
Abstract
Lecanemab (Leqembi®) is approved in the United States for the treatment of Alzheimer’s disease (AD) to be initiated in early AD (mild cognitive impairment [MCI] due to AD or mild AD dementia) with confirmed brain amyloid pathology. Appropriate Use Recommendations (AURs) are intended to help guide the introduction of new therapies into real-world clinical practice. Community dwelling patients with AD differ from those participating in clinical trials. Administration of lecanemab at clinical trial sites by individuals experienced with monoclonal antibody therapy also differs from the community clinic-based administration of lecanemab. These AURs use clinical trial data as well as research and care information regarding AD to help clinicians administer lecanemab with optimal safety and opportunity for effectiveness. Safety and efficacy of lecanemab are known only for patients like those participating in the phase 2 and phase 3 lecanemab trials, and these AURs adhere closely to the inclusion and exclusion criteria of the trials. Adverse events may occur with lecanemab including amyloid related imaging abnormalities (ARIA) and infusion reactions. Monitoring guidelines for these events are detailed in this AUR. Most ARIA with lecanemab is asymptomatic, but a few cases are serious or, very rarely, fatal. Microhemorrhages and rare macrohemorrhages may occur in patients receiving lecanemab. Anticoagulation increases the risk of hemorrhage, and the AUR recommends that patients requiring anticoagulants not receive lecanemab until more data regarding this interaction are available. Patients who are apolipoprotein E ε4 (APOE4) gene carriers, especially APOE4 homozygotes, are at higher risk for ARIA, and the AUR recommends APOE genotyping to better inform risk discussions with patients who are lecanemab candidates. Clinician and institutional preparedness are mandatory for use of lecanemab, and protocols for management of serious events should be developed and implemented. Communication between clinicians and therapy candidates or those on therapy is a key element of good clinical practice for the use of lecanemab. Patients and their care partners must understand the potential benefits, the potential harms, and the monitoring requirements for treatment with this agent. Culture-specific communication and building of trust between clinicians and patients are the foundation for successful use of lecanemab.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lecanemab (Leqembi®), an anti-amyloid monoclonal antibody, is approved by the United States (US) Food and Drug Administration (FDA) for the treatment of Alzheimer’s disease (AD) initiated in early AD (mild cognitive impairment (MCI) or mild dementia due to AD). Treatment candidates should have amyloid pathology as demonstrated by amyloid positron emission tomography (PET) or cerebrospinal fluid (CSF) tests indicative of AD. Lecanemab was approved using the accelerated pathway available to the FDA for drugs that were developed to treat a serious or life threatening illnesses and have an effect on a biomarker considered reasonably likely to predict a clinical response, taking into account the prevalence of the condition and the availability or lack of alternative treatments (1). In the accelerated approval framework, a confirmatory trial is required to demonstrate clinical benefit and the agent can be withdrawn from the market if the confirmatory trial is negative.
In a Phase 2 trial, lecanemab was observed to produce marked lowering of amyloid plaques based on evidence derived from amyloid PET. Lecanemab has completed a confirmatory phase 3 trial (CLARITY-AD) and the FDA will review data from the CLARITY AD to determine if standard approval based on clinical outcomes of the trial is warranted (2). A decision by the FDAregarding standard approval of lecanemab is expected in mid-2023. The accelerated pathway was used recently for the approval of the anti-amyloid monoclonal antibody aducanumab (Aduhelm®) (3).
Marketed lecanemab (Leqembi™) is accompanied by Prescribing Information (PI) from the FDA that describes the indications and usage; dosage, forms, strengths, and administration; contraindications; warnings and precautions; adverse reactions; use in specific populations; development and preparation; clinical pharmacology; nonclinical toxicology; clinical studies; how supplied/storage and handling; and patient counselling information. The PI does not describe how to translate the information into clinical care, adjust clinical practices and workflow to use the agent safely, or present the potential benefits and harms to patients and care partners. Appropriate Use Recommendations (AURs) were developed to assist in guiding the use of new agents such as lecanemab into clinical practice and patient care and to anticipate challenges that may arise with use of a new therapy in real-world settings. Health care systems and advocacy groups may use the AURs to inform use of lecanemab. We describe the appropriate patient for treatment with lecanemab; dosing, administration, and monitoring of lecanemab; apolipoprotein E (APOE) genotyping of lecanemab treatment candidates; discussions with patients and care partners concerning lecanemab treatment; and clinician and workflow considerations regarding lecanemab use in practice. We describe AD-related populations for which there is no available information on the safe use of lecanemab.
The AUR for lecanemab does not discuss efficacy, reimbursement, or accelerated approval. The meaningfulness of the response to lecanemab has been debated, and we do not address that issue as part of use recommendations focusing on provision of lecanemab in community settings (4). The AUR is intended for use by clinicians who have decided to provide lecanemab as a treatment option for patients with early symptomatic AD. AURs are not intended to replace clinician judgment. Clinicians make management choices in the patient’s best interest in conjunction with patients and their care partners; these decisions may vary from the recommendations made in this AUR. All recommendations of the AUR are within the guidelines of the FDA-approved PI and fulfill the requirements of the on-label use of lecanemab. These recommendations may be adjusted as additional data are gathered.
Method
This lecanemab AUR was developed by the Alzheimer’s Disease and Related Disorders Therapeutics Work Group (ADRD TWG) that was formed to address emerging issues in ADRD therapeutics. The ADRD TWG created the AUR for aducanumab (5). Collaborators with special expertise are invited to join TWG projects to augment the expert panel as occurred with the aducanumab AUR update and with the current lecanemab AUR (6). The AUR is based on the PI approved by the FDA, clinical trial data (Phase 2 and Phase 3), information from other drug treatments of dementia, review of the literature on AD and AD treatment, and expert opinion (2, 7–9). The AURs are based on all available data on lecanemab to date including the Phase 3 CLARITY AD trial.
Lecanemab
Lecanemab is a recombinant humanized immunoglobulin gamma 1 (IgG1) anti-amyloid monoclonal antibody that binds to amyloid oligomers, protofibrils, and insoluble fibrils. Protofibrils represent a high molecular weight species of soluble amyloid and are preferentially bound by lecanemab (10). The murine version of lecanemab, mAb158, exhibits a binding preference for amyloid beta-protein (Aβ) protofibrils over Aβ monomers and a less marked binding preference for protofibrils over aggregated fibrillar amyloid. Lecanemab and mAb158 recognize soluble amyloid aggregates in human AD brain extracts (11, 12). The antibody is expressed in a Chinese hamster ovary cell line.
Appropriate Patient for Treatment with Lecanemab
Clinical Diagnosis
Lecanemab has been tested in patients with MCI due to AD and mild AD dementia diagnosed by the National Institute on Aging (NIA)-Alzheimer’s Association (AA) clinical criteria (13, 14) with confirmed amyloid pathology. To ensure that trial participants had mild severity of cognitive impairment, Mini-Mental State Examination (MMSE) (15) scores of 22–30 were required. Lecanemab’s efficacy and safety in less severe (e.g., preclinical AD (16)) and more advanced stages of AD dementia have not been established. We recommend that all patients being considered for lecanemab therapy meet diagnostic criteria for MCI due to AD or probable mild AD dementia with biomarker evidence (amyloid PET or CSF) of the AD pathophysiological process (Table 1).
