
Abstract
The aim of this work was to establish a method for preparing stable and controllable solid self-microemulsifying drug delivery system (S-SMEDDS) by spherical crystallization technique, which was explored for promoting the dissolution, oral bioavailability, and process efficiency. Solubility test, preparation of liquid self-microemulsifying drug delivery system (L-SMEDDS), and the obtained ternary phase diagrams test have been performed to screen and optimize the composition of LSMEDDS. The optimized formulation was used to prepare puerarin solid self-microemulsifying drug delivery system (Pue-SSMEDDS) by spherical crystallization technique. Droplet size and morphological analysis of the optimal Pue-SSMEDDS were determined to evaluate the final formulation. And the Pue-SSMEDDS was also assessed by flowability study, angle of repose, Carr’s index, and flow velocity. Furthermore, the vitro dissolution and pharmacokinetic profile in vivo were analyzed. The study in vitro showed the Pue-SSMEDDS could disperse in the dispersion medium within 60 s and was spherical with the particle size of 19.66 nm and zeta potential of −28.3 mV. It could keep stable at low temperature and seal condition for 3 months. In vivo pharmacokinetic experiments of rats, the mean plasma concentration of self-microemulsion group was much higher than that of conventional tablets and could play a long-lasting efficacy, while there was no significant difference between the LSMEDDS and S-SMEDDS. The results suggested the potential of S-SMEDDS could improve the oral bioavailability of poorly water-soluble drug, such as puerarin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Puerarin is a natural bioactive agent extracted from the dried root of Pueraria lobata and Puerarin thomsonii (1–3) which has a potential in the prevention of hypertension (4), arteriosclerosis (5), cancer (6), and other aspects (7). However, oral bioavailability of puerarin is low mainly due to its poor water solubility of which clinically available aspects are limited nowadays, such as by the gastrointestinal tract. It is necessary for intravenous administration to maintain the blood concentration. The offset for its short half-life in vivo would lead to poor patient compliance on account of multiple doses. In addition, adverse effects may occur in patients, for instance acute intravascular hemolysis, hypersensitivity reaction, and the damage to the liver and kidney (8). Therefore, a new preparation of administration is necessary to overcome these problems.
Methods of improving drug solubility including solid dispersion, nanoemulsions, polymer micelles, liposomes, cyclodextrin (CD) inclusion, and self-emulsifying drug delivery system are adopted to develop the oral drug delivery system (9–14). Self-microemulsifying drug delivery system (SMEDDS), a valid pharmaceutical technology to increase solubility and absorption of poorly water-soluble drugs in vivo, has recently received increasing attention as a kind of new drug delivery systems. It is thermodynamically stable and an isotropic mixture made up of oil, surfactant, co-surfactant, and drug. The system is able to form oil-in-water microemulsion with peristaltic movement in contact with aqueous fluids spontaneously like the movement of the gastrointestinal tract (15–19). The SMEDDS not only improves drug solubilization but also enhances the properties of release and absorption, resulted from the dissolved form of the drug in the formulation and the small droplet size finishing a large interfacial surface area between oil and water. Compared with conventional tablets or capsules, the drug in SMEDDS can be absorbed quickly since the microemulsion may generate into fluids containing dissolved drug small emulsion droplets in contact with the gastrointestinal. The increased surface area and small droplets result in transporting the entrapped drug through the unstirred water layer to the gastrointestinal membrane. This would contribute to a greater increase in drug absorption (19,20). Moreover, the droplets can be rapidly dispersed in the blood as well as lymph (21), and lymphatic transport can avoid the hepatic first-pass effect (22).
Thus, it has been served as a vehicle to improve the absorption of drug and a potential strategy to enhance the oral bioavailability of poorly water-soluble drugs (23). However, SMEDDS is a liquid formulation and has several limitations including low stability and portability during the manufacturing process and interaction of filling the capsule shell (24). To overcome these problems, lipid formulations could show a solid state by using suitable adsorbents. Solid SMEDDS (S-SMEDDS) has been investigated as a candidate, which can be prepared by incorporating the liquid SMEDDS (l-SMEDDS) into the solid dosages, combining the advantages of l-SMEDDS with those of solid dosage forms. This will improve oral bioavailability, storage stability, and patient compliance (25,26).
Recently, there have been some new methods of self-microemulsion solidified, such as spray-drying method (27), solid carriers adsorption (28,29), and so on (30). However, L-SMEDDS consists of oil and surfactants, so the viscosity and adhesion are inevitable. The poor dispersibility and fluidity are not conducive to preparing the following drug dosage forms after adding water, resulting in the low yield of the spray-dried. The adsorption of solid carriers has to add lots of carrier vehicles, which make solid powders sticky and less drug loading into solid powders. In order to improve dissolution and bioavailability of poorly soluble drugs, low-density porous carriers with marvellous surface area composed of porous silica as well as magnesium aluminometasilicate has been used, such as ritonavir (31) and carbamazepine (32) in recent years. Some solid products also need granulation process, which is difficult because of the intrinsic viscosity of water sensitive drug for post-processing. Therefore, a new method of solidification is needed urgently.
The optimal puerarin liquid self-microemulsion had been made, and the purpose of this work was to prepare puerarin solid SMEDDS (Pue-SSMEDDS) by spherical crystallization technique which was a new method. One-step solidification was executed in the liquid phase by the spherical crystallization technology. The method built a good-stability, low-cost, and drug-loading S-SMEDDS in liquid phase meeting a variety of demands of the self-microemulsion as the main medicine component. In this paper, droplet size analysis, morphological analysis, and powder properties were performed to evaluate the optimal formulation S-SMEDDS. In vitro performance of S-SMEDDS of puerarin was studied in hydrochloric acid (0.01 mol L−1) imitating gastric environment, then pharmacokinetics study in the rat was against pueparin suspension composed of conventional tablets and L-SMEDDS.
