Schiele, B. Breitung, A. Mazilkin, S. Schweidler, J. Janek, S. Gumbel, S. Fleischmann, E. Burakowska-Meise, H. Sommer, T. Brezesinski, ACS Omega 3, 16706–16713 (2018)
Q. Ji, X. Gao, Q. Zhang, L. Jin, D. Wang, Y. Xia, S. Yin, S. Xia, N. Hohn, X. Zuo, X. Wang, S. Xie, Z. Xu, L. Ma, L. Chen, G.Z. Chen, J. Zhu, B. Hu, P. Müller-Buschbaum, P.G. Bruce, Y.J. Cheng, Adv. Funct. Mater. 29, 1904961 (2019)
J. Rana, J.K. Papp, Z. Lebens-Higgins, M. Zuba, L.A. Kaufman, A. Goel, R. Schmuch, M. Winter, M.S. Whittingham, W. Yang, B.D. McCloskey, L.F.J. Piper, ACS Energy Lett. 5, 634–641 (2020)
N. Guerrini, L. Jin, J.G. Lozano, K. Luo, A. Sobkowiak, K. Tsuruta, F. Massel, L.C. Duda, M.R. Roberts, P.G. Bruce, Chem. Mater. 32, 3733–3740 (2020)
M. Bianchini, A. Schiele, S. Schweidler, S. Sicolo, F. Fauth, E. Suard, S. Indris, A. Mazilkin, P. Nagel, S. Schuppler, M. Merz, P. Hartmann, T. Brezesinski, J. Janek, Chem. Mater. 32, 9211–9227 (2020)
M.M. Thackeray, S.H. Kang, C.S. Johnson, J.T. Vaughey, R. Benedek, S.A. Hackney, J. Mater. Chem. 17, 3112–3125 (2007)
A.R. Armstrong, M. Holzapfel, P. Novák, C.S. Johnson, S.H. Kang, M.M. Thackeray, P.G. Bruce, J. Am. Chem. Soc. 128, 8694–8698 (2006)
K. Luo, M.R. Roberts, N. Guerrini, N. Tapia-Ruiz, R. Hao, F. Massel, D.M. Pickup, S. Ramos, Y.S. Liu, J. Guo, A.V. Chadwick, L.C. Duda, P.G. Bruce, J. Am. Chem. Soc. 138, 11211–11218 (2016)
K. Luo, M.R. Roberts, R. Hao, N. Guerrini, D.M. Pickup, Y.S. Liu, K. Edström, J. Guo, A.V. Chadwick, L.C. Duda, P.G. Bruce, Nat. Chem. 8, 684–691 (2016)
B. Strehle, K. Kleiner, R. Jung, F. Chesneau, M. Mendez, H.A. Gasteiger, M. Piana, J. Electrochem. Soc. 164, A400–A406 (2017)
T. Teufl, B. Strehle, P. Müller, H.A. Gasteiger, M.A. Mendez, J. Electrochem. Soc. 165, A2718–A2731 (2018)
W. Yin, A. Grimaud, G. Rousse, A.M. Abakumov, A. Senyshyn, L. Zhang, S. Trabesinger, A. Iadecola, D. Foix, D. Giaume, J.M. Tarascon, Nat. Commun. 11, 1252 (2020)
D. Schreiner, T. Zünd, F.J. Günter, L. Kraft, B. Stumper, F. Linsenmann, M. Schüßler, R. Wilhelm, A. Jossen, G. Reinhart, H.A. Gasteiger, J. Electrochem. Soc. 168, 030507 (2021)
X. Cao, H. Li, Y. Qiao, M. Jia, P. He, J. Cabana, H. Zhou, Energy Storage Mater. 38, 1–8 (2021)
X. Cao, H. Li, Y. Qiao, M. Jia, X. Li, J. Cabana, H. Zhou, Adv. Mater. 33, 2004280 (2021)
S. Shen, Y. Hong, F. Zhu, Z. Cao, Y. Li, F. Ke, J. Fan, L. Zhou, L. Wu, P. Dai, M. Cai, L. Huang, Z. Zhou, J. Li, Q. Wu, S. Sun, ACS Appl. Mater. Interfaces 10, 12666–12677 (2018)
W. Huang, C. Lin, M. Zhang, S. Li, Z. Chen, W. Zhao, C. Zhu, Q. Zhao, H. Chen, F. Pan, Adv. Energy Mater. 11, 2102646 (2021)
E. Boivin, N. Guerrini, R.A. House, J.G. Lozano, L. Jin, G.J. Rees, J.W. Somerville, C. Kuss, M.R. Roberts, P.G. Bruce, Adv. Funct. Mater. 31, 2003660 (2021)
