
Abstract
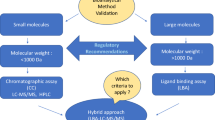
Decades of discussion and publication have gone into the guidance from the scientific community and the regulatory agencies on the use and validation of pharmacokinetic and toxicokinetic assays by chromatographic and ligand binding assays for the measurement of drugs and metabolites. These assay validations are well described in the FDA Guidance on Bioanalytical Methods Validation (BMV, 2018). While the BMV included biomarker assay validation, the focus was on understanding the challenges posed in validating biomarker assays and the importance of having reliable biomarker assays when used for regulatory submissions, rather than definition of the appropriate experiments to be performed. Different from PK bioanalysis, analysis of biomarkers can be challenging due to the presence of target analyte(s) in the control matrices used for calibrator and quality control sample preparation, and greater difficulty in procuring appropriate reference standards representative of the endogenous molecule. Several papers have been published offering recommendations for biomarker assay validation. The situational nature of biomarker applications necessitates fit-for-purpose (FFP) assay validation. A unifying theme for FFP analysis is that method validation requirements be consistent with the proposed context of use (COU) for any given biomarker. This communication provides specific recommendations for biomarker assay validation (BAV) by LC-MS, for both small and large molecule biomarkers. The consensus recommendations include creation of a validation plan that contains definition of the COU of the assay, use of the PK assay validation elements that support the COU, and definition of assay validation elements adapted to fit biomarker assays and the acceptance criteria for both.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Over the past two decades, liquid chromatography coupled with tandem mass spectrometric detection (LC-MS) has been the most widely used bioanalytical tool for pharmacokinetic (PK) and toxicokinetic (TK) analysis in support of pharmaceutical research and development. Regulatory guidance governing this practice for small molecule drugs is well established, largely building upon the landmark FDA Guidance on Bioanalytical Methods Validation (BMV) in 2001 (1). A significant feature of the 2001 FDA BMV Guidance was that chromatographic methods and ligand binding assays (LBA) were incorporated into the same framework for the first time. Although endogenous molecules administered as drugs were included, quantitative analysis of biomarkers was not considered within scope. In the years that followed, both LBA and LC-MS experienced substantial growth in the number of applications to both small and large molecule biomarkers and several papers appeared in the literature offering recommendations for biomarker assay validation (BAV) practices (2,3,4,5,6,7,8,9,10,11,12,13,14). In 2013, the FDA announced in a draft update to the BMV Guidance that biomarker assays would be considered within scope (15). This led to a period of very active discussion (16, 17), culminating in the 2018 BMV Guidance which now regulates biomarker quantitation by either LC-MS or LBA for drug development applications (18). Building from this landmark guidance, the international bioanalytical community has developed a draft version of a global document on bioanalytical validation for both LBA and LC-MS (ICH M10) which includes specific and exhaustive recommendation for the analysis of endogenous compounds (Section 7.1) administered as drugs (19).
LC-MS quantification of biomarkers in biological fluids shares several experimental similarities with PK bioanalysis, particularly for small molecule analytes. However, analysis of biomarkers differs from exogenous xenobiotic analysis because of two additional complexities that influence the approach to quantitation: (a) the presence of target analyte(s) in the control matrices used for calibrator and quality control (QC) sample preparation and (b) greater difficulty in procuring appropriate reference standards. Since the first issue also applies when endogenous compounds are given as drugs, various approaches for handling this problem are described in the current ICH draft M10 guidance (19). The second issue presents a significant challenge to protein quantification as it is difficult to confirm that recombinant derived reference standards truly depict their endogenous counterparts. Owing to this difficulty, among other factors, it is widely acknowledged that it is more challenging for protein biomarker assays to fully abide by the validation constructs developed for PK analysis (17).
Although biomarkers were included in the 2018 FDA Guidance, the intent was not to create a set of prescriptive rules for BAV but to focus the attention on the importance of having reliable biomarker assays when used for regulatory submissions. This realization reflects the situational nature of biomarker applications and the resultant need for fit-for-purpose (FFP) validation (3), a position consistently endorsed by the FDA. A unifying theme for FFP analysis is that method validation requirements be consistent with the proposed context-of-use (COU) for any given biomarker. This concept, along with several other matters related to biomarker method validation in support of drug development tools (DDT) (20), was the subject of a white paper issued in 2019 by the Critical Path Institute (21). This document represents the most comprehensive and current thinking on BAV and is significant since it was co-authored by members from both the pharmaceutical industry and the FDA.
As a follow-on to this seminal work, the present communication seeks to provide specific recommendations for BAV by mass spectrometry, including LC-MS/MS assays for both small and large molecules. Protein assays developed with or without immunoaffinity enrichment are also considered within scope along with methods prepared using high-resolution mass analysis. The recommendations made are the result of extensive discussion by the authors started under the AAPS Bioanalytical Focus Group (currently part of the bioanalytical community) and pertain specifically to biomarker assays used in support of pharmaceutical research and development. While additional MS techniques, notably GC-MS and MALDI-MS, have been used extensively for biomarker analysis, they will not be specifically addressed, although the recommendations will apply to these techniques in most cases. The recommendations made herein are intended to satisfy the requirements for fully validated methods used to deliver biomarker data for regulatory decisions; however, the information provided may also find use in guiding exploratory methods validated by sponsors to inform pre-regulatory decisions. Accordingly, we will use the terms exploratory versus full validation throughout the manuscript to highlight this distinction.