MATERIALS AND METHODS
The puerarin (99.4%) was purchased from Wanxi Pharmaceutical Co., Ltd. (Hennan, China), and conventional tablets were purchased from Tongrentang Pharmaceutical Technology Development Co., Ltd. (Beijing, China). Polyoxyethylene hydrogenated castor oil (Cremophor RH40) was supplied as a gift sample by BASF, Germany. 1,2-Propanediol and ethanol were obtained from Fengjingqiu commerce limited liability company (Beijing, China) and Pilot Chemical Co., Ltd. (Shanghai, China), respectively. Castor oil was bought from Damao Chemical Reagent (Tianjian, China). Ethyl cellulose (EC 10cp) and silica powder were both kindly given from Sunward Pharmaceutical Excipients Co., Ltd. (Anhui, China), as a sample. Liquid paraffin was charged from Sinopharm Chemical Reagent Co., Ltd. Other chemicals were of high-performance liquid chromatography (HPLC) or analytical grade.
Solubility Test
To select a suitable oil and cosolvent for SMEDDS formulation, the solubility studies of puerarin in different oils and co-solvents were carried out. The samples have been handled according to the preliminary study of our research group. After a certain amount of supernatant was weighed, the content was dissolved and fixed to the scale of the flask by flow phase dilution. Following verification of specificity, linearity, precision, accuracy, and recovery, the method has been successfully applied in solubility studies.
Preparation of Puerarin L-SMEDDS
The puerarin L-SMEDDS containing castor oil, Cremophor RH40, and 1,2-propanediol has been obtained. The final content of puerarin in the formulations was fixed at 7% (w/w) (33). First, puerarin was dissolved in an appropriate amount of a mixture of 9% castor oil and 38% 1,2-propanediol. Then, appropriate amount of surfactant was used in the melted state before preheating at 37°C. The mixture was vortexed until a clear solution to ensure homogeneity.
Influence of Dilution Times, pH, and Ionic Strength on L-SMEDDS Behavior
Before L-SMEDDS was solidified, the required amount of drug was incorporated with L-SMEDDS basis. One milliliter of this mixture used in the optimized formulation was added to a glass beaker containing different volumes (9, 49, 99, and 199 mL) of different solutions (0.9% normal saline solution, hydrochloric acid(0.1 mol· L−1), and pH 6.8 phosphate buffer), and every mixture was stored at ambient temperature for 24 h (34,35). The dilution times were determined in accordance with the droplet size analysis that could not be measured by the equipment at high amount of water, e.g., 100–500-fold (22). The visual inspection of the resultant microemulsion was done under a white background.
Preparation of Puerarin Solid Self-Microemulsion
The amount of poor solvent (liquid paraffin) was measured in a beaker around the water bath. The L-SMEDDS and the silica powder were added to the 7 mL good solvent and bridging agent containing 0.5 g ethylcellulose at room temperature and uniformly dispersed. Then, add the above solution to the poor solvent under fixed stirring condition (400 r/min, paddle type agitator with four blades). The stirring continued to obtain agglomerates (about 30 min), which were filtered and dried overnight. The dried products were stored in screw-crapped jars at room temperature before using.
Droplet Size of Self-Microemulsion
Ten microliters of the L-SMEDDS formulation was diluted with 10 mL water and gently mixed with a magnetic stirrer. The droplet size of the microemulsions loaded with puerarin was determined by photo correlation spectroscopy (Nano ZS90, Malvern Instruments, UK). The solid SMEDDS (400 mg) was dispersed in 20 mL water and gently vortex-mixed. After mixing, an aliquot of the mixture was filtered through a membrane filter (0.45 μm; nylon syringe filter). The size of the droplet in the filtrate was also measured by photo correlation spectroscopy (Nano ZS90, Malvern Instruments, UK). The results were compared with previous liquid SMEDDS droplet size analysis results. All studies were repeated three times and the average value was used.
Morphological Analysis
The outer macroscopic structure of the S-SMEDDS was investigated using the Phoenix microscopic image processing and analysis software (Phmias 2008 Cs ver 3.0 Demo) installed on the computer. The system was attached to the XSZ-G biological microscope (Mike Audi Industrial Group Co., Ltd.) containing the five million pixels electro camera eyepiece (MC-D 500 U, Phoenix Optical Group).
Particle Size and Size Distribution
The particle size and distribution could be determined by sieving analysis method using the standard sieve stipulated in the Chinese Pharmacopoeia 5 min on the oscillator; the weight of the substance on every sieve was obtained to calculate the percentage of different ranges of size.
Flowability
In this study, the angle of repose, compressibility index, and flow velocity were determined to evaluate the fluidity of the Pue-SSMEDDS.
Angle of Repose
This is the most commonly used test for powder flowability by means of angle of repose (36). The angle of repose was measured by a fixed funnel method (37). The end of a funnel was placed 2 cm above the flat base and the powder was released from the funnel. From the height of the cone (h) and the radius of the flat base (r), the angle of repose (α) was determined by using the following equation:
Carr’s Index
Carr’s index reflects the compressibility of the powder. There is a correlation between the compressibility index and the flowability of the crystals (38). The bulk and tapped densities were used to calculate Carr’s index (CI). The CI of the samples could be computed from the bulk and tapped densities by the following equation:
where ρ t and ρ b are tapped density and bulk density of powder, respectively.