J. Zhang, Q. Zhang, D. Wong, N. Zhang, G. Ren, L. Gu, C. Schulz, L. He, Y. Yu, X. Liu, Nat. Commun. 12, 3071 (2021)
Y. Shin, W.H. Kan, M. Aykol, J.K. Papp, B.D. McCloskey, G. Chen, K.A. Persson, Nat. Commun. 9, 4597 (2018)
T. Wang, C. Zhang, S. Li, X. Shen, L. Zhou, Q. Huang, C. Liang, Z. Wang, X. Wang, W. Wei, ACS Appl. Mater. Interfaces 13, 12159–12168 (2021)
J.M. Zheng, Z.R. Zhang, X.B. Wu, Z.X. Dong, Z. Zhu, Y. Yang, J. Electrochem. Soc. 155, A775–A782 (2008)
Q. Li, D. Ning, D. Zhou, K. An, G. Schuck, D. Wong, W. Kong, C. Schulz, G. Schumacher, X. Liu, Chem. Mater. 32, 9404–9414 (2020)
S. Maiti, H. Sclar, R. Sharma, N. Vishkin, M. Fayena-Greenstein, J. Grinblat, M. Talianker, L. Burstein, N. Solomatin, O. Tiurin, Y. Ein-Eli, M. Noked, B. Markovsky, D. Aurbach, Adv. Funct. Mater. 31, 2008083 (2021)
E. Evenstein, Rosy, S. Haber, H. Sclar, L. Houben, K. Leung, M. Leskes, M. Noked, Energy Storage Mater. 19, 261–269 (2019)
Rosy, S. Taragin, E. Evenstein, S. Maletti, D. Mikhailova, M. Noked, ACS Appl. Mater. Interfaces 13, 44470–44478 (2021)
B. Sun, M. El Kazzi, E. Müller, E.J. Berg, J. Mater. Chem. A 6, 17778–17786 (2018)
T. Teufl, D. Pritzl, P. Krieg, B. Strehle, M.A. Mendez, H.A. Gasteiger, J. Electrochem. Soc. 167, 110505 (2020)
F. Wu, G.T. Kim, T. Diemant, M. Kuenzel, A.R. Schür, X. Gao, B. Qin, D. Alwast, Z. Jusys, R.J. Behm, D. Geiger, U. Kaiser, S. Passerini, Adv. Energy Mater. 10, 2001830 (2020)
J.G. Han, J.B. Lee, A. Cha, T.K. Lee, W. Cho, S. Chae, S.J. Kang, S.K. Kwak, J. Cho, S.Y. Hong, N.S. Choi, Energy Environ. Sci. 11, 1552–1562 (2018)
J. Xu, M. Sun, R. Qiao, S.E. Renfrew, L. Ma, T. Wu, S. Hwang, D. Nordlund, D. Su, K. Amine, J. Lu, B.D. McCloskey, W. Yang, W. Tong, Nat. Commun. 9, 947 (2018)
Y. Yu, P. Karayaylali, S.H. Nowak, L. Giordano, M. Gauthier, W. Hong, R. Kou, Q. Li, J. Vinson, T. Kroll, D. Sokaras, C.J. Sun, N. Charles, F. Maglia, R. Jung, Y. Shao-Horn, Chem. Mater. 31, 7864–7876 (2019)
Y. Yu, P. Karayaylali, D. Sokaras, L. Giordano, R. Kou, C.J. Sun, F. Maglia, R. Jung, F.S. Gittleson, Y. Shao-Horn, Energy Environ. Sci. 14, 2322–2334 (2021)
F. Ning, B. Li, J. Song, Y. Zuo, H. Shang, Z. Zhao, Z. Yu, W. Chu, K. Zhang, G. Feng, X. Wang, D. Xia, Nat. Commun. 11, 4973 (2020)
E. McCalla, M.T. Sougrati, G. Rousse, E.J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novák, J.M. Tarascon, J. Am. Chem. Soc. 137, 4804–4814 (2015)
E. McCalla, A.S. Prakash, E. Berg, M. Saubanère, A.M. Abakumov, D. Foix, B. Klobes, M.-T. Sougrati, G. Rousse, F. Lepoivre, S. Mariyappan, M.-L. Doublet, D. Gonbeau, P. Novák, G. Van Tendeloo, R.P. Hermann, J.-M. Tarascon, J. Electrochem. Soc. 162, A1341–A1351 (2015)
M. Ting, M. Burigana, L. Zhang, Y.Z. Finfrock, S. Trabesinger, A. Jonderian, E. McCalla, Chem. Mater. 32, 849–857 (2020)