The current white paper is intended to complement current regulatory guidance documents and existing biomarker validation literature. Herein, we define essential features for biomarker methods validation (i.e., validation elements) for consideration in this ongoing dialog in a framework that encompasses both small and large molecule biomarkers measured by LC-MS/MS. Specific emphasis is placed on the importance of a well-articulated validation plan to define the approach taken to validation and to defend any deviations taken from the 2018 FDA BMV Guidance. Given the situational nature of biomarker analysis, we fully endorse the concept of fit-for-purpose methods validation but maintain that this can only be understood through the context of use of the assay, which must be clearly defined.
CREATE A VALIDATION PLAN
In an effort to bring clarity and harmonization to BAV, several additional articles (22,23,24,25,26) were published on potential acceptance criteria, culminating in the official regulatory agency position on the topic in the 2018 FDA BMV (18). However, specific BAV acceptance criteria are still not included for good reason. While PK defines a specific COU, the context of use for a biomarker can be much more varied and complex. Hence, the extent of BAV is a topic of active discussion for regulators and bioanalytical practitioners alike.
The lack of prescriptive guidance describing acceptance criteria will require a different and practical approach to assay validation for biomarkers starting from the better-known PK/TK assay validation. The COU concept comprises not only the biological variation but also variation in the types of measurements and assays that could be part of a biomarker thus making it very difficult to provide broadly applicable, prescriptive acceptance criteria. Indeed, it becomes difficult to even provide a detailed list of what experiments should be part of a generally acceptable BAV.
The recommended best practice at the time of this writing is to develop a validation plan for each assay intended to be part of biomarker measurement that fully takes into account the COU. As a best practice, we recommend that the FDA BMV guidance form the basis of the validation plan and that changes to the experiments and acceptance criteria be supported scientifically and thoroughly explained and documented. Additional experiments should be added when a claim about the performance or use of the assay is not supported by the basic BMV experiments. For example, such a claim might be the discrimination between known proteoforms (27) as the readout for the assay. In this case, validation experiments should be conducted to demonstrate the claimed discrimination between protein species. Acceptance criteria might be addressed in a similar fashion by considering the intended use of the results, and the types and magnitude of the errors permissible while still providing actionable results (21). Even though biomarkers used in pharmaceutical research and development bring new challenges to assay validation, it is possible to define a core set of validation elements that are nearly universal (Table I) for assays conducted by LC-MS. While not complete for any given assay, the elements in the table form the baseline set of experiments that will be required as best practice to validate any LC-MS assay.
It is important within the context of drug development to understand the use of the assay relative to the stage of the project in the pipeline. Biomarker assays used for making regulatory decisions (fully validated) should be validated using the same acceptance criteria as the BMV guidance wherever possible. However, for those biomarkers “intended to support early drug development” (exploratory), only elements required to demonstrate data reliability should be included in the assay validation. The FDA has acknowledged that some BMV parameters may not be applicable for making regulatory decisions for biomarker assays and that additional considerations regarding acceptance criteria may be appropriate in some cases.
Although BAV will have similarities with PK assay validation, differences in acceptance criteria inherent to biomarker methods require case-by-case establishment. Importantly, criteria and rationale should be documented a priori in the validation plan.
CONTEXT OF USE
The first section of the validation plan should define the context of use (21) for the biomarker assay. The context of use provides the framework to justify which assay validation elements are essential, as well as acceptance criteria for the validation.
Biology Under Investigation
A clear understanding of the biology studied and how biomarker measurements enable decisions is fundamental to determining why certain changes from the BMV are required. This section of the validation plan provides justification for the validation experiments and associated acceptance criteria. This section also helps define the type of assay readout to be used, the matrices to be investigated, limitations, and a prediction of the expected changes in the results with disease and treatment. This section is a critical element for communicating the expectations of the assay design and performance with regulators, as well as investigators and project teams, who ultimately use and interpret the results.
Establishing the assay performance characteristics based on the biology prior to method development and pre-validation is essential to a successful outcome. Complex sample preparation involving multiple extractions, digestions, or derivatizations may, while achieving the requisite sensitivity, introduce variability and result in precision that prevents differentiating biomarker levels observed in disease patients from those in a healthy population. However, where the biological differences are large between disease patients and the healthy population, an assay having broader acceptance criteria will be acceptable. Therefore, understanding the biology of the biomarker will enable method developers to assess methods against their intended use and through pre-validation testing of normal and disease samples demonstrate that the assay is likely to succeed in validation. Conversely, the pre-validation testing of normal and disease samples may show that the assay is not currently adequate and require additional work to meet the required performance standards. The authors are of the opinion though that if an assay can achieve a better precision than called for based on the biology and COU, it is inappropriate to arbitrarily use wider acceptance criteria.
Expected Analyte Behavior
A description of the expected behavior of the biomarker in the studies it is supporting helps to define validation elements such as precision and accuracy, or total allowable error where appropriate. When available, important features such as a normal range or disease-state cut points should be described as part of the expected changes in biomarker results. In a fully validated assay, much will be known about the behavior of the biomarker in the studies it supports. This section of a validation plan helps to explain why changes in the basic elements of an assay validation are justified relative to the measurement being made and the decisions those measurements support.
Decisions to Be Made from the Experimental Results
The decisions made from the biomarker data are another important element in defining the context of use. The types of decisions made in drug discovery and development from biomarker data are listed in Table II. Understandably, greater validation is required according to the importance of the decision to regulators and ultimately to patients. Typically, decisions made early in development, where the risk is primarily to the sponsor, have lower regulatory risk and may be undertaken with an assay having a lower level of validation. Moreover, such decisions are often verified during the development of a drug candidate. In contrast, subsequent safety or efficacy studies to support a drug registration must produce the high-quality results expected for regulatory submission.