Flow Velocity
A certain amount of Pue-SSMEDDS was weighted and made to outflow the funnel uniformly. Then, record the time and the speed was calculated by the equation v = m/t. The results presented were the mean value of six determinations.
In Vitro Dissolution
Dissolution profile of Pue-SSMEDDS was performed in a dissolution tester according to the Chinese Pharmacopoeia Appendix Method II (the big glass method). The dissolution tester was equipped with an outer water-bath in order to maintain the constant temperature and sink conditions for a while. The dissolution test was performed at 37°C with 900 mL hydrochloric acid (0.01 mol· L−1) as the dissolution medium. An appropriate amount of six self-made S-SMEDDSs was taken in dissolution cup, and samples were collected at predetermined intervals of 0.083, 0.17, 0.33, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 10, 12, 14, and 24 h. An equivalent volume (5 mL) of fresh dissolution medium was added to compensate for any loss due to sampling immediately. The collected samples were filtrated through the membrane filter (0.45 μm; nylon syringe filter). The concentration of puerarin in the resulting solution (20 μL) was analyzed by HPLC and converted into different time of the cumulative dissolution percentage by calculating formula. A plot of percent cumulative release of puerarin against time was constructed to illustrate the drug release profile. The drug release curve was fitted by zero-order model, first-order model, and Higuchi model, respectively. The correlation coefficient (R) was close to 1, which indicated that the choice of the model was better than other fitted equations. Cumulative dissolution percentage was calculated according to the following equation:
where C t referred to the content of each sampling point, V 0 is the volume of dissolution medium, V is the sample volume, and W is the total amount of the dissolution medium.
In order to compare the difference among each other, L-SMEDDS was contrasted with S-SMEDDS in vitro dissolution at the same coordinate. The value of every point of profiles represented the mean ± SD of six experiments.
Stability Study
Pue-SSMEDDSs were sealed in a bottle at storage condition of different temperature (4, 25, and 60°C) at intervals of 1 and 3 months. The samples were protected from light to evaluate for their physical stability, including the macroscopic appearance of the samples and droplet size of the microemulsion within 100 nm. The size of the droplet was measured by photo correlation spectroscopy (Nano ZS90, Malvern Instruments, UK).
In Vivo Pharmacokinetics Study
In pharmacokinetics study, specific pathogen-free grade male Sprague–Dawley rats, purchased from the Medical Animal Test Center of Anhui Medical University (Anhui. China), were used for the experiment. The animal experiment was approved by the institutional animal ethical committee and in compliance with the requirements of the National Act on the use of experimental animals (People’s Republic of China).
The rats were housed under normal laboratory conditions of temperature, relative humidity, and light. They were fasted for 24 h with free access to water prior to the experiments. Randomized design was applied to divide the rats into three groups of six animals. Three formulations, puerarin suspension, Pue-L-SMEDDS, and Pue-S-SMEDDS, were orally administered to the three groups at a dose of 35 mg/kg, respectively. Puerarin suspension, referring to Yufengningxin tablet as a conventional Chinese medicine preparation, is composed of the chemical ingredient pueraria. The suspension preparation was considered as a control group.
Blood samples (0.3 mL) were collected by retro-orbital puncture at predetermined time points, which was 0.17, 0.33, 0.5, 1, 2, 4, 6, 8, 12, and 24 h after oral administration. Plasma was collected by centrifuging at 3000 r· min−1 for 10 min, and transferred to a clean tube to store at −20°C until analysis. Then, 200 μL plasma sample was placed in a centrifugal tube and 20 μL of 6% perchloric acid added earlier to vortex for 2 min. The mixture was centrifuged at 12000 r· min−1 for 10 min to separate the denatured protein precipitate. The collected supernatants were used for HPLC (SIL-20AHT, Shimadzu®, Japan) was equipped with a UV detector and an automatic sampling system (SPD-10 M20A and SIL-20A, Shimadzu, Japan). The mobile phase was composed of methanol and water (25:75, volume ratio). The eluent was monitored at wavelength 250 nm with a flow rate of 1.0 mL min−1, which was accomplished on a reversed phase Kromosil C18 column (4.6 × 250 mm, 5 μm) at a flow rate of 1.0 mL min−1. The column temperature was kept at 25°C.
The DAS 2.1.1 pharmacokinetics software (issued by the State Food and Drug Administration of China for pharmacokinetics study) was used for calculating pharmacokinetics parameters such as area under concentration versus time curve (AUC0-t), clearance, and volume of distribution. The peak plasma concentration of the drug (C max) and the time taken to reach C max (T max) were obtained directly from the plasma concentration versus time profiles. All results were expressed as mean and standard deviation (mean ± SD) of six consecutive measurements for each sample. The pharmacokinetics parameters from different formulations were compared for statistical significance by one-way ANOVA.
RESULTS AND DISCUSSION
Solubility Test
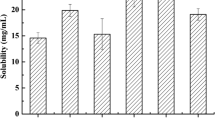
SMEDDS consists of oil, surfactant, and co-surfactant, so the vehicles should have a high solubilizing capacity for the drug. The solubility of puerarin in different vehicles is shown in Fig. 1. Puerarin’s solubility was little in different oil phases and the sequence was by castor oil > EO > oleic acid > GTCC. The sequence of the solubility of puerarin in different emulsifiers was by Tween 80 > RH40 > HS15 > EL. In many cases, the solubility of different co-emulsifiers was distinct significantly, which the solubility of 1,2-propanediol was significantly higher than that of the others. So this study defined the maximum solubility of 1,2-propylene glycol as co-emulsifier and it is to ensure adequate drug loading. However, the constitution of castor oil and Tween 80 exterior presents with light yellow and cream in water, respectively. When the amount of RH40 was too high, the system would form a gel after dilution with water and thus affecting the efficiency of self-emulsifying. Therefore, the co-emulsifier was added to the emulsifier to enhance emulsifying capacity and reduce the amount of emulsifier in this study. Moreover, according to ternary phase diagram test and formulation optimization of L-SMEDDS by central composite design-response surface methodology, castor oil, Cremophor RH40, and 1,2-propylene glycol have been chosen to prepare the best formation self-microemulsion optimization for the further research (33).