Clinical Features Relevant to Lecanemab
The lecanemab phase 2 and phase 3 studies excluded patients outside a specific age range (50–90 years old). Participants with immunologic disorders who were not adequately controlled or require therapy with immunoglobulins, systemic monoclonal antibodies, systemic immunosuppressants or plasmapheresis were excluded in the phase 3 CLARITY AD lecanemab study. Participants with other unstable medical conditions as well as those with stroke or transient ischemic attacks, bleeding disorders, or seizures in the previous 12 months were excluded in the phase 3 trial. Women who were pregnant or lactating were not be treated with lecanemab; no information is available on use in these situations. Individuals with depression who scored >8 on the Geriatric Depression Scale or had a body mass index (BMI) greater than 35 or less than 17 were excluded from both the phase 2 and 3 studies. We recommend adopting these same criteria while allowing medical judgment for individual circumstances when the impact of these modifications has been considered (Table 2). CLARITY AD excluded patients who had seizures within the past 12 months. A history of seizures may be related to symptoms of more severe amyloid related imaging abnormalities (ARIA; discussed in more detail below) including seizures and status epilepticus (6). We recommend excluding patients with any history of seizures until additional data are available. Cerebral amyloid angiopathy-related inflammation/amyloid beta-related angiitis (CAA-ri/ABRA) increase the risk for ARIA (discussed below) and should exclude patients as treatment candidates.
Concomitant Medications
Lecanemab trials allowed the concurrent administration of symptomatic anti-dementia therapies (cholinesterase inhibitors and memantine). These agents can be allowed as concomitant medications by practitioners providing lecanemab. Patients receiving treatment for medical or psychiatric illnesses may be considered for treatment with lecanemab if the medical or psychiatric condition and the relevant medication doses are stable at the time of treatment initiation and if the patient is expected to be adherent to the schedule of infusions, scans, and clinical assessments required.
Aducanumab (Adulhelm®) is an anti-amyloid monoclonal antibody approved for the treatment of early AD. It is in the same class of agents as lecanemab and the treatment population is the same. The administration, titration, and magnetic resonance imaging (MRI) monitoring of aducanumab differ from those of lecanemab (6). Patients should not be given lecanemab if they are receiving aducanumab or had severe or recurrent ARIA with use of aducanumab.
Patients on anticoagulants are at higher risk for macrohemorrhage associated with lecanemab therapy. We recommend excluding patients from treatment with lecanemab if they are on warfarin, vitamin K antagonists, or direct oral anticoagulants (dabigatran, rivaroxaban, edoxaban, apixaban, betrixaban), or heparin until more evidence has accrued regarding the safety of administering lecanemab to patients on anticoagulants in the real-world practice setting. Severe, multi-focal brain hemorrhages leading to death were reported in a patient treated with tPA for acute stroke who had received lecanemab during an open-label extension (17). Lecanemab may increase the risk of hemorrhage from concomitant administration of thrombolytics (intravenous or intra-arterial), and we recommend that patients on lecanemab not be treated with acute thrombolytics until safety evidence of their combined use is available. Participants with clotting disorders should be excluded. Participants in lecanemab trials were allowed to continue or initiate treatment with aspirin. We recommend allowing patients on standard doses of aspirin (up to 325 mg/day) or other antiplatelet agents (clopidogrel, prasugrel, ticagrelor; at standard therapeutic doses) to be considered for treatment with lecanemab if they meet other criteria for therapy. Patients who are homozygous for the APOE4 gene are at increased risk for ARIA with lecanemab administration, (see below) and the risk may be increased with antiplatelet agents.
Imaging Recommendations
A positive amyloid biomarker — either elevated amyloid on PET or elevated phosphorylated tau and low Aβ42 level (increased p-tau/Aβ42 ratio) in the CSF — is required prior to initiating treatment with lecanemab to establish that abnormal amyloid, the target of anti-amyloid MABs, is present. Positive tau PET is indicative of the presence of AD, but individuals may have a positive amyloid PET and a negative tau PET (18). Given this lack of concordance in some patients, we recommend use of amyloid PET to demonstrate the presence of the amyloid target when imaging is the biomarker modality used for confirmation. Reliable and approved blood biomarkers for AD pathology — elevated plasma phospho-tau, decreased Aβ 42/40 ratio, abnormal profiles from combinations of biomarkers — may become fully validated soon but are not currently considered adequate to identify appropriate patients for treatment. High negative predictive values of some blood-based biomarkers suggests that negative results might be the basis for deciding which individuals are very unlikely to have AD and do not require further assessment (19).
A non-contrast MRI, utilizing TI fluid-attenuated inversion recovery (FLAIR) and T2*-weighted Gradient Recalled Echo (GRE) or equivalent sequences (such as susceptibility weighted imaging (SWI)), and diffusion-weighted imaging (DWI), preferably on a 3T magnet should be obtained to determine if an individual is a candidate for lecanemab therapy. If a pre-existing MRI with the aforementioned sequences obtained within 12 months prior to initiating therapy is available, it can be used to approve the patient for consideration for lecanemab therapy if abnormal amyloid status is confirmed and other recommendations are met. MRI-based exclusion criteria of the lecanemab phase 3 (CLARITY AD) study included a history of any CNS macrohemorrhage >10 mm in diameter, more than 4 microhemorrhages (<10 mm in diameter), evidence of superficial siderosis, evidence of brain vasogenic edema, significant white matter hyperintensities, multiple lacunar strokes, or any cerebral strokes involving a major vascular territory (20). Evidence of cerebral contusion, encephalomalacia, brain aneurysms or other vascular malformations, central nervous system (CNS) infection, and brain tumors other than meningioma or arachnoid cysts excluded patients from phase 3 trial participation. These same restrictions should apply when considering patients for treatment with lecanemab (Table 2). MRI evidence of underlying CAA-ri/ABRA or other conditions placing patients at risk for ARIA as well as more serious forms of ARIA should exclude patients as treatment candidates. Table 3 presents the criteria for probable CAA-ri with the corresponding MRI description (21).
Eligible patients must have a screening MRI within 12 months of treatment initiation to establish the potential safety of lecanemab therapy and MRI is also used to monitor for ARIA (discussed below). Those unable to undergo MRI due to claustrophobia, pacemaker, defibrillator, or metal implants are not eligible for lecanemab therapy. Computerized tomography (CT) is not adequate to detect the pathology that can render a patient inappropriate for lecanemab therapy or to monitor for ARIA once treatment is initiated.
Appropriate Use of APOE Genotyping or Proteotyping When Considering Treatment with Lecanemab
The APOE4 genotype represents the strongest genetic risk factor for sporadic AD (22–24). APOE in humans has three alleles: APOE ε3 (APOE3) is most common; APOE ε2 (APOE2) is least common and reduces the risk of AD; while APOE ε4 (APOE4), present in ∼20–25% of the population, increases the risk of clinical AD in a dose-dependent manner. There is a significant interaction with sex, with female APOE4 carriers at higher risk for AD than males, particularly at younger ages (25). APOE4 genotype increases the risk of CAA and common AD co-pathologies such as cerebrovascular disease (22). Most patients with younger onset AD have at least one APOE4 allele. In the CLARITY AD trial, 69% of participants had at least one allele; 53% were heterozygotes and 16% were homozygotes (2). The APOE gene produces the APOE protein, and some laboratory tests determine APOE status by assessing the patient’s proteotype.
APOE4 carriers (especially homozygotes) are at increased risk for ARIA, symptomatic ARIA, and recurrent ARIA (discussed below; shown in Table 5). They are at increased risk for CAA-ri/ABRA, an additional risk factor for ARIA. We recommend APOE genotyping of all treatment candidates before initiating lecanemab therapy. This information will inform risk discussions and help guide safety considerations.