Assay Readout
Biomarkers cover a wide range of measurement types not common to the more familiar PK assays. The value reported from the measurement, the assay readout, can vary even for the same molecule when used to address different questions at different stages of drug discovery and development. Table III lists several different forms of assay readout for a biomarker. Many of these readouts will be specific to a particular use while others may find application across many different situations. Therefore, defining the readout for the biomarker is a critical part of defining the context of use.
Sample Type
The type of sample to be analyzed is also part of the COU and helps to define the biomarker. Sample types with respect to matrix and pre-analytical considerations of sample collection, transfer, and storage stability are well known in the bioanalytical field. When working with non-liquid matrices, additional considerations are required beyond those for liquid matrices.
A biomarker study sample is collected, treated, and stored before even reaching the analytical laboratory. The procedures that are followed to effectively create the sample are often described as pre-analytical factors and include the sample-collection protocol and the sample storage conditions. When practical, analyte stability should be demonstrated in native matrix samples that mimic the expected study samples. Ideally, the stability determination would be done with endogenous analyte in the native sample matrix. A fortified sample matrix is less desirable but may be the only practical alternative.
Various papers have dealt with the issue of pre-analytical considerations (8, 28, 29). An assessment of the appropriateness of the matrix (serum, plasma, CSF, etc.) should be conducted far in advance of the actual study. Changes to the matrix source or collection paradigm during a study should be avoided and if changes are required, appropriate bridging studies should be conducted to demonstrate the equivalence to matrices employed during validation. Variability in biomarker sample collection should be minimized. Procedural controls to standardize sample collection, especially across clinical sites should be implemented (8, 28, 29) and include standardizing vials, anticoagulants, or dividing samples into multiple aliquots to aid in repeat testing while minimizing effects such as may be caused by freeze-thaw cycles.
The validation plan should contain as much information on the pre-analytical procedures as possible and should be updated if additional information is developed during method development or validation.
Reference Material
The preceding discussions and several referenced white papers make it clear that one of the most significant differences between a PK assay and a biomarker assay is the availability of suitable reference material. Indeed, much of the fit-for-purpose justification is formed around the materials that can be obtained for calibration, quality control, and critical assay reagents. The quality and consistent availability of the reference materials relates directly to the context of use, which in turn depends on the risk that can be tolerated in terms of longitudinal assay performance, traceable accuracy, and robust assay performance between laboratories and analytical techniques. The validation plan should address both the supply and the characterization of reference materials to be used during assay validation, as well as throughout a study. The relationship between the intended use of the assay being validated and the reagent quality required to meet that need should be described. In addition, it may be necessary to detail procedures for bridging changes in supply, such as lot and source of the reference materials to ensure continuity of the biomarker results.
VALIDATION ELEMENTS AND ACCEPTANCE CRITERIA
Validation Elements from the Current Guidance Document
As discussed previously, the bulk of the elements of any validation are consistent and covered in the FDA BMV guidance. The validation plan should call out the validation elements that will be unchanged from the guidance, including the acceptance criteria to be used in conjunction with each. Table I lists these validation elements.
Validation Elements Changed or Added to Support the Context of Use
Equally important is to clearly document changes from or additions to the validation elements in the FDA guidance and to include the scientific basis for changes from the recommended validation practices. Elements such as matrix equivalence or parallelism, for example, are not commonly performed with drug assays but are common elements of a BAV. Similarly, the use of an endogenous QC and the system for tracking longitudinal assay performance may be critical to the use of the biomarker assay but not included for a drug assay. Changes to the experiments performed under a given element should also be included in this part of the validation plan. For example, proof that an assay for an exogenous compound is selective can be accomplished by running blank matrix samples, while an endogenous biomarker will require a different set of experiments to show that the assay is selectively making the required measurement. Likewise, changes in acceptance criteria should be listed and scientifically justified based on the COU and supporting data from method development, or previous versions of the assay used for a different COU such as exploratory assays. It is critical that objective evidence be provided to support the changes from the prescribed validation. Ultimately, it must be clear from the proposed changes to the validation elements and the COU that changes in the validation elements support appropriate interpretation of the biomarker results.
Validation Elements in the Current Guidance that Will Not Be Included
Finally, the validation plan should include a section detailing the elements from the current guidance that will not be conducted. The COU supported by the preceding sections of the validation plan should make it clear why elements of an assay validation are not being conducted as described in the guidance; i.e., a sound scientific or technical reason for the change should be included in the assay validation plan. If a core element (Table I) is to be excluded from the assay validation for scientific reasons, rationale or better yet data should be provided to support that decision. If elements of the prescribed assay validation are not to be followed for technical reasons, a risk assessment should be included in place of data to justify why the biomarker results will be considered valid.
Validation Elements Most Often Changed for a Biomarker Assay
The most common differences between biomarker assays and PK assays are definition of the context of use, the quality, and availability of reference material, and the lack of analyte-free, sample-matched matrix for selectivity, and spiking experiments. The working group spent significant time discussing practical approaches to developing and validating reliable biomarker assays. The following are suggestions on how to manage the most difficult questions in biomarker assay validations and are specifically intended for assays used to support regulatory decisions.