Solubility data of puerarin
Properties of Puerarin Liquid Self-Microemulsion
The appearance of puerarin liquid self-microemulsion was observed by visual method under a white background. The visual method was based on the observation that a coarse emulsion phase was formed easily on addition of small amounts of water (39). The addition of about 1 g L-SMEDDS at stirring speed 50 r min−1 in a beaker of distilled water 100 mL at temperature 37°C, which was clear, transparent, and slightly opalescent. The time of self-emulsification was recorded by turbidity that became unchanged when self-emulsification reached equilibrium (40). The time for uniform dispersion of the microemulsion was less than 60 s and transparent clarification with opalescence. The particle size and the potential were 20.37 nm and −23.5 mV, respectively.
At the progress of preparing the L-SMEDDS, the ternary phase diagram has been constructed (33). The particle size first decreased and grew with the increase of K m (the ratio of surfactant and cosurfactant), when the weight percent of oil was lower than 25%. In the prescription with the increase of surfactant concentration, viscosity and emulsifying ability increased in the system. The emulsifying ability dominated when the system of viscosity was lower and the particle size decreased. However, when the weight percent of oil increased to a higher level (<25%), certain obstacles to the emulsifying ability and the particle size increased again. When the weight percent of oil was higher than 25%, particle size decreased with the increase of the proportion of medium oil phase or K m . It concluded that the inherent viscosity of oil reached the maximum extent and the viscosity of the whole system could not significantly influence the effect of the emulsion. Then, the particle size decreased with the increasingly emulsifying ability. The emulsification time increased with the increase of the ratio of oil phase or K m (The relative decrease of emulsifying ability is the fact that the percent of surfactant is lower compared with oil) (13).
Influence of Dilution Times, pH, and Ionic Strength on L-SMEDDS Behavior
The results showed that there was no precipitation or phase separation that occurred in different dispersion mediums. It may be deduced that dilution times, pH, and ionic strength did not influence L-SMEDDS’s behavior from the test. There was little effect on the system after L-SMEDDS was diluted infinitely following by diluting 100-fold, but the zeta potential of the system is too small to maintain stability. So the droplet size was determined by diluting 100-fold in subsequent experiments. The results indicated the L-SMEDDS preparation was stable to a certain extent and of their robustness to dilution (41).
Preparation of Puerarin Solid Self-microemulsion
Ethanol was used as the good solvent system (the good solvent and the bridging agent) and liquid paraffin as the non-solvent. The addition of the bridging agent promoted the transfer of the drug to the emulsified phase in which crystal agglomerates were condensed and grew spherically (41). When the drug solution was added to the poor solvent, the affinity of the drug with good solvent and bridging agent was strong. There was not enough time for the good solvent spreading to the poor solvent and emulsion droplets forming in a moment by the shear force of stirring. Then, the emulsion droplet in the good solvent gradually diffused to the poor solvent; emulsion droplets of the drug continue to crystallize forming spherical particles under the help of bridging agent. In this study, four solvents (CH2Cl2, CHCl3, acetone, and absolute ethanol) were used to choose from. The microspheres were prepared with the individually or mixed good solvent. The appearance, CI, and angle of repose were studied and used for selection of the best solvent. The results were shown in Table I. From the results, we could suggest that a good solvent system, such as absolute ethanol, acetone, or the mixture of them, and the mixture of CH2Cl2 and ethanol, was conducive to the formation of the spherical particles. Given acetone and CH2Cl2 were more toxic than ethanol, then the flowability and compressibility of the product were poor; thus, the ethanol was selected as the good solvent.
In this work, we investigated the effect of the water medium (water solution containing 0.08% SDS) and non-aqueous medium (liquid paraffin) on the results. We found that the water solvent as the poor solvent was not able to make self-microemulsion cured successfully, but the liquid paraffin was able to make it, which might be insoluble for ingredients in the liquid paraffin, especially for the self-microemulsion composition. Finally, 125 mL of liquid paraffin as the poor solvent and 7 mL of ethanol as the good solvent were chosen as the composition of microspheres.
Droplet Size of Solid Self-Microemulsion
To evaluate the reconstitution property of the S-SMEDDS formulation, the droplet size of the reconstituted microemulsion was measured compared with the L-SMEDDS formulation (Fig. 2). The mean droplet size of the L-SMEDDS formulation was 20.37 nm, and that of the S-SMEDDS formulation had a peak appearing at the range from 10 to 100 nm. There was the second peak of size between 100 and 1000 nm, and the phenomenon was probably due to the fact that ethyl cellulose (10cp) was used as the carrier in the experiment and ethyl cellulose (10cp) had stickiness. So some of the emulsion droplets were gathered in the process of solidification. Interfacial tension effects and capillary action showed that the bridging agent (wetting agent) plays a role in blocking drug crystallization (42). Since bridging agent and poor solvent are immiscible with preferential wetting solution crystallization, so crystalline deposit precipitates immediately under agitation. What is more, the zeta potential of the S-SMEDDS had no significant difference compared with the L-SMEDDS. From these achievements, the solidified method did not seem to have a remarkable effect on the droplet size and the zeta potential, which almost indicated that the Pue-SSMEDDS was made successfully by spherical crystallization technique.