Q. Jacquet, A. Iadecola, M. Saubanère, L. Lemarquis, E.J. Berg, D. Alves Dalla Corte, G. Rousse, M.L. Doublet, J.M. Tarascon, Chem. Mater. 30, 7682–7690 (2018)
H. Li, S. Ramakrishnan, J.W. Freeland, B.D. McCloskey, J. Cabana, J. Am. Chem. Soc. 142, 8160–8173 (2020)
N. Li, M. Sun, W.H. Kan, Z. Zhuo, S. Hwang, S.E. Renfrew, M. Avdeev, A. Huq, B.D. McCloskey, D. Su, W. Yang, W. Tong, Nat. Commun. 12, 2348 (2021)
J. Lee, A. Urban, X. Li, D. Su, G. Hautier, G. Ceder, Science 343, 519–522 (2014)
M.A. Cambaz, A. Urban, S.A. Pervez, H. Geßwein, A. Schiele, A.A. Guda, A.L. Bugaev, A. Mazilkin, T. Diemant, R.J. Behm, T. Brezesinski, M. Fichtner, Chem. Mater. 32, 3447–3461 (2020)
J. Lee, J.K. Papp, R.J. Clément, S. Sallis, D.H. Kwon, T. Shi, W. Yang, B.D. McCloskey, G. Ceder, Nat. Commun. 8, 981 (2017)
J. Lee, D.A. Kitchaev, D.H. Kwon, C.W. Lee, J.K. Papp, Y.S. Liu, Z. Lun, R.J. Clément, T. Shi, B.D. McCloskey, J. Guo, M. Balasubramanian, G. Ceder, Nature 556, 185–190 (2018)
Z. Lun, B. Ouyang, D.A. Kitchaev, R.J. Clément, J.K. Papp, M. Balasubramanian, Y. Tian, T. Lei, T. Shi, B.D. McCloskey, J. Lee, G. Ceder, Adv. Energy Mater. 9, 1802959 (2019)
Y. Yue, N. Li, Y. Ha, M.J. Crafton, B.D. McCloskey, W. Yang, W. Tong, Adv. Funct. Mater. 31, 2008696 (2021)
K. Zhou, S. Zheng, F. Ren, J. Wu, H. Liu, M. Luo, X. Liu, Y. Xiang, C. Zhang, W. Yang et al., Energy Storage Mater. 32, 234–243 (2020)
M.J. Crafton, Y. Yue, T.Y. Huang, W. Tong, B.D. McCloskey, Adv. Energy Mater. 10, 2001500 (2020)
R. Satish, L. Wichmann, M.J. Crafton, R. Giovine, L. Li, J. Ahn, Y. Yue, W. Tong, G. Chen, C. Wang, R.J. Clement, R. Kostecki, ChemElectroChem 8, 3982–3991 (2021)
T.Y. Huang, M.J. Crafton, Y. Yue, W. Tong, B.D. McCloskey, Energy Environ. Sci. 14, 1553–1562 (2021)
W.H. Kan, D. Chen, J.K. Papp, A.K. Shukla, A. Huq, C.M. Brown, B.D. McCloskey, G. Chen, Chem. Mater. 30, 1655–1666 (2018)
Q. Jacquet, A. Iadecola, M. Saubanère, H. Li, E.J. Berg, G. Rousse, J. Cabana, M.L. Doublet, J.M. Tarascon, J. Am. Chem. Soc. 141, 11452–11464 (2019)
D.A. Kitchaev, Z. Lun, W.D. Richards, H. Ji, R.J. Clément, M. Balasubramanian, D.H. Kwon, K. Dai, J.K. Papp, T. Lei, B.D. McCloskey, W. Yang, J. Lee, G. Ceder, Energy Environ. Sci. 11, 2159–2171 (2018)
D. Chen, J. Wu, J.K. Papp, B.D. McCloskey, W. Yang, G. Chen, Small 16, 2000656 (2020)
Y. Yue, Y. Ha, T.Y. Huang, N. Li, L. Li, Q. Li, J. Feng, C. Wang, B.D. McCloskey, W. Yang, W. Tong, ACS Nano 15, 13360–13369 (2021)
M.A. Cambaz, B.P. Vinayan, H. Geßwein, A. Schiele, A. Sarapulova, T. Diemant, A. Mazilkin, T. Brezesinski, R.J. Behm, H. Ehrenberg, M. Fichtner, Chem. Mater. 31, 4330–4340 (2019)
B. Breitung, Q. Wang, A. Schiele, Đ. Tripković, A. Sarkar, L. Velasco, D. Wang, S.S. Bhattacharya, H. Hahn, T. Brezesinski, Batter. Supercaps 3, 361–369 (2020)