Definition of the Measured Quantity and the Detected Chemical Species
Small molecule pharmacokinetic PK assays have a clear, well-defined measured entity, the drug. Many biomarker assays are not as well-defined. For example, endogenous protein quantification aims to measure the concentration of protein X in a sample, while the entity measured in bottom-up LC-MS assays is the surrogate peptide. Another example is measurement of total cholesterol after hydrolysis of the plasma sample. The assay detects cholesterol, but the sample contains many species of conjugated cholesterol, as well as the cholesterol molecule itself. Definition of the quantity measured and the chemical entity measured is required to define the biomarker assay. Therefore, it is recommended that for clarity, an assay title contains both the measured quantity and the chemical entity detected such as “determination of the molar concentration of protein X by detection of peptide Y”. In this example, detection of a different peptide from protein X could lead to a different concentration for protein X.
Definition of Reference Material for Creation of Standards and Quality Controls
Availability and quality of critical materials are two challenges to the translation of a biomarker assay as it transitions from exploratory use into a clinical setting where the assay may function as a pivotal driver for efficacy, safety, patient segregation, or disease diagnosis. Critical materials, which include reference standards, internal standards, and critical reagents should be monitored, beginning in method development. The minimum characterization required for each should be predefined and appropriate for the intended stage of drug development. Criteria may change based on supply or assay requirements, but this does not obviate the need for a definitive characterization of each material.
A variety of guidelines and white papers describe critical materials for small and large molecule therapeutics, and these serve as a starting point (3, 6, 8, 18, 22, 30, 31). These generally represent the most conservative approach and often require materials of known identity and purity for which the source, lot number, expiration date, and certificates of analysis are available. The guidelines sometimes list similar, but more relaxed requirements for either internal standards or non-critical reagents, or emphasize a fit-for-purpose approach, which acknowledges the difficulty often experienced in supplying reference materials for biomarkers. This is especially true for biomolecules where identity and purity sometimes cannot be assured, as recombinant protein may not closely mimic endogenous protein, or protein variants may interfere with the assay (e.g., post-translational modifications (PTMs), 3D conformers, truncated proteins) (32, 33).
Biomarkers amenable to mass spectrometry–based measurement include simple small molecules, complex small molecules (peptides, lipids, bile acids, oligos, etc.), and proteins. Obtaining appropriate critical materials, and characterizing them, becomes harder with increasing molecular complexity and, therefore requirements cannot be pre-specified. This is also true of stable isotope-labeled (SIL) internal standards. SIL internal standards are usually considered the best option for analyzing both simple and complex small molecules, and in using SIL protein internal standards, there is currently limited but growing experience (34). It is critical that the bioanalytical scientist develops a clear understanding of the attributes of critical materials and their impact on assay integrity.
Several characteristics can be evaluated as part of reference material certification, governed by the type of reference material and its inherent liabilities (Table IV). These elements determine their effect on accuracy, precision, and repeatability of the assay. A certificate of analysis should accompany any standardized material and may include structural information, post-translational modifications, lot normalization for concentration or activity, excipients, stability, preparation, test and retest dates, storage conditions, identity, purity, and appearance.
Important information is often missing from commercial materials; therefore, additional characterization may be required. For example, if purity is low, potential impurities may need to be identified, or if a compound is recognized as hygroscopic, water content and storage conditions to prevent hydration may be necessary. Other ancillary techniques can be applied to further characterize and ensure material quality, and these techniques may be added over time as a biomarker progresses through development. Care should be taken for biomarkers used for making regulatory decisions to assure the stability of critical reagents is known and tracked. In cases where the reagent is difficult to obtain, a re-certification procedure may be necessary.
The validation plan must include the requirements for critical materials with particular attention given to reference material used for assay calibration and quality control. The minimum requirement should be a certificate of analysis proving identity and purity. Further discussion regarding best practices for selecting and characterizing protein reference materials based on the intended use of the assay can be found in Cowan et al. (32) with multiple case studies presented to illustrate some of the challenges.
Considerations for Bottom-Up Protein Biomarker Quantification by MS
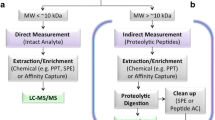
The vast majority of LC-MS/MS-based protein biomarker measurements are currently made on surrogate peptide analytes released by digestion from target proteins of interest using trypsin or an alternate enzyme. Because SIL versions for surrogate peptides are easy to produce, quantitative LC-MS/MS analysis of protein mixtures is a common practice and forms the basis of targeted proteomics. Unfortunately, stoichiometric inference of protein concentrations from peptide data can be highly inaccurate due to variation in peptide recovery, a phenomenon that has been widely studied (34, 36,37,38).
Various reagents and practices have been introduced to address this problem and are summarized in Table V. The incorporation of internal standards released by digestion offers the means to track digestion and are available in several formats: winged peptides (extended sequences added to N- and C-terminus of target peptide (39, 40)), concatenated peptides (mixture of target peptides expressed in a linear sequence (41)), recombinant SIL proteins (42,43,44). Although the pros and cons of these approaches have been reviewed (45), a consensus has emerged that digestible internal standards offer improved accuracy when compared to non-digestible peptides (34, 39, 40). Moreover, recombinant full-length proteins with SIL incorporation clearly provide the best analytical performance (34, 37,38,39, 43).
The use of protein calibrants is also highly endorsed to improve assay accuracy and performance. Because it is difficult to routinely prepare SIL proteins, external calibration curves made from recombinant protein standards offer a practical alternative. While there are acknowledged limitations to this approach, it is generally preferred over methods relying on peptide calibrants (34). Another alternative is to prepare calibration standards from characterized endogenous matrix control samples (36, 46). This approach provides the most authentic representation of the target protein but is often limited by specimen availability and the need for proper characterization.