Appropriate Dosing, Administration and Monitoring of Lecanemab
Administration of Lecanemab
Lecanemab is administered intravenously every other week. No titration of lecanemab is required. Dosing is weight-adjusted, and patients are administered 10 milligrams per kilogram of body weight. Lecanemab is provided in vials of 500 mg/5 mL (100 mg/mL) or 200 mg/2 mL (100 mg/mL). It is added to an infusion bag containing 250 mL of 0.9% sodium chloride injection and administered through an intravenous line with a terminal low-protein binding 0.2 micron in-line filter. The infusion requires approximately 1 hour. As infusion reactions may occur (described below), patients should be observed for 3 hours after the first infusion with a follow-up telephone call later that day, as delayed infusion reactions are occasionally observed. The postinfusion observation period may be reduced to 2 hours for the second and third infusions and to 30 minutes for subsequent infusions if no infusion reactions have occurred. If an infusion is missed, administer the next dose as soon as possible and every two weeks thereafter.
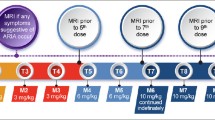
The principal side effects of lecanemab are infusion reactions and ARIA. Post-treatment monitoring of patients is directed principally at detecting ARIA and guiding management decisions that minimize the likelihood of worsening or recurrence of ARIA. We recommend obtaining MRIs after the 5th, 7th, and 14th infusions as outlined in the PI. We recommend an additional week 52 (i.e., before the 26th infusion) MRI scan, especially for APOE4 carriers and those with evidence of ARIA on earlier MRIs. This safety monitoring should be supplemented by unscheduled safety MRIs, obtained in response to the occurrence of symptoms potentially caused by ARIA. Symptoms of ARIA may be non-specific, such as headache and dizziness and may overlap with symptoms of AD, such as increasing confusion. Table 4 presents a list of the symptoms most commonly observed in patients with symptomatic ARIA. Deciding whether an unscheduled safety MRI is indicated requires clinical judgment based on the quality and intensity of the symptoms and the likelihood that they are produced by ARIA. Heightened vigilance for ARIA is warranted in APOE4 carriers, especially homozygotes. After 12 months of treatment, obtaining MRIs should be guided by patient symptoms and prior MRI findings.
Amyloid-Related Imaging Abnormalities
ARIA is a common side effect of treatment with amyloid-lowering monoclonal antibodies. Two types of ARIA occur: ARIA-E with edema and ARIA-H with hemorrhagic changes. ARIA is typically mild-moderate radiographically, asymptomatic, or mildly symptomatic clinically, and self-limited (reversible). ARIA occurred in 12.6% of all participants based on MRI in the CLARITY AD (phase 3) trial; 2.8% were symptomatic. Of the Phase 2 participants, 9.9% on the 10 mg/kg biweekly dose exhibited radiographic ARIA (i.e., the dose administered in the CLARITY AD phase 3 trial). Radiographic ARIA rates and the rates of ARIA with symptoms were substantially higher among patients with an APOE ε4 (APOE4) genotype, especially those homozygous for APOE4, compared to noncarriers (Table 5). In the CLARITY AD trial, the rate of ARIA in APOE4 noncarriers was 5.4%, in APOE4 heterozygotes it was 10.9%, and in APOE4 homozygotes the rate was 32.6%. Rates of symptomatic ARIA were 1.4%, 1.7%, and 9.2%, respectively. These observations support APOE genotyping of all patients (who agree) to inform risk discussions with treatment candidates and their care partners (discussed in more detail below). Table 5 shows the rates of ARIA-E and ARIA-H for the Phase 2 and the Phase 3 (CLARITY AD) lecanemab trials (2, 9).
ARIA Monitoring
We recommend obtaining MRI scans prior to the 5th, 7th, and 14th infusions. We also suggest a week 52 (prior to the 26th infusion) MRI scan, especially in those who are APOE4 carriers or had evidence of ARIA (whether with or without symptoms) on earlier MRIs (Figure 1). In CLARITY AD, ARIA-E tended to occur early, with most episodes (71%) detectable on the first or second MRIs obtained at weeks 9 and 13. Eighty-one percent resolved spontaneously within 4 months of radiographic detection. Initial episodes of ARIA-E continued to occur on safety MRIs at 24 and 52 weeks in APOE4 carriers but were infrequent in non-carriers after week 13.

MRI monitoring for lecanemab
Cerebral Macrohemorrhage
A cerebral macrohemorrhage represents a major central nervous system event often with enduring neurological deficits. A macrohemorrhage is more likely to occur when patients with pre-existing microbleeds or CAA-ri/ABRA are given anticoagulants (26, 27). Though numbers are small, the rate of cerebral macrohemorrhage in the CLARITY AD double blind and available open label periods was higher in patients on anticoagulation and in those on anticoagulation and lecanemab (Table 6). ARIA events with serious morbidity or mortality with lecanemab treatment are infrequent; three fatalities have been reported in the CLARITY AD open label extension which the site principal investigators attributed to lecanemab. One fatality occurred in an elderly APOE4 gene non-carrier with cardiovascular disease on anticoagulation who developed a macrohemorrhage; a second death occurred in a patient homozygous for APOE4 with a large vessel occlusion and pathologically confirmed severe CAA and vasculitis with multi-focal hemorrhage following tPA; a third death occurred in a patient homozygous for APOE4 with severe ARIA-E and ARIA-H and a clinical syndrome resembling CAA-ri (16) (Table 6 (28)). We recommend that patients receiving anticoagulants not be treated with lecanemab (detailed above). If patients on lecanemab require treatment with anticoagulants, we recommend stopping the lecanemab infusions. Lecanemab therapy can be reinstated if anticoagulation is no longer medically indicated.
Taken together, APOE4 gene carrier status, especially APOE4 homozygosity, CAA-ri/ABRA, and anticoagulation are all risk factors for intracerebral bleeding. Caution should be used when considering lecanemab for patients who have these risk factors.
Management of ARIA
Our recommendations for the management of ARIA with lecanemab are based on the ARIA management protocol in the CLARITY AD study and the management of ARIA with other amyloid-lowering antibodies (Figure 2). To detect ARIA early and mange it safely, radiologists and treating clinicians must recognize and interpret the pattern of MRI changes seen with ARIA-E and ARIA-H, know how to apply the ARIA management protocol in Figure 2, and recognize and manage the range of symptoms that may occur. Dosing can continue for individuals with radiographically mild asymptomatic ARIA recognized only on MRI (Table 7) with careful monitoring for symptoms and monthly non-contrast MRI scans until ARIA-E has resolved and ARIA-H has stabilized (Table 8). For those with moderate or severe radiographic ARIA or those with symptoms, lecanemab infusions should be suspended. Patients should receive careful clinical monitoring, management of symptoms, and monthly non-contrast MRI until ARIA-E resolves or ARIA-H stabilizes (Table 8). Once ARIA symptoms and radiographic ARIA-E changes resolve, restarting lecanemab can be considered based on a discussion of risks and benefits with the patient and family. Recurrent ARIA can occur. Decisions regarding ARIA management and when to discontinue treatment require consideration of the severity of the radiographic changes, presence and severity of any symptoms, patient’s APOE genotype, co-morbidities, and concurrent medications.