The particle size distribution and the zeta potential of a the puerarin liquid self-microemulsion and b the puerarin solid self-microemulsion
Morphological Analysis
Morphological characterization of the optimal solid self-microemulsion was presented in Fig. 3. According to the microscopic image (Fig. 3), the particles of solid self-microemulsion were well separated. The particles showed a satisfactory regular spherical shape with shine and shallow dents. The micro porous in the matrix and on the surface might form channels for water to infiltrate, which would be conducive to the formation and dispersion of microemulsion. The results of the self-microemulsion above illustrated that the particle size and potential of the solid self-microemulsion were close to the liquid self-microemulsion. Judged by this phenomenon, the solid self-microemulsion was successfully solidified.

Optical microphotographs of puerarin solid self-microemulsion at a ×4 magnification and b ×10 magnification
Particle Size and Size Distribution
From the particle size distribution of the agglomerated product, it was noticed that the smaller solid particles still existed. But the particles size distributed mainly between 450 and 600 μm, which was very concentrated (Fig. 4). To sum up, in vitro reconstitution study and characterization ensured that the S-SMEDDS had good quality.

Particle size distribution of puerarin solid self-microemulsion
Flowability Study
Table II showed the flowability and compressibility of the microparticles that were represented in terms of the angle of repose, outflow speed, and Carr’s index. Materials with CI value below 15% indicated good fluidity and value above 25% indicated poor fluidity (43). The angle of repose was below 30° proving a very good fluidity. All microparticles generally showed fluidity and compressibility and might be attributable to the spherical shape, since they are of contacts in the spherical-shaped microparticles. They were smaller than those in other shaped conventional crystals. The results indicated that they might be directly compressible.
In Vitro Dissolution
In this paper, the determination method on puerarin dissolution of the material and hydrochloric acid (0.01 mol L−1) was not interfered with each other. The specific properties of in vitro dissolution method were good, and the standard curve was y = 97231x + 46839 (R = 0.9999) with the linear range 2–140 μg· mL−1. The RSD of the low (5 μg· mL−1), middle (20 μg· mL−1), and high (100 μg· mL−1) concentration of puerarin solutions was 1.33, 1.79, and 1.67%, respectively (n = 5). The RSD of stability and percent recovery were 2.07 and 2.43% (n = 5) indicating that puerarin was stable in 24 h and the method was reasonable.
An excessive amount of puerarin was added to different dissolution mediums of 37°C hydrochloric acid (0.1 moL· L−1), distilled water, and pH 6.8 phosphate buffer. The solubility of puerarin in different mediums by means of the determination of the peak area was calculated according to the standard curve. The results of hydrochloric acid (0.1 moL· L−1), distilled water, and pH 6.8 phosphate buffer were 2.7, 3.6, and 5.6 mg· mL−1, respectively, all of which could reach the drain condition. In general, the vivo environment of oral preparations is simulated on the basis of gastric environment. So hydrochloric acid (0.1 moL·L−1) was chosen as the dissolution medium.
The sequence of in vitro release fitted model was by Higuchi model > first-order model > zero-order model by calculating (Table III). Therefore, the best prescription and preparation technique of the aggregates accorded with the Higuchi model. The release of puerarin from S-SMEDDS was substantially complete 100% within 24 h. The ability of Pue-S-SMEDDS had the tendency to emulsify very lowly compared to Pue-L-SMEDDS and was reflected by the rate of dissolution. The dissolution curve of liquid self-microemulsion and solid self-microemulsion were also compared in the experiment. The drug release curve was found to be very fast that the 10 min was close to 80% and was completely released as to liquid self-microemulsion while 10% released within 30 min as to solid self-microemulsion (Fig. 5). Faster dissolution may be due to the formation of nano-size droplets of resultant microemulsion. However, Pue-S-SMEDDS needed to be dissolved to form a microemulsion in vitro, and it would be the rate of limiting step for puerarin release in vitro. So it had a better effect on the release of the drug than liquid self-microemulsion Fig. 6.

The cumulative drug released of puerarin solid self-microemulsion in hydrochloric acid (0.01 mol L−1) (mean ± SD, n = 6)

Comparison of the dissolution curves of liquid self-microemulsion and solid self-microemulsion (mean ± SD, n = 6)
Stability Study
There were no obvious changes with the appearance and the content in different storage conditions. In this study, the evaluation of temperature stability of solid self-microemulsion, suggesting that the optimal formulation has no remarkable changes under all conditions except the temperature of 60°C on account of oil spill on their macroscopic appearance at the end of the experiment, which was due to both the temperature-induced denaturation of the Cremophor RH 40 and the gradual increase of particle size of the microemulsion (data not shown).