Based on the existing body of literature, the following recommendations are made for fully validated LC-MS/MS-based protein assays involving surrogate peptide analytes: (1) digestion recovery and reproducibility should occur as part of method development and should be well documented; (2) peptide SIL internal standards should be spiked prior to digestion to track potential instability; (3) protein calibration standards should be used whenever possible (labeled or unlabeled). In cases where this is not possible, clear justification should be provided for using an alternate calibration method; (4) in situations where protein calibrators cannot be used, a suitable in-study digestion control should be incorporated into all batches to verify that acceptable digestion occurred. A well-characterized endogenous QC sample is one option for consideration.
Procedure for Addressing the Endogenous Levels of Biomarker in Control Matrix
A fundamental challenge inherent to biomarker analysis is that control matrices used to prepare calibrator and QC samples will likely contain the target analyte(s). Given the presence of the endogenous analyte in untreated matrix, characterization of the selectivity, accuracy, and precision in a biomarker assay validation cannot be practically accomplished via the same spiking experiments performed for PK assay validation.
In certain cases, it is possible to screen pools of control matrix to identify lots containing low levels of the target analyte (47). However, unless suitable quantities of a control matrix can be found with levels sufficiently below the intended LLOQ, superposition of the endogenous, and spiked analyte will occur to confound quantitative analysis. There are several methods that have been successfully applied to the problem: the use of surrogate matrix, surrogate analyte, standard addition, and background subtraction (5, 19). The exact method chosen will depend on the assay context of use. In the sections that follow, these assay formats for LC-MS biomarker quantification are briefly introduced.
Surrogate Matrix
The surrogate matrix method is the most common approach to biomarker quantification in pharmaceutical research and involves replacing the endogenous control matrix with a matrix variant devoid of the target analyte(s) (19, 48,49,50). Several different procedures for creation and use of a surrogate matrix have been successfully implemented in different situations. The recommended best practice is to use the matrix closest in composition and analytical behavior to the sample matrix. Generally, this means testing for surrogate matrix during method development starting with the most complex and moving to the least complex.
-
1.
Dilute control matrix matched to the sample. In cases where the endogenous level of the biomarker is low, dilution of the control matrix may allow for the required LLOQ to be measured and validated using the typical spiking experiments outlined in the bioanalytical method validation guidance.
-
2.
In the case of protein biomarkers, it is possible to use control matrix from an alternate species having a different sequence from the target analyte.
-
3.
Analyte depletion strategies, particularly with immunoaffinity capture of the analyte, reduce the concentration of endogenous biomarker while preserving much of the remaining native matrix. Modification of the endogenous analyte by selective chemical or enzymatic reaction is another possible method for depletion of analyte from biological matrix. Charcoal-stripped matrix is often used as the surrogate matrix for the preparation of calibrators in clinical diagnostic assays. In all cases, every attempt should be made to preserve as much of the original matrix composition as possible while reducing the amount of endogenous analyte to levels below detection in the assay.
-
4.
The final general procedure involves preparing the surrogate matrix entirely by synthetic means or purchasing a synthetic material. A common example of the former is the use of bovine serum albumin/phosphate-buffered saline mixtures as surrogates for serum or plasma. Synthetic matrices such as artificial CSF and urine can be purchased from commercial suppliers. Such completely synthetic matrices are not close in composition to the sample matrix and may not perform similarly to control matrix owing to the absence of endogenous binding partners which may influence recovery. An example of this phenomenon is found in LC-MS/MS assays for beta-amyloid peptides (51, 52). Synthetic matrices should only be used when a better alternative is not practically available. Additionally, while synthetic matrices tend to be more consistent in performance over time, the absence of the complexity of the native matrix may increase the potential for non-specific binding of the analyte to surfaces used in sample preparation.
The performance or suitability of a surrogate matrix is typically established during method development using experiments described in the subsequent section on parallelism. In many cases, it can be shown that surrogate matrices give acceptable performance by comparing the stable isotope-labeled calibration curve spiked in native matrix to the unlabeled compound calibration curve spiked in surrogate matrix, because the stable isotope-labeled (SIL) internal standard behaves as the unlabeled analyte in terms of recovery and matrix effects (48). This is especially important to demonstrate when using surrogate matrices derived from synthetic origin.
Stable Isotope as a Surrogate Analyte
A surrogate analyte method uses a stable isotope-labeled version of the target analyte which can be spiked into sample-matched native matrix. This method (19, 53,54,55,56,57,58,59) is unique to mass spectrometry and takes advantage of the mass differential between the labeled compound and the endogenous analyte. An advantage of this approach is that native matrices may be used to prepare calibrator and QC samples as is done for xenobiotic bioanalysis.
A fundamental assumption in surrogate analyte methods is that equimolar amounts of the native analyte and its stable isotope-labeled analog will produce equal signals in the mass spectrometer. To ensure that this situation holds in practice, a procedure referred to as “response balance” must be conducted. Any difference observed between signals generated from equimolar amounts of unlabeled and labeled analyte must be corrected for in each analytical run either by applying a correction factor shown to be consistent between instruments and over time or by de-tuning the higher signal on the system to be used for analysis of samples (48).
Standard Addition
The method of standard addition is the practice of adding multiple levels of reference material directly to different aliquots of the sample being measured. The advantage of this method is that each sample has its own calibration curve measured directly in the sample (19, 48). The objective of standard addition is to compensate for matrix effects in each sample. Standard addition has utility in value assignment of QC pools. The downsides are the large number of samples generated to measure the concentration of each unknown and the volume of sample needed to do so. Additionally, small deviations from linearity can introduce bias at low concentrations.