MRI monitoring for lecanemab
Management of Serious and Severe ARIA
The decision to provide lecanemab therapy requires institutional preparedness for the management of serious and severe ARIA. Before patients are treated with lecanumab, a written protocol for the management of serious and severe ARIA should be developed, and the team of individuals possibly required for patient care should be informed about the lecanemab therapy program, the rare occurrence of severe or serious ARIA, and the availability of the written protocol. Table 9 lists the resources needed to ensure that the medical facility is prepared to manage patients with major ARIA consequences. Though rare, providers should be prepared to recognize and manage serious cases of ARIA, which resemble the syndrome of CAA-ri (29). Such cases typically manifest as large areas of edema with high signal on non-contrast FLAIR MRI sequences. Tissue swelling with mass effect from edema may be present and numerous microhemorrhages may be observed. APOE4 carriers and especially APOE4 homozygotes are at the highest risk for these events. No specific tests have been identified to predict which patients will experience this severe inflammatory form of ARIA. Patients may be asymptomatic, mildly symptomatic, or severely symptomatic. Seizures, status epilepticus, encephalopathy or stupor may occur, and lateralizing neurological signs may be observed. Severe ARIA may be reversible (30) or result in permanent disability or death (17, 31). When patients exhibit such severe symptoms and signs, an MRI scan should be obtained promptly to identify ARIA changes. The imaging should include DWI sequences to exclude an ischemic stroke. Admission to a hospital or critical care unit with personnel experienced in the management of cerebral edema may be needed. Early initiation of high-dose glucocorticoid treatment (e.g., methylprednisolone 1g intravenously per day for 5 days followed by an oral steroid taper over several weeks) should be considered. Monitoring for seizures and treatment, if they occur, should be part of the management protocol.
Stopping Lecanemab for ARIA-Related Observations or Severe Infusion Reactions
We recommend discontinuing treatment with lecanemab for severe radiographic ARIA, severe symptomatic ARIA, any macrohemorrhage, one area of superficial siderosis, more than 10 microhemorrhages occurring since initiation of treatment, more than two episodes of ARIA or development of any medical condition that requires treatment with anticoagulants (Figure 2). We recommend considering discontinuation of lecanemab if patients have grade 3 or higher infusion reactions (discussed below; Table 10).
Management of Infusion Reactions
Infusion reactions occurred in 26.4% of participants on lecanemab in the CLARITY AD study and were typically mild to moderate in severity. Infusion reactions usually occurred during the first 2 treatments and were seen during the infusion or up to several hours after the infusion. Infusion reaction symptoms typically resolve within 24 hours and can usually be managed at home. Symptoms include fever, chills, headache, rash, nausea, vomiting, abdominal discomfort, and elevated blood pressure. For more severe reactions (grade 2; Table 10 (32, 33)), the infusion should be stopped, and the patient treated with diphenhydramine and acetaminophen in milder cases and with oral dexamethasone (0.75 mg/day for 2–3 days) or oral methylprednisolone (80 mg twice per day for 2–3 days) when significant symptoms are present. The diphenhydramine or acetaminophen may be repeated every 4–6 hours until symptoms fully resolve. A call should be placed to the patient or care partner later that day to assess symptom resolution. For patients with grade 3 or higher infusion reactions (Table 10) the lecanemab infusion should be discontinued. Following an infusion reaction, patients should be pretreated with oral diphenhydramine 25–50 mg and oral acetaminophen 650 mg–1,000 mg 30 minutes prior to the next infusion. Low dose oral dexamethasone (0.75 mg 6 hours before infusion) or oral methylprednisolone (80 mg 6 hours prior to infusion) can be used for prophylaxis or treatment if diphenhydramine and acetaminophen are ineffective. Antihypertensive treatment may be needed for management of elevated blood pressure. Continue this prophylactic regimen until the patient remains asymptomatic in clinic and at home following 2–4 infusions. If a new infusion reaction occurs, oral diphenhydramine 25–50 mg and oral acetaminophen 650 mg-1,000 mg can be repeated every 4–6 hours postinfusion as needed with a contact later that day and the next day to determine if further treatment is needed. Although severe reactions are rare, clinics should be prepared with bronchodilators, oxygen, and epinephrine if needed.
The clinician can consider diphenhydramine or a topical corticosteroid cream for mild-moderate skin hypersensitivity reactions.
Appropriate Discussions with Patients and Care Partners Concerning Treatment with Lecanemab
Patients with AD identified by their physicians as potential candidates for treatment with lecanemab must understand the potential benefits and potential harms of treatment. Care partners and family members of patients considering treatment with lecanemab must understand the benefits being sought, the occurrence of ARIA and its possible consequences, the possibility of infusion reactions, the need for twice monthly intravenous infusions, and the requirement for MRIs at baseline (if not done within the past 12 months) and 3 or 4 scans in the first year of therapy for the safe management of ARIA. Patients seeking treatment with lecanemab will be informed that APOE genotyping is recommended, there must be confirmation of amyloid pathology in the brain with either amyloid PET or CSF studies, and an MRI scan taken within the past 12 months must be available or obtained to ensure that they do not have vascular or other types of pathology incompatible with lecanemab therapy (described above). Patients with early AD have sufficiently preserved cognitive capacities to engage in and understand discussions regarding potential benefits, risks, and care requirements, yet they are challenged when there is substantial situational uncertainty (34). This must be considered in counseling patients about treatment with lecanemab. A variety of informational aids and decision-making tools have been identified and shown to be useful to assist the understanding of patients with cognitive impairment (35).
Treatment expectations for disease modifying therapies differ from those of symptomatic therapies. Cognitive enhancing agents such as the cholinesterase inhibitors and memantine temporarily improve function above baseline or temporarily postpone cognitive decline. They do not slow the rate of disease progression. The goal of disease-modifying therapies like lecanemab is slowing of the disease progression and cognitive decline; they are not expected to improve cognition or function.
The primary outcome of the phase 3 trial of lecanemab was the Clinical Dementia Rating-Sum of Boxes (CDR-SB) (36, 37). The CDR-SB is unfamiliar to patients and must be translated into terms that are more meaningful such as preserving cognition and function and remaining less dependent on others for longer periods of time. These are therapeutic goals that have been articulated as clinically meaningful by patients and their care partners (4, 38, 39). Potential slowing of loss of function, delayed progression from one global stage of AD to another, delay in disease milestones, delayed loss of quality of life for the patient, as well as reduced burden or loss of quality of life for the care partner may be more meaningful information than numerical CDR-SB scores (40).
APOE genotyping informs the risk of treatment with lecanemab (described above), and genotyping is recommended to inform discussions of lecanemab therapy. Genotyping of a treatment candidate that reveals the patient to be an APOE4 gene carrier has implications for all first-degree relatives as they might might share the genetic risk. Counseling regarding genotyping and its ramifications has an important role in appropriate treatment discussions (41, 42).
In the phase 3 CLARITY AD trial, patients of Hispanic ethnicity comprised 12.5% and 12.3% of the lecanemab and placebo groups respectively. Asian patients represented 17.1% and 16.9% of the lecanemab and placebo participants. Black individuals represented only 2.3% and 2.7% of the lecanemab and placebo groups (2). There may be ethnic and racial differences in treatment responses, therapy preferences, adverse events, treatment adherence, and patient-care partner relationships. Differences in the frequency of amyloid positivity (e.g., less common in Asian, Hispanic, and Black compared to White individuals (43)) and differences in the relationship between APOE4 and dementia between African Americans and White Americans (44) suggest that generalizations across ethnic and racial groups are not warranted. The modest number of racial and ethnic minority individuals included in the lecanemab trials provide limited insight into potential ethnic/racial differences in trial outcomes. This issue is particularly important in the US, where lack of diversity has been identified as a major challenge to research and treatment equity. Transparency concerning the small number of minority participants in lecanemab trials is important to acknowledge in discussions with minority individuals considering lecanemab therapy.