Oral Bioavailability Study
In order to evaluate the bioavailability of the Pue-SSMEDDS formulation, in vivo pharmacokinetic study was performed in rats. Figure 5 showed the time course of puerarin blood concentration after the oral administration, and the pharmacokinetics parameters were presented in Table IV. Relative bioavailability of microemulsion was dramatically improved in comparison with puerarin suspension. Compared with the suspension, the AUC and C max from L-SMEDDS and S-SMEDDS were both significantly higher (P < 0.001). From L-SMEDDS and S-SMEDDS, there was approximately 20–30-fold and 1.9–2.5-fold increase in AUC and C max, respectively. The phenomenon that was observed might be due to the fact that the SMEDDS could have facilitated its absorption through the lymphatic pathway. It has been already reported that lymphatic pathway might play an important role in improving bioavailability by lipid-based delivery system for drugs with low bioavailability (44). Puerarin could be absorbed fast but not completely after oral absorption, which made it low bioavailability. In the human body, its wide distribution, fast metabolism, and accumulate easily, and absorption half-life, elimination half-life, and mean residence time of 10.3 min, 74.0 min, and 1.28 h, respectively (45). Pharmacokinetics study for Yufengningxin tablets in rabbits (46), the absolute bioavailability of puerarin of only 5.45%, indicated its absorption and low bioavailability compared with injection. But it can maintain a long time in the target organ heart-brain and low drug concentration of metabolic organs (47). The administration of puerarin by oral route is still relatively a good choice. So future research would be focused on improving its extent of absorption. It could be observed that the mean value of T max from L-SMEDDS had no statistical difference with suspension (P > 0.05), but the mean value of T max from S-SMEDDS was statistically different with suspension, which was 4-fold of that of pueparin suspension (P < 0.001). The delayed T max from S-SMEDDS might be due to the sustained release of puerarin from solid self-microemulsion system. What is more, no significant difference was observed between the L-SMEDDS and S-SMEDDS for AUC(0-t), which showed that existing puerarin in the form of S-SMEDDS kept the absorption enhanced as high as with L-SMEDDS. It have been explained that SMEDDS, a lipid-based formulation, is thought to be partially absorbed via the lymphatic route. That might reduce the chance for pre-systemic drug metabolism and hepatic first pass metabolism, therefore raising the bioavailability of drugs (48). Constantinides (49) also revealed that the drug compound in o/w microemulsions reached the capillaries incorporated within the oil droplets, which can protect the drug from chemicals as well as enzymatic degradation, increase the residence time, and improve the bioavailability in vivo. The T1/2 of L-SMEDDS and S-SMEDDS were 2.4-fold and 4.7-fold of that of suspension, respectively. From the results of the experiment, it could be inferred that the solid self-microemulsifying components had significant effects on lengthening the time of maximum plasma concentrations, decreasing the rate of elimination of pueparin and enhancing the bioavailability of poor water soluble drug Fig. 7. In conclusion, the increase of bioavalability offered by SMEDDS was maybe owing to the absorption and the degradation process in vivo, and S-SMEDDS has a significant impact.

Blood concentration-time profile of puerarin in rats after oral administration of puerarin in the formulation as (black circle) suspension, (black square) L-SMEDDS, and (black triangle) S-SMEDDS. Every point represents the mean ± SD of six experiments
Bimodal Phenomena of Drug-Time Curve
Irrigating the stomach of puerarin solid self-microemulsion and liquid self-microemulsion, blood drug concentration peaks appeared at 30 min after administration. The blood drug concentration increased again. The second peak appeared and was lower than the first peak after 8 h of intragastric administration, indicating the hepatoenteral circulation of pueparin. This phenomenon was similar to the reported results of AJS (50). The second peak concentration might be associated with the intestinal lymphatic transport because drug of high oleophilicity has been reported as a substrate for lymphatic transport (51). We guessed the possibilities of bimodal phenomena below:
-
1.
Gastrointestinal circulation: The drug was absorbed across membrane transport by the stomach. The concentration was relatively high to the presence of ionic form in the stomach. Entering the small intestine, it would be absorbed to form biabsorption once again.
-
2.
Double site absorption: The drug in the gastrointestinal tract of different parts with two absorption points, of which intestinal mucosal permeabilities for the drug were not same, making different parts of the absorption time and rate not consistent. Then, the absorption of the drug could be superimposed on the concentration of blood that was bimodal phenomenon.
-
3.
Drug distribution: The drug was distributed around the tissue rapidly after drug absorption and blood concentration decreased. After that, the organization of the drug was again distributed into the blood.
-
4.
Effects of metabolites of the drug: The drug would be the formation of glucuronic acid conjugates in the liver. The conjugates turned into prototype drugs after glucuronic acid hydrolases with the excretion of bile into the intestine to reabsorb. The protein competition could also become double peaks as the protein binding of metabolites was higher than drug protein binding. Of course, all suppositions should need proving by experiments.
CONCLUSIONS
S-SMEDDS drug delivery system containing puerarin was prepared successfully. The quality and bioavailability of the optimal formulation were assessed and executed in vitro and in vivo. It was explored to find a suitable medium with a good solubilizing capacity through the solubility of pueparin in different oils, emulsifiers, and co-emulsifiers. According to the construction of the ternary phase diagram and formulation optimization of SMEDDS by the central composite design-response surface methodology, the final formulation has been obtained (33). To improve the stability and overcome the drawbacks of the liquid formulation, the S-SMEDDS was prepared by the spherical crystallization technique. Liquid paraffin for 125 mL as the poor solvent and ethanol for 7 mL as the good solvent were chosen as the composition of microspheres by optimizing. Furthermore, droplet size, morphological analysis, and powder properties of the optimal Pue-SSMEDDS were determined at the same time. Both the particle size and zeta potential measurements suggested that puerarin in the solid SMEDDS was the similar to puerarin in the liquid SMEDDS. Dissolution and stability were conducted for further evaluation in vitro. The solid SMEDDS could better control the release of the drug compared with the liquid SMEDDS. As mentioned in this paper, dissolution of the solid SMEDDS was the main limiting factor of the drug release. So the solid SMEDDS could reduce the loss of drug in vivo and further improved the bioavailability of the drug. The plasma concentration of pueparin in rats was determined by HPLC, and the pharmacokinetics behavior of microemulsion was compared with the suspension. The AUC of L-SMEDDS and S-SMEDDS after oral administration increased 23.23-fold and 27.03-fold, respectively. In vivo studies demonstrated that the solid SMEDDS might be a useful tool for enhancing the absorption of puerarin. Thus, this study would contribute to the application of the S-SMEDDS by the spherical crystallization technique and providing a useful solid dosage form for oral poorly water-soluble drug like pueparin in the modernization research of Chinese medicine.