A modified version of standard addition with an external calibration can be used with native matrix containing endogenous analyte to measure baseline levels of endogenous compounds. The method overcomes the concerns associated with standard addition involving measurement of precision and accuracy of the extrapolated values for standards and quality controls, while removing the need for surrogate matrix or surrogate analyte in many biomarker applications. In this approach, the control native matrix is spiked with analyte to create a calibration curve. Calibration in the native matrix is constrained to a linear-through-zero fit with 1/x weighting and excludes lowest level calibrators exceeding accuracy criteria. The resulting calibration curve has concentrations well above the endogenous level. Endogenous analyte concentration in the native matrix and the measurement precision is determined from replicate blanks (n ≥ 6). QCs below the endogenous level are calculated from the difference between the spike readback and the measured endogenous level (48, 60). The difficulty with the latter approach is the need to extrapolate below the native baseline which the current FDA BMV guideline for PK assays strongly discourages. Additionally, this approach is not recommended for experiments where levels of analyte are expected to decrease (e.g., inhibition experiments, downregulation experiments, knockout experiments) or in cases where the disease state exhibits lower levels of endogenous analyte than those present in the available native matrix. However, for biomarker assays, it is currently the best way to measure an endogenous substance in the same matrix as the sample.
Biomarker Assay Quality Controls
As defined in the BMV, quality controls (QC) are samples created much like a calibrator by spiking a control matrix with a known amount of analyte. They are designed to mimic the study samples and used during assay validation to demonstrate the precision of the assay both within and between runs. While similar controls should be made for biomarker assay validation using one of the methods described above to prepare spiked samples with known concentrations, it is also critical to include native sample controls to assure stability of the assay over time (see Table IX). These controls have been referred to as endogenous quality controls (eQCs) (61). The eQCs do not have analyte added and are meant to demonstrate the behavior of the assay during sample analysis relative to assay validation. Ideally, multiple levels of eQCs are chosen and are limited to those in healthy individuals and any available disease-state matrix. Endogenous QCs could be chosen from several different samples or a heterogenous pool that have varying levels of endogenous analyte spanning the dynamic range of the assay.
The eQC may also be called longitudinal controls and are used for control charting of the assay over time. The practice is common in diagnostic applications and may also apply to biomarkers used in pharmaceutical research depending on the assay context of use (8, 62).
Unlike QCs used to characterize precision and accuracy during assay validation, the eQC must stay consistent over time, not just within a run. There are two practical approaches when commercial native matrix is not available from a supplier for use as eQCs. The first is to create a pool of matrix from as many healthy individuals as practical. A pool containing portions from hundreds of individuals is large enough to use over a long period of time, and potentially stable through time thus providing a source of QC material that can be replenished. Using a large pool might also be a practice that is simple to standardize across laboratories (46).
When only smaller pools of disease matrix or in cases where only individual subject material is available, the procedure for value assigning the eQC and for bridging between lots of matrix should be established prior to initiating assay validation and should be thoroughly documented as part of the method and validation. Value assignment is essential to establish the accepted control concentration ranges for the performance of the assay. A predefined number of individual measurements over a defined number of analytical runs, such as 5 measures in each of 4 runs for a total of 20, is used to assess the mean value for the eQC and the expected deviations from the mean. Endogenous QC samples run during analysis of unknowns are included in a moving average, and typical control charting practices are applied to identify drift and out-of-control events. (62)
Incurred Sample Reanalysis Applied to Biomarkers
Incurred sample reanalysis (ISR) is performed in PK studies to ensure reproducibility of analysis, sample stability, and correct, consistent execution of the method. During method validation, spiked samples are used for the determination of accuracy and precision. These spiked samples may not adequately mimic study samples from dosed subjects due to a number of reasons, e.g., extraction recovery, sample inhomogeneity, matrix effects, back conversion of unstable metabolites. To ensure reproducibility of analysis, a subset of incurred samples is reanalyzed. The repeated result is compared with the initial results which provide a measure of the analysis reproducibility (63,64,65,66,67,68).
Since an endogenous analyte is by definition already in a study sample, the concept of an incurred sample in biomarker analysis is different from that in exogenous compound bioanalysis; the incurred sample is available during method development. The purpose of ISR for biomarkers then is in ensuring assay robustness and endogenous sample stability and can be addressed differently from the process used for PK sample analysis.
To address biomarker assay needs, biomarker ISR should focus on proving the ability to quantitate a biomarker in an incurred sample, store that sample under a controlled condition, and then obtain a statistically similar concentration upon reanalysis. Biomarker ISR can be effectively evaluated by means of measurement of eQC(s), incurred sample parallelism, and incurred sample stability. The use of a large batch of eQCs (longitudinal QCs) at one or more concentrations and assay trending can help verify assay performance over time and ensure an assay is under control. Additionally, the use of eQCs in each run can provide a check on calibrator preparation and sample stability. For the latter, repeat analysis of the eQCs or one or more study samples over an extended period of time and trending analysis may be used to demonstrate stability of the biomarker in the native matrix and assay robustness. The assessment of biomarker incurred sample stability (ISS) may include disease subject samples (63). Thus, incurred sample re-analysis is fulfilled when eQCs are run with each analysis and fall within established acceptance criteria.