Z. Lun, B. Ouyang, D.H. Kwon, Y. Ha, E.E. Foley, T.Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B.D. McCloskey, W. Yang, R.J. Clément, H. Ji, G. Ceder, Nat. Mater. 20, 214–221 (2021)
A.-Y. Kim, F. Strauss, T. Bartsch, J.H. Teo, T. Hatsukade, A. Mazilkin, J. Janek, P. Hartmann, T. Brezesinski, Chem. Mater. 31, 9664–9672 (2019)
F. Strauss, J.H. Teo, J. Maibach, A.-Y. Kim, A. Mazilkin, J. Janek, T. Brezesinski, ACS Appl. Mater. Interfaces 12, 57146–57154 (2020)
Y. Ma, J.H. Teo, F. Walther, Y. Ma, R. Zhang, A. Mazilkin, Y. Tang, D. Goonetilleke, J. Janek, M. Bianchini, T. Brezesinski, Adv. Funct. Mater. (2022). https://doi.org/10.1002/adfm.202111829
Y. Ma, J.H. Teo, D. Kitsche, T. Diemant, F. Strauss, Y. Ma, D. Goonetilleke, J. Janek, M. Bianchini, T. Brezesinski, ACS Energy Lett. 6, 3020–3028 (2021)
J.H. Teo, F. Strauss, Đ. Tripković, S. Schweidler, Y. Ma, M. Bianchini, J. Janek, T. Brezesinski, Cell Rep. Phys. Sci. 2, 100465 (2021)
C. Sångeland, B. Sun, D. Brandell, E.J. Berg, J. Mindemark, Batter. Supercaps 4, 785–790 (2021)
F. Strauss, J.H. Teo, A. Schiele, T. Bartsch, T. Hatsukade, P. Hartmann, J. Janek, T. Brezesinski, ACS Appl. Mater. Interfaces 12, 20462–20468 (2020)
J.H. Teo, F. Strauss, F. Walther, Y. Ma, S. Payandeh, T. Scherer, M. Bianchini, J. Janek, T. Brezesinski, Mater. Futures 1, 015102 (2022)
K. Nie, X. Wang, J. Qiu, Y. Wang, Q. Yang, J. Xu, X. Yu, H. Li, X. Huang, L. Chen, ACS Energy Lett. 5, 826–832 (2020)
L. Seidl, R. Grissa, L. Zhang, S. Trabesinger, C. Battaglia, Adv. Mater. Interfaces 9, 2100704 (2022)
B. Li, Q. Su, C. Liu, Q. Wang, M. Zhang, S. Ding, G. Du, B. Xu, J. Power Sources 496, 229835 (2021)
A.A. Delluva, J. Kulberg-Savercool, A. Holewinski, Adv. Funct. Mater. 31, 2103716 (2021)
Y.N. Yang, Y.X. Li, Y.Q. Li, T. Zhang, Nat. Commun. 11, 5519 (2020)
J.M. Tarascon, Joule 4, 1616–1620 (2020)
L. Zhang, C. Tsolakidou, S. Mariyappan, J.M. Tarascon, S. Trabesinger, Energy Storage Mater. 42, 12–21 (2021)
U. Maitra, R.A. House, J.W. Somerville, N. Tapia-Ruiz, J.G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M.A. Pérez-Osorio, F. Massel, D.M. Pickup, S. Ramos, X. Lu, D.E. McNally, A.V. Chadwick, F. Giustino, T. Schmitt, L.C. Duda, M.R. Roberts, P.G. Bruce, Nat. Chem. 10, 288–295 (2018)
R.A. House, U. Maitra, L. Jin, J.G. Lozano, J.W. Somerville, N.H. Rees, A.J. Naylor, L.C. Duda, F. Massel, A.V. Chadwick, S. Ramos, D.M. Pickup, D.E. McNally, X. Lu, T. Schmitt, M.R. Roberts, P.G. Bruce, Chem. Mater. 31, 3293–3300 (2019)
R.A. House, U. Maitra, M.A. Pérez-Osorio, J.G. Lozano, L. Jin, J.W. Somerville, L.C. Duda, A. Nag, A. Walters, K.J. Zhou, M.R. Roberts, P.G. Bruce, Nature 577, 502–508 (2020)
E. Boivin, R.A. House, M.A. Pérez-Osorio, J.J. Marie, U. Maitra, G.J. Rees, P.G. Bruce, Joule 5, 1267–1280 (2021)