In intragastric administration of puerarin-mixed suspension of rats, the drug curve did not appear bimodal phenomena that may be related to the preparation. It is necessary to study on whether there are one or two absorptions for microemulsion.
References
Wang C, Song Z. In vitro monitoring of nanogram levels of puerarin in human urine using flow injection chemiluminescence. Bioorg Med Chem Lett. 2004;14(16):4127–30.
Chen CC, Chan WH. Impact effects of puerarin on mouse embryonic development. Reprod Toxicol. 2009;28(4):530–5.
Luo CF, Yuan M, Chen MS, Liu SM, Zhu L, Huang BY, et al. Pharmacokinetics, tissue distribution and relative bioavailability of puerarin solid lipid nanoparticles following oral administration. Int J Pharm. 2011;410(1–2):138–44.
Song XP, Chen PP, Chai XS. Effects of puerarin on blood pressure and plasma renin activity in spontaneously hypertensive rats. Zhongguo Yao Li Xue Bao. 1988;9(1):55–8.
Yan LP, Chan SW, Chan AS, Chen SL, Ma XJ, Xu HX. Puerarin decreases serum total cholesterol and enhances thoracic aorta endothelial nitric oxide synthase expression in diet-induced hypercholesterolemic rats. Life Sci. 2006;79(4):324–30.
Wang Y, Ma Y, Zheng Y, Song J, Yang X, Bi C, et al. In vitro and in vivo anticancer activity of a novel puerarin nanosuspension against colon cancer, with high efficacy and low toxicity. Int J Pharm. 2013;441(1–2):728–35.
Zhang Y, Wang R, Wu J, Shen Q. Characterization and evaluation of self-microemulsifying sustained-release pellet formulation of puerarin for oral delivery. Int J Pharm. 2012;427(2):337–44.
Liu SD, Mo HP. Occurrence and prevention of adverse reaction induced by puerarin injection. Chin J Intern Med. 2005;25(9):852–5.
Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12(23–24):1068–75.
Chen Y, Lu Y, Chen J, Lai J, Sun J, Hu F, et al. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int J Pharm. 2009;376(1–2):153–60.
He W, Tan Y, Tian Z, Chen L, Hu F, Wu W. Food protein-stabilized nanoemulsions as potential delivery systems for poorly water-soluble drugs: preparation, in vitro characterization, and pharmacokinetics in rats. Int J Nanomedicine. 2011;6:521–33.
Lian R, Lu Y, Qi J, Tan Y, Niu M, Guan P, et al. Silymarin glyceryl monooleate/poloxamer 407 liquid crystalline matrices: physical characterization and enhanced oral bioavailability. AAPS PharmSciTech. 2011;12(4):1234–40.
Hu X, Lin C, Chen D, Zhang J, Liu Z, Wu W, et al. Sirolimus solid self-microemulsifying pellets: formulation development, characterization and bioavailability evaluation. Int J Pharm. 2012;438(1–2):123–33.
Wang D, Li H, Gu J, Guo T, Yang S, Guo Z, et al. Ternary system of dihydroartemisinin with hydroxypropyl-beta-cyclodextrin and lecithin: simultaneous enhancement of drug solubility and stability in aqueous solutions. J Pharm Biomed Anal. 2013;83:141–8.
Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45(1):89–121.
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11 Suppl 2:S93–8.
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–82.
Kang BK, Lee JS, Chon SK, Jeong SY, Yuk SH, Khang G, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1–2):65–73.
Grove M, Mullertz A, Nielsen JL, Pedersen GP. Bioavailability of seocalcitol II: development and characterisation of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur J Pharm Sci. 2006;28(3):233–42.
Shahnaz G, Hartl M, Barthelmes J, Leithner K, Sarti F, Hintzen F, et al. Uptake of phenothiazines by the harvested chylomicrons ex vivo model: influence of self-nanoemulsifying formulation design. Eur J Pharm Biopharm. 2011;79(1):171–80.
Amri A, Le Clanche S, Therond P, Bonnefont-Rousselot D, Borderie D, Lai-Kuen R, et al. Resveratrol self-emulsifying system increases the uptake by endothelial cells and improves protection against oxidative stress-mediated death. Eur J Pharm Biopharm. 2014;86(3):418–26.
Weerapol Y, Limmatvapirat S, Kumpugdee-Vollrath M, Sriamornsak P. Spontaneous emulsification of nifedipine-loaded self-nanoemulsifying drug delivery system. AAPS PharmSciTech. 2015;16(2):435–43.
Kim CK, Cho YJ, Gao ZG. Preparation and evaluation of biphenyl dimethyl dicarboxylate microemulsions for oral delivery. J Control Release. 2001;70(1–2):149–55.
Qi X, Qin J, Ma N, Chou X, Wu Z. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation. Int J Pharm. 2014;472(1–2):40–7.
Wang Z, Sun J, Wang Y, Liu X, Liu Y, Fu Q, et al. Solid self-emulsifying nitrendipine pellets: preparation and in vitro/in vivo evaluation. Int J Pharm. 2010;383(1–2):1–6.