Appropriate Adjustments and Workflow Considerations Regarding Lecanemab Use in Clinical Practice
Most clinicians have limited experience with lecanemab or other monoclonal antibody therapies for AD. These AURs are intended to help clinicians anticipate, plan, and implement the clinical practice and workflow alterations needed to accommodate lecanemab therapy. Clinician education regarding AD, cognitive assessment, amyloid confirmation with PET or CSF, ARIA detection and management, infusion and infusion reactions, APOE genotyping, and patient communication are foundational for success in lecanemab treatment programs. Table 11 describes the resources needed to implement and maintain a lecanemab treatment program. Development of a protocol for management of severe and serious ARIA is recommended for clinicians and institutions implementing a therapy program with lecanemab (described above; Table 9).
Clinicians should serially assess the mental status of all patients in their practice with MCI or dementia (45). Lecanemab is appropriate for patients with early AD like those included in the phase 2 and phase 3 clinical trials. Assessment of patients with evidence of cognitive impairment, complaints of cognitive decline, or observations from caregivers consistent with progressive cognitive impairment are triggers for determining if the patient is a candidate for further assessment and possible treatment (46).
Considering a patient for lecanemab therapy involves excluding non-AD causes of cognitive impairment, assessing a baseline or recent MRI for possible exclusionary factors, and confirming amyloid pathology with amyloid PET or CSF amyloid and tau assays (described above). Safe use of lecanemab requires periodic MRI imaging and urgent MRI studies if patients develop symptoms consistent with ARIA (described above). Clinical skills in assessing symptoms that may be indicative of ARIA and access to MRIs that can be made available rapidly are necessary for lecanemab treatment programs. Communication channels between patients and their caregivers and clinical staff must be established to allow rapid communication regarding symptoms suggestive of ARIA, infusion reactions, or other side effects. Guidance for patient counseling is available in the PI (www.accessdata.fda.gov).
Once treatment is initiated, supportive evidence of effectiveness may include less-than-expected decline on standard rating instruments performed by clinicians or change of trajectory of cognitive decline observed by family members (47). Confirmation of efficacy of lecanemab in the clinical setting is not feasible since the change in the rate of decline is relatively subtle, a change in rate of decline cannot be detected without multiple comprehensive trial-like assessments collected longitudinally, trajectories vary among and within patients, and comparison with a placebo group is not available.
Lecanemab is administered intravenously every other week and access to infusion centers, AD-specific infusion resources, in-office infusion settings, or home infusion opportunities is required. Infusion reactions are common with lecanemab administration; serious infusion reactions were uncommon (2, 9). Pre-infusion discussions with patients and care partners about the possibility of infusion reactions, institution of ameliorative therapy when reactions occur, and vigilance regarding possible serious reactions will be standard procedures for clinicians providing lecanemab therapy.
Communication with patients and their families regarding potential benefits and potential harms of treatment with lecanemab is essential for those administering this agent. Communication with informed professionals has become more urgent as social and electronic media have emerged as common sources of patient information, and misinformation found on the internet or purveyed through social media may influence patient decisions (48). Potential harms are balanced by discussions of potential treatment benefits of a disease-modifying agent like lecanemab and the implications of choosing no treatment (49). Communication that respects the cultural background of the therapy candidate is increasingly recognized as a key aspect of the patient-clinician information exchange. Lack of trust in a health care provider increases the likelihood of non-disclosure of health problems by the patient and could jeopardize appropriate management of lecanemab side effects (50). Comprehensive, transparent communication and building of trust between patients and clinicians will enhance the likelihood of successful and safe use of lecanemab in real-world clinical practice.
Discussion
The goal of this AUR is to provide clinicians with information that facilitates the safe use of lecanemab in community practices. Patients participating in clinical trials differ from AD patients who are not included in trials. Trial participants are usually younger, healthier, better educated, have fewer comorbidities, and are less diverse than real-world patients. Use of lecanemab in older, less healthy, less well educated, and more diverse populations may result in efficacy and safety outcomes that differ from those observed in trials. Patients in trials are recruited and treated in expert centers and are closely monitored by expert clinicians with experience using lecanemab and other monoclonal antibodies. Trials use centralized readings of amyloid PET and MRI that are not available in community settings. These differences between providing lecanemab in a closely supervised clinical trial and providing this agent in community care practices indicate the need for vigilance to ensure the safe and effective use of lecanemab (51, 52). AURs are meant to anticipate the challenges to implementing lecanemab in practice. Post-marketing surveillance of putative side effects and the use of registries for patients receiving lecanemab or other monoclonal antibodies will facilitate data collection and guide any necessary use adjustments. We recommend that all patients receiving lecanemab be enrolled in the Alzheimer’s Network for Treatment and Diagnostics (ALZ-NET; www.alz-net.org) or similar registries as they become available. Education regarding use of lecanemab and other anti-amyloid monoclonal antibodies and emerging information regarding these agents will be available through ALZ-NET.
The efficacy and safety of lecanemab are known only for the type of patients that were included in clinical trials manifesting early AD with confirmed amyloid positivity (Tables 1 and 2). These AURs recommend patients be selected for initiation of treatment based on criteria like those of the completed lecanemab clinical trials; this is the population in whom efficacy and safety have been demonstrated.
The efficacy and safety of lecanemab for patients with AD dementia more severe than those included in the trials are not known, and recommendations for stopping lecanemab therapy when patients progress beyond mild AD dementia will depend on accruing more information. Key aspects of this discussion will depend on effectiveness observations collected by the clinician suggesting continuing disease slowing, safety observations regarding ARIA, and discussions with the patient and family members regarding the lack of trial information for this aspect of the illness and their impression of the potential benefits of continuing treatment.
Patients included in the lecanemab trials were in the early symptomatic stages of late age-of-onset AD, yet there are other populations of amyloid-bearing patients for whom treatment with anti-amyloid monoclonal antibodies may be useful. There is limited experience with early age-of-onset AD (EOAD), traditionally defined as symptom onset at age <65 years. Only 166 out of 859 lecanemab treated patients in CLARITY AD were 50 to 64 years of age at the time of enrollment. Such patients can be placed on lecanemab provided they meet the other treatment recommendations, but caution must be exercised as the clinical trials were not powered to provide safety or efficacy information in EOAD.
There are other populations of amyloid-bearing patients for whom treatment with anti-amyloid monoclonal antibodies may be useful. Patients with autosomal dominant AD (ADAD) were not specifically excluded from the lecanemab trials, and a small number may have been included. The specifics of safety and efficacy in these patients are unknown. Some ADAD-causing mutations are associated with a higher rate of CAA and patients with these CAA-related ADAD mutations should be excluded from current treatment with lecanemab. Information regarding which mutations are associated with CAA is available in the mutation database of Alzforum (www.alzforum.org). Clinical trials specifically designed for individuals with ADAD are available (53).
Persons with Down syndrome develop AOAD and are amyloid positive. There is an increased occurrence of CAA in patients with Down syndrome and they should be excluded from treatment with lecanemab (54). Clinical trials for patients with Down syndrome are under consideration and additional data including information that may guide the use of lecanemab in this population are expected (55).
Patients included in the lecanemab trials had relatively typical forms of memory-predominant AD. Patients with atypical AD syndromes including logopenic variant primary progressive aphasia, posterior cortical atrophy, or behavioral or dysexecutive AD have positive amyloid studies and may be candidates for lecanemab treatment. Patients with these syndromes were not specifically excluded from the lecanemab trials if they met the inclusion criteria for participation. The safety and efficacy of treatment in these patients has not been studied. The absence of treatment-related information for patients with atypical AD should be acknowledged in discussions with the therapy candidate and their care partner.
