
Abstract
OBJECTIVE
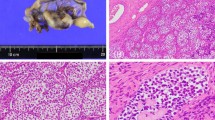
Frasier syndrome (FS) phenotype in 46,XY patients usually consists of female external genitalia, gonadal dysgenesis, high risk of gonadoblastoma and the development of end stage renal failure usually in the second decade of life. FS is caused by heterozygous de novo intronic splice site mutations of the Wilms’ tumor suppressor gene 1 (WT1), although a few cases with typical exonic WT1 Denys-Drash mutations that resemble an FS phenotype have been described. The aim of this study was to present further data on the spectrum of FS phenotypes through the evaluation of a 29-year-old patient with a predominantly male phenotype and coexistence of Sertoli cell tumor and gonadoblastoma.
RESULTS
Genetic analysis using standard methods for DNA sequencing confirmed FS due to a WT1 gene mutation, IVS9+4C>T.
CONCLUSIONS
This very rare case illustrates the natural course of FS over many years due to the neglect by the patient to address his need for follow-up, while adding further data on the spectrum of FS phenotypes associated with IVS9+4 C>T mutations. The coexistence of the rare Sertoli cell tumor and gonadoblastoma emphasizes that early clinical recognition and molecular identification facilitates appropriate patient management, especially with respect to the high risk of gonadal malignancy.
Article PDF
Similar content being viewed by others


References
Frasier SD, Bashore RA, Mosier HD, 1964 Gonado-blastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr 64: 740–745.
Moorthy AV, Chesney RW, Lubinsky M, 1987 Chronic renal failure and XY gonadal dysgenesis: Frasier syndrome—a commentary on reported cases. Am J Med Genet 3: 297–302.
Jeanpierre C, Denamur E, Henry I, et al, 1998 Identification of constitutional WT1 mutations in patients with isolated diffuse mesangial sclerosis and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am J Hum Genet 62: 824–833.
Royer-Pokora B, Beier M, Henzler M, 2004 Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms’ tumor development. Am J Med Genet 127A: 249–257.
Saylam K, Simon P, 2003 WT1 gene mutation responsible for male sex reversal and renal failure: the Frasier syndrome. Eur J Obstet Gynecol Reprod Biol 110: 111–113.
Scharnhorst V, van der Eb AJ, Jochemsen AG, 2001 WT1 proteins: functions in growth and differentiation. Gene 273: 141–161.
Rauscher FJ 3rd, Morris JF, Tournay OE, et al, 1990 Binding of the Wilms’ tumor locus zinc finger protein to the EGR-1 consensus sequence. Science 250: 1259–1262.
Wang ZY, Qiu QQ, Deuel TF, 1993 The Wilms’ tumor gene product WT1 activates or suppresses transcription through separate functional domains. J Biol Chem 268: 9172–9175.
Haber DA, Sohn RL, Buckler AJ, et al, 1991 Alternative splicing and genomic structure of the Wilms’ tumor gene WT1. Proc Nat Acad Sciences USA 88: 9618–9622.
Barbaux S, Niaudet P, Gubler MC, et al, 1997 Donor splice site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17: 467–470.
Fallat ME & Donahoe PK, 2006 Intersex genetic anomalies with malignant potential. Curr Opin Pediatr 18: 305–311.
Megremis S, Mitsioni A, Fylaktou I, et al, 2011 Broad and unexpected phenotypic expression in Greek children with steroid-resistant nephrotic syndrome due to mutations in the Wilms’ tumor 1 (WT1) gene. Eur J Pediatr 170: 1529–34. Epub ahead of print, Apr 16.
Klamt B, Koziell A, Poulat F, et al, 1998 Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 _/_ KTS splice isoforms. Hum Molec Genet 7: 709–714.
Chan WK, To KF, But WM, et al, 2006 Frasier syndrome: a rare cause of delayed puberty. Hong Kong Med J 12: 225–227.
Melo KF, Martin RM, Costa EM, 2002 An unusual phenotype of Frasier syndrome due to IVS9+4C>T mutation in the WT1 gene: Predominantly male ambiguous genitalia and absence of gonadal dysgenesis. J Clin Endocrinol & Metabol 87: 2500–2505.
Cooper DN and Youssoufian H, 1988 The CpG dinucleotide and human genetic disease. Hum Genet: 78: 151–155.
Schumacher V, Gueler B, Looijenga LH, et al, 2008 Characteristics of testicular dysgenesis syndrome and decreased expression of SRY and SOX9 in Frasier syndrome. Molec Reprod Devel 75: 1484–1494.
Denamur E, Bocquet N, Baudouin V, et al, 2000 WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental glomerulosclerosis. Nat Kidney Int 57: 1868–1872.
Auber F, Jeanpierre C, Denamur E, et al, 2009 Management of Wilms tumors in Drash and Frasier syndromes. Pediatr Blood & Cancer 52: 55–59.
Kohsaka T, Tagawa M, Takekoshi Y, et al, 1999 Exon 9 mutations in the WT1 gene, without influencing KTS splice isoforms, are also responsible for Frasier syndrome. Hum Mut 14: 466–470.
Tajima T, Sasaki S, Tanaka Y, et al, 2003 46,XY phenotypic male with focal segmental glomerulosclerosis caused by theWT1 splice site mutation. Horm Res Pediatr 60: 302–305.
Lee EK, Holzbeierlein JM, 2009 Management of the contralateral testicle in patients with unilateral testicular cancer. World J Urol 27: 421–426.
Looijenga LH, Hersmus R, Oosterhuis JW, Cools M, Drop SL, Wolffenbuttel KP, 2007 Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 21: 480–495.
Koziel A, Charmandari E, Hindmarsh PC, et al, 2000 Frasier syndrome, part of the Denys Drash continuum or simply a WT1 gene associated disorder of intersex and nephropathy? Clin Endocrinol 52: 519–524.
Gomez-García I, Romero Molina M, López-García Moreno A, et al, 2010 Sertoli cell tumor, a rare testicular tumor, our experience and review of the literature. Arch Espanol Urol 63: 392–395.
Koziell AB & Grundy R, 1999 Frasier and Denys-Drash syndromes: different disorders or part of a spectrum. Arch Dis Childhood 81: 365–369.
Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al, 1999 The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms’ tumor. Hum Mut 13: 146–153.
Chernin G, Vega-Warner V, Schoeb DS, et al, Members of the GPN Study Group, 2010 Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clin J Am Soc Nephrol 5: 1655–1662.
Author information
Authors and Affiliations
Corresponding author
Additional information
These authors contributed equally
Rights and permissions
About this article
Cite this article
Kitsiou-Tzeli, S., Deligiorgi, M., Malaktari-Skarantavou, S. et al. Sertoli cell tumor and gonadoblastoma in an untreated 29-year-old 46,XY phenotypic male with Frasier syndrome carrying a WT1 IVS9+4C>T mutation. Hormones 11, 361–367 (2012). https://doi.org/10.14310/horm.2002.1366
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.14310/horm.2002.1366