C. Zhao, Q. Yang, F. Geng, C. Li, N. Zhang, J. Ma, W. Tong, B. Hu, ACS Appl. Mater. Interfaces 13, 360–369 (2021)
A. Kulka, C. Marino, K. Walczak, C. Borca, C. Bolli, P. Novák, C. Villevieille, J. Mater. Chem. A 8, 6022–6033 (2020)
E. Marelli, C. Marino, C. Bolli, C. Villevieille, J. Power Sources 450, 227617 (2020)
Q. Wang, S. Mariyappan, G. Rousse, A.V. Morozov, B. Porcheron, R. Dedryvère, J. Wu, W. Yang, L. Zhang, M. Chakir, M. Avdeev, M. Deschamps, Y.S. Yu, J. Cabana, M.L. Doublet, A.M. Abakumov, J.M. Tarascon, Nat. Mater. 20, 353–361 (2021)
Y. Yu, D. Ning, Q. Li, A. Franz, L. Zheng, N. Zhang, G. Ren, G. Schumacher, X. Liu, Energy Storage Mater. 38, 130–140 (2021)
N. Voronina, N. Yaqoob, H.J. Kim, K.S. Lee, H.D. Lim, H.G. Jung, O. Guillon, P. Kaghazchi, S.T. Myung, Adv. Energy Mater. 11, 2100901 (2021)
Y. Ma, Y. Ma, S.L. Dreyer, Q. Wang, K. Wang, D. Goonetilleke, A. Omar, D. Mikhailova, H. Hahn, B. Breitung, T. Brezesinski, Adv. Mater. 33, 2101342 (2021)
B. Jache, P. Adelhelm, Angew. Chem. Int. Ed. 53, 10169–10173 (2014)
M. Goktas, C. Bolli, E.J. Berg, P. Novák, K. Pollok, F. Langenhorst, M.V. Roeder, O. Lenchuk, D. Mollenhauer, P. Adelhelm, Adv. Energy Mater. 8, 1702724 (2018)
M. Goktas, C. Bolli, J. Buchheim, E.J. Berg, P. Novák, F. Bonilla, T. Rojo, S. Komaba, K. Kubota, P. Adelhelm, ACS Appl. Mater. Interfaces 11, 32844–32855 (2019)
W. Liu, X. Chen, C. Zhang, H. Xu, X. Sun, Y. Zheng, Y. Yu, S. Li, Y. Huang, J. Li, ACS Appl. Mater. Interfaces 11, 23207–23212 (2019)
B. Qin, A. Schiele, Z. Jusys, A. Mariani, T. Diemant, X. Liu, T. Brezesinski, R.J. Behm, A. Varzi, S. Passerini, ACS Appl. Mater. Interfaces 12, 3697–3708 (2020)
H. Kim, D.H. Seo, A. Urban, J. Lee, D.H. Kwon, S.H. Bo, T. Shi, J.K. Papp, B.D. McCloskey, G. Ceder, Chem. Mater. 30, 6532–6539 (2018)
N.H. Bashian, M. Zuba, A. Irshad, S.M. Becwar, J. Vinckeviciute, W. Rahim, K.J. Griffith, E.T. McClure, J.K. Papp, B.D. McCloskey, D.O. Scanlon, B.F. Chmelka, A. Van Der Ven, S.R. Narayan, L.F.J. Piper, B.C. Melot, Chem. Mater. 33, 5757–5768 (2021)
L. Wang, Z. Cao, P. Zhuang, J. Li, H. Chu, Z. Ye, D. Xu, H. Zhang, J. Shen, M. Ye, ACS Appl. Mater. Interfaces 13, 13338–13346 (2021)
N. Schlüter, P. Novák, D. Schröder, Adv. Energy Mater 12, 2200708 (2022)
A. Schürmann, B. Luerßen, D. Mollenhauer, J. Janek, D. Schröder, Chem. Rev. 121, 12445–12464 (2021)
Acknowledgments
This work was supported by BASF SE. We thank J. H. Teo for insightful discussion.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dreyer, S.L., Kondrakov, A., Janek, J. et al. In situ analysis of gas evolution in liquid- and solid-electrolyte-based batteries with current and next-generation cathode materials. Journal of Materials Research 37, 3146–3168 (2022). https://doi.org/10.1557/s43578-022-00586-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43578-022-00586-2