Individuals in the preclinical phase of AD who are cognitively normal and have positive amyloid PET or CSF studies were excluded from the completed lecanemab trials. An ongoing trial will determine if treatment with lecanemab in the preclinical phase of AD will prevent or delay the emergence of cognitive symptoms (56).
Excluding patients with some comorbidities and treatments including anticoagulants, severe cerebrovascular disease, and others may result in disproportionately excluding patients from underrepresented groups including Black/African Americans, Latinos, Asians, Native Americans and Pacific Islanders, and others with adverse determinants of health from being considered as candidates for lecanemab therapy. These individuals were under-represented in the lecanemab trials, but they may still qualify for lecanemab treatment if they meet the treatment criteria detailed herein. Transparent acknowledgement of the limited information available regarding treatment responses in these patients should be acknowledged in discussions with the patient and caregiver.
Spontaneous ARIA-like events occur in patients with CAA and ABRA (20, 29, 57). CAA/ABRA is associated with cerebral microbleeds which are a risk factor for ARIA (20, 29, 57, 58). Patients with underlying CAA-ri may be at particularly high risk for ARIA (59). Evolving information may improve our ability to identify patients who are at particularly high risk for ARIA events.
Summary
This AUR presents information derived from the lecanemab clinical trials and from research studies and care experience with AD to assist practitioners who have chosen to provide patients with the option of treatment with lecanemab. We recommend that patients treated with lecanemab be like those included in lecanemab trials where safety and efficacy have been shown. Exclusion of patients with more than four microhemorrhages or other evidence of cerebrovascular disease is thought to reduce the risk of ARIA associated with lecanemab therapy. APOE genotyping is recommended to identify patients who are APOE4 gene carriers, especially those homozygous for APOE4 who are at higher risk for the occurrence of ARIA. We recommend that patients requiring treatment with anticoagulants not be given lecanemab and that thrombolytic therapy for ischemic stroke not be administered to lecanemab recipients. Lecanemab is given intravenously every other week and requires no titration. The AURs suggest an MRI be obtained at baseline and prior to the 5th, 7th, and 14th infusions and also after 52 weeks of therapy (for APOE4 carriers or those with a history of ARIA) in order to monitor for asymptomatic radiographic ARIA. An MRI is obtained if patients have symptoms suggestive of an ARIA event. Infusion reactions are relatively common and may require prophylactic management with anti-inflammatory therapies. Thorough discussion of the expected therapeutic benefit of this disease- modifying therapy as well as the potential for harm are key elements in good clinical practice for the use of lecanemab.
References
U.S. Food & Drug Administration. Guidance for Industry, Expedited Programs for Serious Conditions — Drugs and Biologics. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER), May 2014.; 2014.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 202210.1056/NEJMoa2212948.
Dunn B, Stein P, Temple R, Cavazzoni P. An appropriate use of accelerated approval — aducanumab for Alzheimer’s disease. N Engl J Med. 2021;385(9):856–857; https://doi.org/10.1056/NEJMc2111960.
Petersen RC, Aisen PS, Andrews JS, Atri A, Matthews BR, Rentz DM, et al. Expectations and clinical meaningfulness of randomized controlled trials. Alzheimers Dement. 202310.1002/alz.12959.
Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398–410; https://doi.org/10.14283/jpad.2021.41.
Cummings J, Rabinovici GD, Atri A, Aisen P, Apostolova LG, Hendrix S, et al. Aducanumab: appropriate use recommendations update. J Prev Alzheimers Dis. 2022;9(2):221–230; https://doi.org/10.14283/jpad.2022.34.
Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, et al. Safety and tolerability of BAN2401— a clinical study in Alzheimer’s disease with a protofibril selective Abeta antibody. Alzheimers Res Ther. 2016;8(1):14; https://doi.org/10.1186/s13195-016-0181-2.
Satlin A, Wang J, Logovinsky V, Berry S, Swanson C, Dhadda S, et al. Design of a Bayesian adaptive phase 2 proof-of-concept trial for BAN2401, a putative disease-modifying monoclonal antibody for the treatment of Alzheimer’s disease. Alzheimers Dement (N Y). 2016;2(1):1–12; https://doi.org/10.1016/j.trci.2016.01.001.
Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res Ther. 2021;13(1):80; https://doi.org/10.1186/s13195-021-00813-8.
Soderberg L, Johannesson M, Nygren P, Laudon H, Eriksson F, Osswald G, et al. Lecanemab, aducanumab, and gantenerumab — binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurotherapeutics. 202210.1007/s13311-022-01308-6.
Tucker S, Moller C, Tegerstedt K, Lord A, Laudon H, Sjodahl J, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis. 2015;43(2):575–588; https://doi.org/10.3233/JAD-140741.
Johannesson M, Sahlin C, Soderberg L, Basun H, Falting J, Moller C, et al. Elevated soluble amyloid beta protofibrils in Down syndrome and Alzheimer’s disease. Mol Cell Neurosci. 2021;114:103641; https://doi.org/10.1016/j.mcn.2021.103641.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–279; DOI:https://doi.org/10.1016/j.jalz.2011.03.008.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269; https://doi.org/10.1016/j.jalz.2011.03.005.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198; https://doi.org/10.1016/0022-3956(75)90026-6.
Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2016;12(3):292–323; https://doi.org/10.1016/j.jalz.2016.02.002.
Reish NJ, Jamshidi P, Stamm B, Flanagan ME, Sugg E, Tang M, et al. Multiple cerebral hemorrhages in a patient receiving lecanemab and treated with t-PA for stroke. N Engl J Med. 202310.1056/NEJMc2215148.
Pontecorvo MJ, Devous MD, Sr., Navitsky M, Lu M, Salloway S, Schaerf FW, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140(3):748–763; https://doi.org/10.1093/brain/aww334.
Hampel H, O’Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14(11):639–652; https://doi.org/10.1038/s41582-018-0079-7.
Salvarani C, Morris JM, Giannini C, Brown RD, Jr., Christianson T, Hunder GG. Imaging findings of cerebral amyloid angiopathy, Abeta-Related Angiitis (ABRA), and cerebral amyloid angiopathy-related inflammation: a single-institution 25-year experience. Medicine (Baltimore). 2016;95(20):e3613; https://doi.org/10.1097/MD.0000000000003613.
Auriel E, Charidimou A, Gurol ME, Ni J, Van Etten ES, Martinez-Ramirez S, et al. Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2016;73(2):197–202; https://doi.org/10.1001/jamaneurol.2015.4078.
Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron. 2019;101(5):820–838; https://doi.org/10.1016/j.neuron.2019.01.056.
Martens YA, Zhao N, Liu CC, Kanekiyo T, Yang AJ, Goate AM, et al. ApoE cascade hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron. 2022;110(8):1304–1317; https://doi.org/10.1016/j.neuron.2022.03.004.
Koutsodendris N, Nelson MR, Rao A, Huang Y. Apolipoprotein E and Alzheimer’s disease: findings, hypotheses, and potential mechanisms. Annu Rev Pathol. 2022;17:73–99; https://doi.org/10.1146/annurev-pathmechdis-030421-112756.
Neu SC, Pa J, Kukull W, Beekly D, Kuzma A, Gangadharan P, et al. Apolipoprotein E genotype and sex risk factors for Alzheimer disease: a meta-analysis. JAMA Neurol. 2017;74(10):1178–1189; https://doi.org/10.1001/jamaneurol.2017.2188.