Xiao L, Yi T, Liu Y. A new self-microemulsifying mouth dissolving film to improve the oral bioavailability of poorly water soluble drugs. Drug Dev Ind Pharm. 2013;39(9):1284–90.
Yi T, Wan J, Xu H, Yang X. A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur J Pharm Biopharm. 2008;70(2):439–44.
Jang DJ, Jeong EJ, Lee HM, Kim BC, Lim SJ, Kim CK. Improvement of bioavailability and photostability of amlodipine using redispersible dry emulsion. Eur J Pharm Sci. 2006;28(5):405–11.
Cho W, Kim MS, Kim JS, Park J, Park HJ, Cha KH, et al. Optimized formulation of solid self-microemulsifying sirolimus delivery systems. Int J Nanomedicine. 2013;8:1673–82.
Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13–14):606–12.
Deshmukh A, Kulkarni S. Solid self-microemulsifying drug delivery system of ritonavir. Drug Dev Ind Pharm. 2014;40(4):477–87.
Milovic M, Djuris J, Djekic L, Vasiljevic D, Ibric S. Characterization and evaluation of solid self-microemulsifying drug delivery systems with porous carriers as systems for improved carbamazepine release. Int J Pharm. 2012;436(1–2):58–65.
Ye L, Hu RF, Wang XH, Tang JH. Optimization of puerarin self-microemulsifying system using central composite design-response surface methodology. Chin Tradit Pat Med. 2014;3:514–9.
Kim JY, Ku YS. Enhanced absorption of indomethacin after oral or rectal administration of a self-emulsifying system containing indomethacin to rats. Int J Pharm. 2000;194(1):81–9.
Atef E, Belmonte AA. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS). Eur J Pharm Sci. 2008;35(4):257–63.
Jallo LJ, Ghoroi C, Gurumurthy L, Patel U, Dave RN. Improvement of flow and bulk density of pharmaceutical powders using surface modification. Int J Pharm. 2012;423(2):213–25.
Garala K, Patel J, Patel A, Raval M, Dharamsi A. Influence of excipients and processing conditions on the development of agglomerates of racecadotril by crystallo-co-agglomeration. Int J Pharm Investig. 2012;2(4):189–200.
Nokhodchi A, Maghsoodi M. Preparation of spherical crystal agglomerates of naproxen containing disintegrant for direct tablet making by spherical crystallization technique. AAPS PharmSciTech. 2008;9(1):54–9.
Schussele A, Bauer-Brandl A. Note on the measurement of flowability according to the European Pharmacopoeia. Int J Pharm. 2003;257(1–2):301–4.
Venkatesh G, Majid MI, Mansor SM, Nair NK, Croft SL, Navaratnam V. In vitro and in vivo evaluation of self-microemulsifying drug delivery system of buparvaquone. Drug Dev Ind Pharm. 2010;36(6):735–45.
Varshosaz J, Tavakoli N, Salamat FA. Enhanced dissolution rate of simvastatin using spherical crystallization technique. Pharm Dev Technol. 2011;16(5):529–35.
Kawashima Y, Okumura M, Takenaka H. The effects of temperature on the sphericalcrystallization of Salicylic acid. Powder Technol. 1984;39:41–7.
Parida R. Evaluation parameters for spherical agglomerates formed by spherical crystallisation technoque. Int J Pharm Bio Sci. 2010;3:1–10.
Iosio T, Voinovich D, Perissutti B, Serdoz F, Hasa D, Grabnar I, et al. Oral bioavailability of silymarin phytocomplex formulated as self-emulsifying pellets. Phytomedicine. 2011;18(6):505–12.
Jin XL, Chen GF, Zhu XY, et al. Puerarin dynamics pharmacokinetics in healthy volunteers. Chin J Clin Pharmacol. 1991;2:115–8.
Zhang ZR, You XJ, Wei ZP, He Q, Li SW. Yufengningxin capsule in pharmacokinetics and bioavailability studies in rabbits. Chin Pharm J. 1997;4:32–4.
Wu YH, Su ZR, Chen JN, Lin J, Lai XP. The evaluation of puerarin route of administration by distribution from mice plasma concentration-time curve and organizational characteristics.Tradit Chin Drug Res Pharmacol. 2005(2):112-15.
Woo JS, Lee CH, Shim CK, Hwang SJ. Enhanced oral bioavailability of paclitaxel by coadministration of the P-glycoprotein inhibitor KR30031. Pharm Res. 2003;20(1):24–30.
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–72.
Wu L, Qiao Y, Wang L, Guo J, Wang G, He W, et al. A self-microemulsifying drug delivery system (SMEDDS) for a novel medicative compound against depression: a preparation and bioavailability study in rats. AAPS PharmSciTech. 2015;16:1051–8. doi:10.1208/s12249-014-0280-y. 1–8.
Wu C, Wang Z, Zhi Z, Jiang T, Zhang J, Wang S. Development of biodegradable porous starch foam for improving oral delivery of poorly water soluble drugs. Int J Pharm. 2011;403(1–2):162–9.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81274100).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The animal experiment was approved by the institutional animal ethical committee and in compliance with the requirements of the National Act on the use of experimental animals (People’s Republic of China).
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Cheng, G., Hu, R., Ye, L. et al. Preparation and In Vitro/In Vivo Evaluation of Puerarin Solid Self-Microemulsifying Drug Delivery System by Spherical Crystallization Technique. AAPS PharmSciTech 17, 1336–1346 (2016). https://doi.org/10.1208/s12249-015-0469-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0469-8