Specificity (Interference Testing)
A biomarker assay must demonstrate specific detection of the analyte under conditions reasonably anticipated to be encountered within the defined context of use. There is consensus among the working group that proper demonstration of specificity for a biomarker assay may require an in-depth knowledge of the biology of the entity being measured. Therefore, it is important that input be sought from subject matter experts on the likely types of interference that could be part of the biological system under study. During method validation, confirming lack of analyte interference is part of the requirements to demonstrate assay specificity. Emphasis in this section is on the types of analytical specificity testing that might be applied to biomarkers that are not usually applied to exogenous analytes.
Known Isoforms, Conjugates, and Modified Forms
The problem posed by analysis of a defined single chemical substance as a biomarker is that the biological system being measured may not relate to one single molecule. The importance of interference testing is to demonstrate that the assay result is not confounded by contributions to the detected signal by non-target chemical species also present in the sample. The problem comes in determining which different chemical species might interfere analytically and affect the biomarker results. Often, both active and inactive isoforms (i.e., total) for protein biomarkers are included in hybrid ligand binding-LC-MS/MS assays. The recommended approach is to understand what is being measured by the biomarker assay to avoid the risk of diluting the effect of the active form by detecting multiple isoforms and proteoforms (69). Lipids represent another example owing to the high number of isomers present for a given analyte coupled with the limited availability of isomeric standards for interference testing. Because of these issues, a universal approach to demonstrating a complete lack of endogenous interference is extremely difficult and may not be necessary depending on how the interfering molecules affect the biomarker result. It is, therefore, important that thought be given to what interferences are most likely to confound the intended biomarker assay result.
One of the most common interferences comes from biological modification of the target analyte or species being measured. Proteins exemplify this, where care in selection of surrogate peptides must be taken to avoid isoforms, unwanted cleavage products, precursor proteins, and post-translational modifications when such chemical entities are anticipated or known to have a negative impact on the biomarker result. Practically speaking, interferences that need to be avoided must be identified and addressed in method development. For validation, it will be necessary to demonstrate specific detection of the analyte in the presence of the interference much in the way a drug assay validation would approach a glucuronide conjugate. In the case of lipid or small molecule metabolite assays, it is important where possible to obtain standards for isomers of the target analyte in order to confirm selective detection, typically by chromatographic separation.
Other species in addition to proteins will have various biological forms that may be found to interfere with the biomarker result. Steroids and sterols provide an example since in some cases, a large reservoir of analyte may be in the sample as a conjugate. Similarly, protein-binding partners are also known to act as reservoirs or impact recovery of the analytes or generation of surrogate peptides. Degradation of the conjugate during sample handling and even analysis could confound the biomarker result.
As part of demonstrating specific detection during validation, the assay should be shown to be unchanged in the presence of the known interfering materials. Two approaches are recommended. The first is to fortify samples or surrogate matrix with the interference at concentrations relevant to the samples to be analyzed and measure the analyte at the QC levels used in the assay (46). The QC read-back values should match expected values within accuracy and precision limits defined in the validation plan. A second approach may be used when the interference is not easily added to the sample. In most cases, the compound will not be available, or it will not be a single compound easily added to QC samples. In this case, samples that are known to contain the interference are added to the QC or the QC is prepared in the samples with the interference (46). Proper read-back concentrations of blanks and QCs demonstrate the assay is not affected by the interference.
Matrix Effects
Almost all LC-MS-based bioanalytical assays require sample preparation prior to analysis. During method development, it is critical to optimize the recovery of the analyte to ensure that the extraction procedure is efficient and reproducible (70, 71). Evaluating recovery for small molecule biomarkers is much more straightforward when compared to protein biomarkers, which often have multiple preparation steps including immuno-capture and digestion (72). This is especially the case because full-length SIL internal standards are often not available for protein biomarkers.
In this section, “analyte” refers to either a small molecule biomarker or a signature peptide for a protein biomarker. Typically, recovery is measured by comparing the analyte responses from extracted QC samples to those of extracts of “blank” native matrix or surrogate matrix spiked with an equivalent amount of analyte after extraction. Recovery experiments should be performed for at least two concentration levels, with at least three replicates for each concentration level. Extraction recovery should be assessed in disease and normal patient samples, as well as hemolyzed or lipemic plasma. There are no acceptance criteria for absolute values of recovery; however, recovery of the extraction method should be consistent and reproducible.
In LBA, matrix effect is associated with the binding interaction between the assay reagents, the analyte, and other matrix components, while in LC-MS assays, matrix effect refers to visible and non-visible interferences. The primary sources of matrix effect in LC-MS assays are differential ion formation and differential extraction recovery of the analyte from sample to sample (see the previous paragraph). Matrix interference of ionization efficiency should be investigated for all LC-MS assays regardless of the ionization technique used (61). Where an immunocapture step is used during sample preparation of protein biomarkers, consideration should also be given to the factors noted above for LBA “matrix effect” with regard to the matrix interfering with the interaction between the protein and capture reagent.
Determination of the matrix factor (73, 74) can be a useful technique when it is performed in a minimum of six lots of “blank” matrix. Usually, analyte is spiked at low and high concentrations along with internal standard into a matrix extract that has been processed using the full sample preparation procedure and then determining the absolute and internal standard normalized peak responses relative to the responses in solvent (61). It is recommended that the internal standard normalized matrix effects should not exceed 20% CV, but ultimately this should be determined as required to meet the requisite performance for the context of use (COU) for the biomarker assay. The MF approach may be limited by the biomarker chemotype (small vs large molecule) and the ability to identify true blank matrix samples for these experiments. The concept of a matrix effect factor (MEF), which uses SIL analytes to estimate matrix effects, was introduced by Zhou et al. as a potential solution to this problem (75). In addition, alternate approaches for handling this issue have been reviewed elsewhere (61).