Tsivgoulis G, Zand R, Katsanos AH, Turc G, Nolte CH, Jung S, et al. Risk of symptomatic intracerebral hemorrhage after intravenous thrombolysis in patients with acute ischemic stroke and high cerebral microbleed burden: a meta-analysis. JAMA Neurol. 2016;73(6):675–683; https://doi.org/10.1001/jamaneurol.2016.0292.
An SJ, Kim TJ, Yoon BW. Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: an update. J Stroke. 2017;19(1):3–10; 10.5853/jos.2016.00864.
Irizarry M, van Dyck C, Sabbagh M, Bateman RJ, S. C, editors. Clarity AD: A phase 3 placebo-controlled, double-blind, parallel-group, 18-month study evaluating lecanemab in early Alzheimer’s disease. 15th Clinical Trials on Alzheimer’s Disease Conference; 2022; San Francisco, CA USA.
Antolini L, DiFrancesco JC, Zedde M, Basso G, Arighi A, Shima A, et al. Spontaneous ARIA-like events in cerebral amyloid angiopathy-related inflammation: a multicenter prospective longitudinal cohort study. Neurology. 2021;97(18):e1809–e1822; https://doi.org/10.1212/WNL.0000000000012778.
VandeVrede L, Gibbs DM, Koestler M, La Joie R, Ljubenkov PA, Provost K, et al. Symptomatic amyloid-related imaging abnormalities in an APOE epsilon4/epsilon4 patient treated with aducanumab. Alzheimers Dement (Amst). 2020;12(1):e12101; https://doi.org/10.1002/dad2.12101.
Piller C. Scientists tie third clinical trial death to experimental Alzheimer’s drug. Science. 2022.
Lenz HJ. Management and preparedness for infusion and hypersensitivity reactions. Oncologist. 2007;12(5):601–609; https://doi.org/10.1634/theoncologist.12-5-601.
Doessegger L, Banholzer ML. Clinical development methodology for infusion-related reactions with monoclonal antibodies. Clin Transl Immunology. 2015;4(7):e39; https://doi.org/10.1038/cti.2015.14.
Zhang Y, Wang J, Sun T, Wang L, Li T, Li H, et al. Decision-making profiles and their associations with cognitive performance in mild cognitive impairment. J Alzheimers Dis. 2022;87(3):1215–1227; https://doi.org/10.3233/JAD-215440.
Ho MH, Chang HR, Liu MF, Chien HW, Tang LY, Chan SY, et al. Decision-making in people with dementia or mild cognitive impairment: a narrative review of decision-making tools. J Am Med Dir Assoc. 2021;22(10):2056–2062 e2054; https://doi.org/10.1016/j.jamda.2021.06.034.
Williams MM, Storandt M, Roe CM, Morris JC. Progression of Alzheimer’s disease as measured by Clinical Dementia Rating Sum of Boxes scores. Alzheimers Dement. 2013;9(1 Suppl):S39–44; https://doi.org/10.1016/j.jalz.2012.01.005.
O’Bryant SE, Waring SC, Cullum CM, Hall J, Lacritz L, Massman PJ, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer’s research consortium study. Arch Neurol. 2008;65(8):1091–1095; https://doi.org/10.1001/archneur.65.8.1091.
DiBenedetti DB, Slota C, Wronski SL, Vradenburg G, Comer M, Callahan LF, et al. Assessing what matters most to patients with or at risk for Alzheimer’s and care partners: a qualitative study evaluating symptoms, impacts, and outcomes. Alzheimers Res Ther. 2020;12(1):90; https://doi.org/10.1186/s13195-020-00659-6.
Assuncao SS, Sperling RA, Ritchie C, Kerwin DR, Aisen PS, Lansdall C, et al. Meaningful benefits: a framework to assess disease-modifying therapies in preclinical and early Alzheimer’s disease. Alzheimers Res Ther. 2022;14(1):54; https://doi.org/10.1186/s13195-022-00984-y.
Cohen S., editor Clarity AD results in context. 2022 CLARITY AD CTAD Presentations. Clinical Trials in Alzheimer’s Disease (CTAD annual meeting). San Francisco, California, November 29th, 2022.2022.
Green RC, Christensen KD, Cupples LA, Relkin NR, Whitehouse PJ, Royal CD, et al. A randomized noninferiority trial of condensed protocols for genetic risk disclosure of Alzheimer’s disease. Alzheimers Dement. 2015;11(10):1222–1230; https://doi.org/10.1016/j.jalz.2014.10.014.
Roberts JS, Patterson AK, Uhlmann WR. Genetic testing for neurodegenerative diseases: Ethical and health communication challenges. Neurobiol Dis. 2020;141:104871; https://doi.org/10.1016/j.nbd.2020.104871.
Wilkins CH, Windon CC, Dilworth-Anderson P, Romanoff J, Gatsonis C, Hanna L, et al. Racial and ethnic differences in amyloid PET positivity in individuals with mild cognitive impairment or dementia: a secondary analysis of the Imaging Dementia-Evidence for Amyloid Scanning (IDEAS) Cohort Study. JAMA Neurol. 2022;79(11):1139–1147; https://doi.org/10.1001/jamaneurol.2022.3157.
Naslavsky MS, Suemoto CK, Brito LA, Scliar MO, Ferretti-Rebustini RE, Rodriguez RD, et al. Global and local ancestry modulate APOE association with Alzheimer’s neuropathology and cognitive outcomes in an admixed sample. Mol Psychiatry. 2022;27(11):4800–4808; https://doi.org/10.1038/s41380-022-01729-x.
Atri A. The Alzheimer’s disease clinical spectrum: diagnosis and management. Med Clin North Am. 2019;103(2):263–293; https://doi.org/10.1016/j.mcna.2018.10.009.
Liss JL, Seleri Assuncao S, Cummings J, Atri A, Geldmacher DS, Candela SF, et al. Practical recommendations for timely, accurate diagnosis of symptomatic Alzheimer’s disease (MCI and dementia) in primary care: a review and synthesis. J Intern Med. 2021;290(2):310–334; https://doi.org/10.1111/joim.13244.
Dansson HV, Stempfle L, Egilsdottir H, Schliep A, Portelius E, Blennow K, et al. Predicting progression and cognitive decline in amyloid-positive patients with Alzheimer’s disease. Alzheimers Res Ther. 2021;13(1):151; https://doi.org/10.1186/s13195-021-00886-5.
Tan SS, Goonawardene N. Internet health information seeking and the patient-physician relationship: a systematic review. J Med Internet Res. 2017;19(1):e9; https://doi.org/10.2196/jmir.5729.
Tarn DM, Wenger A, Good JS, Hoffing M, Scherger JE, Wenger NS. Do physicians communicate the adverse effects of medications that older patients want to hear? Drugs Ther Perspect. 2015;31(2):68–76; https://doi.org/10.1007/s40267-014-0176-7.
Nong P, Williamson A, Anthony D, Platt J, Kardia S. Discrimination, trust, and withholding information from providers: Implications for missing data and inequity. SSM Popul Health. 2022;18:101092; https://doi.org/10.1016/j.ssmph.2022.101092.
Martin K, Begaud B, Latry P, Miremont-Salame G, Fourrier A, Moore N. Differences between clinical trials and postmarketing use. Br J Clin Pharmacol. 2004;57(1):86–92; https://doi.org/10.1046/j.1365-2125.2003.01953.x.
Franzen S, Smith JE, van den Berg E, Rivera Mindt M, van Bruchem-Visser RL, Abner EL, et al. Diversity in Alzheimer’s disease drug trials: The importance of eligibility criteria. Alzheimers Dement. 2022;18(4):810–823; https://doi.org/10.1002/alz.12433.
Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement. 2017;13(1):8–19; https://doi.org/10.1016/j.jalz.2016.07.005.
Carmona-Iragui M, Balasa M, Benejam B, Alcolea D, Fernandez S, Videla L, et al. Cerebral amyloid angiopathy in Down syndrome and sporadic and autosomal-dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1251–1260; https://doi.org/10.1016/j.jalz.2017.03.007.
Rafii MS. Alzheimer’s Disease in Down Syndrome: progress in the design and conduct of drug prevention trials. CNS Drugs. 2020;34(8):785–794; https://doi.org/10.1007/s40263-020-00740-6.
Rafii MS, Sperling RA, Donohue MC, Zhou J, Roberts C, Irizarry MC, et al. The AHEAD 3–45 Study: Design of a prevention trial for Alzheimer’s disease. Alzheimers Dement. 202210.1002/alz.12748.
Salvarani C, Hunder GG, Morris JM, Brown RD, Jr., Christianson T, Giannini C. Abeta-related angiitis: comparison with CAA without inflammation and primary CNS vasculitis. Neurology. 2013;81(18):1596–1603; https://doi.org/10.1212/WNL.0b013e3182a9f545.
Haller S, Vernooij MW, Kuijer JPA, Larsson EM, Jager HR, Barkhof F. Cerebral microbleeds: imaging and clinical significance. Radiology. 2018;287(1):11–28; https://doi.org/10.1148/radiol.2018170803.
Kirshner HS, Bradshaw M. The inflammatory form of cerebral amyloid angiopathy or “Cerebral Amyloid Angiopathy-Related Inflammation” (CAARI). Curr Neurol Neurosci Rep. 2015;15(8):54; 10.1007/s11910-015-0572-y.
Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol. 1987;149(2):351–356; https://doi.org/10.2214/ajr.149.2.351.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
The authors adhered to the publication ethics specified by the Journal of Prevention of Alzheimer’s Disease. This is an original work and all authors have contributed importantly to its creation. The authors have verified the accuracy of the content of the article.
Additional information
Disclosure and Conflicts of Interest
JC has provided consultation to Acadia, Actinogen, Acumen, AlphaCognition, Aprinoia, AriBio, Artery, Biogen, BioVie, Cassava, Cerecin, Diadem, EIP Pharma, Eisai, GemVax, Genentech, GAP Innovations, Janssen, Jocasta, Karuna, Lilly, Lundbeck, LSP, Merck, NervGen, Novo Nordisk, Oligomerix, Optoceutics, Ono, Otsuka, PRODEO, Prothena, ReMYND, Roche, Sage Therapeutics, Signant Health, Simcere, Sunbird Bio, Suven, SynapseBio, TrueBinding,,Vaxxinity, and Wren pharmaceutical, assessment, and investment companies. JC is supported by NIGMS grant P20GM109025; NINDS grant U01NS093334; NIA grant R01AG053798; NIA grant P20AG068053; NIA grant P30AG072959; NIA grant R35AG71476; Alzheimer’s Disease Drug Discovery Foundation (ADDF); Ted and Maria Quirk Endowment; and the Joy Chambers-Grundy Endowment. LGA has provided consultation to Eli Lilly, Biogen, Two Labs, FL Dept Health, Genentech, NIH Biobank, Eli Lilly, GE Healthcare, Eisai, Roche Diagnostics, and Alnylam. LGA receives the following research support: NIA U01 AG057195, NIA R01 AG057739, NIA P30 AG010133, Alzheimer Association LEADS GENETICS 19-639372, Alzheimer Association SG-23-1061716, Roche Diagnostics RD005665, AVID Pharmaceuticals, Life Molecular Imaging. LGA has received honoraria for participating in independent data safety monitoring boards and providing educational CME lectures and programs. LGA has stock in Cassava Sciences. GDR has provided consultation to Eli Lilly, Eisai, Roche, GE Healthcare, Johnson & Johnson and Genentech pharmaceutical companies. He receives grant support from NIH, Alzheimer’s Association, American College of Radiology, Rainwater Charitable Foundation, Genentech (via the Alliance for Therapies in Neuroscience) and from Avid Radiopharmaceuticals, GE Healthcare and Life Molecular Imaging (view the New IDEAS study). AA has received honoraria for consulting; participating in independent data safety monitoring boards; providing educational lectures, programs, and materials; or serving on advisory boards for AbbVie, Acadia, Allergan, the Alzheimer’s Association, Axovant, AZ Therapies, Biogen, Eisai, Grifols, Harvard Medical School Graduate Continuing Education, JOMDD, Lundbeck, Merck, Roche/Genentech, Novo Nordisk, Qynapse, Sunovion, Suven, and Synexus. AA receives book royalties from Oxford University Press. AA receives institutional research grant/contract funding from NIA/NIH 1P30AG072980, AZ DHS CTR040636, Washington University St Louis, and Gates Ventures. His institution receives/received funding for clinical trial grants, contracts and projects from government, consortia, foundations, and companies for which he serves/served as contracted site-PI. AA served as site-PI for the EMERGE study at his previous institution. PA has received research funding from NIA, FNIH, the Alzheimer’s Association, Janssen, Lilly and Eisai, and personal fees from Biogen, Merck, Roche, Abbvie, ImmunoBrain Checkpoint, Rainbow Medical and Shionogi. SS was a site PI and co-chair of the investigator steering committee for the ENGAGE trial and he receives research support and consultancy fees from Lilly, Biogen, Avid, Eisai, Genentech, and Roche. SMG is on the Data Safety Monitoring Board for DIAN-TU and has been a consultant for Eli Lilly. SH is owner of and a full-time employee of Pentara Corporation which consults for both Biogen and Eisai and many other AD clients. DS is a director and consultant of Prothena Biosciences and serves on an Advisory Board for Eisai. MW has served on Advisory Boards for Eli Lilly, Cerecin/Accera, Roche, Alzheon, Inc., Merck Sharp & Dohme Corp., Nestle/Nestec, PCORI/PPRN, Dolby Family Ventures, National Institute on Aging (NIA), Brain Health Registry and ADNI. He serves on Editorial Boards for Alzheimer’s & Dementia, TMRI and MRI. He has provided consulting and/or acted as a speaker to Cerecin/Accera, Inc., BioClinica, Nestle/Nestec, Roche, Genentech, NIH, The Buck Institute for Research on Aging, FUJIFILM-Toyama Chemical (Japan), Garfield Weston, Baird Equity Capital, University of Southern California (USC), Cytox, and Japanese Organization for Medical Device Development, Inc. (JOMDD), Peerview Internal Medicine, Vida Ventures, Medscape, Eisai, Korean Dementia Society, China Association for Alzheimer’s Disease (CAAD, and T3D Therapeutics. He holds stock options with Alzheon, Inc., Alzeca, and Anven. RCP has served as a consultant for Roche, Inc.; Genentech, Inc.; Eisai, Inc.; Eli Lilly, Inc.; and Nestle, Inc. he receives grant support from the National Institutes of Health. SS was a site PI for CLARITY and ENGAGE and co-chair of the investigator steering committee for the ENGAGE trial and his hospital receives research support from Lilly, Biogen, Avid, Eisai, Genentech, Roche and he receives consultancy fees from Lilly, Biogen, Avid, Eisai, Genentech, Roche, Novo Nordisk, Acumen and Prothena.
Rights and permissions
Open Access : This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Cummings, J., Apostolova, L., Rabinovici, G.D. et al. Lecanemab: Appropriate Use Recommendations. J Prev Alzheimers Dis 10, 362–377 (2023). https://doi.org/10.14283/jpad.2023.30
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.14283/jpad.2023.30