Ionization effects may include additional evaluations with appropriate matrix samples from disease subjects, hemolyzed samples, or administered agents when there is a scientific rationale for suspecting that they may cause matrix interference. Other approaches to evaluate matrix effects, including measuring recovery and precision of over-spiked samples or preparation of QC’s with a variety of matrix lots (representing disease subjects, etc.), have been applied to qualify the impact of endogenous materials on assay quality (59, 76).
Selection of appropriate sample extraction techniques, elimination of non-specific or specific binding, and the use of stable isotope-labeled internal standards is critical to minimize assay matrix variability. In the absence of appropriate internal standards (such as when using analog small molecules or non-surrogate peptide internal standards) (16), the impact of matrix effects can be significantly different from sample to sample resulting in erroneous results. This situation can be further exacerbated if the internal standard does not chromatographically co-elute with the analyte.
Parallelism (Surrogate Matrix/Surrogate Analyte Selection)
The term parallelism in LC-MS assays describes the degree to which the observed response change for a given change in analyte concentration is similar between the surrogate and control matrix. Clearly, concordance between control and surrogate matrix is essential for assay accuracy. For small-molecule LC-MS biomarker assays, differential recovery and matrix effects are the primary reasons for deviations from concordance. Fortunately, SIL internal standards may be used to track differences in recovery or ion suppression/enhancement between surrogate and control matrices. (13, 77). Chemically equivalent reference standards are often available for small molecules and may routinely be spiked to prepare standards and QCs since matrix binding partners are readily disrupted by the methods used for extraction. These factors allow synthetically prepared surrogate matrices to be viable options in many cases for small molecule biomarkers.
The linear calibration curves associated with LC-MS assays confer significant advantages for parallelism assessment (13, 77). Figure 1 displays a graphical representation of standard curves prepared in either surrogate or authentic sample matrix using a common set of analyte-spiking solutions (48). Three methods for parallelism assessment are illustrated that may be used to qualify a surrogate matrix for use in validation: (a) spike-recovery, (b) dilutional linearity, and (c) standard addition. Application of these tools for LC-MS was described by Houghton et al. (5) and more recently reviewed by Jenkins (78).

Three complementary experiments may be used during method development to assess the ability of a surrogate matrix to perform in a manner equivalent to the biological control matrix over the intended range of analysis. Concordance in this behavior is referred to as parallelism. Prior to assessment, a pool of the control matrix is prepared along with a series of analyte-spiking solutions. These solutions are spiked both into the biological matrix (solid circles, upper curve) and the surrogate matrix (lower curve) along with internal standard and analyzed. The open circle on the y-axis corresponds to the response from unspiked control matrix whose concentration is estimated by interpolation from the standard curve prepared in the surrogate matrix (depicted by arrows). The first method, a spike recovery, is used to assess parallelism at concentrations above the pool concentration (spiked samples indicated by solid squares). One-hundred percent recovery implies that the concentration calculated for the spiked sample by interpolation from the surrogate matrix calibration curve is equal to the sum of the derived pool concentration and the amount spiked. Recovery may be calculated from each of the points on the upper curve. The second method, b dilutional linearity, is used to assess parallelism at concentrations below the biological pool concentration and is assessed by diluting the pool with surrogate matrix and comparing the measured interpolated concentration to the theoretical value calculated from the dilution factor (diluted samples indicated by solid triangles). Ideally, dilutional linearity is performed using a pool of high analyte concentration and, in such cases, analyte spiking may not be necessary. The third method, c standard addition, offers a third method for parallelism assessment and makes use of the fact that extrapolation of the upper curve through the negative x-axis, provides an estimate of the biological pool concentration. Parallelism exists when this extrapolated concentration is close in value to the concentration interpolated from the surrogate matrix curve. Adapted from (60) with permission of Future Science Group
Parallelism is generally assumed for surrogate analyte methods since SIL calibrants are spiked into native matrices in a manner similar to PK assays. Despite using native biological matrix for calibration, parallelism between the SIL calibrant and analyte should be demonstrated. Specifically, we recommend that a QC sample spiked at the ULOQ be serially diluted over the range of analysis using native matrix and calculating dilution-corrected recovery. Demonstrating the ability to quantitate QC samples prepared by spiking endogenous analyte is offered as a further demonstration of parallelism.
Parallelism assessment for large molecules is inherently more complex than for small-molecule biomarkers (79, 80). Although the same experimental techniques apply, protein biomarkers encompass greater method diversity (bottom-up vs. top-down; hybrid assay vs. conventional extraction) and opportunities for differential recovery. This latter point, influenced by the interactions with matrix binding partners, accounts for the greater diversity in internal standard types (protein SIL vs. peptide SIL; extended vs. truncated peptide). For LBA, parallelism is often achieved by identifying a minimum required dilution (MRD) (79, 81). This technique may be applied to LC-MS assays not limited by sensitivity.
In addition to these factors, a fundamental concern for proteins is the inability of recombinant standards to adequately depict endogenous protein analytes due to differences in post-translational modifications (PTMs) and protein folding (i.e., 3-D structure). These differences may cause differential immuno-recognition (in the case of hybrid methods) (8, 82, 83) or other factors, such as differential digestion, leading to differential recovery (34). Unfortunately, because these issues are difficult to track and quantify, only relative accuracy is possible for protein biomarkers.
Stability
Sample stability is a core expectation of every bioanalytical experiment, and a clear understanding of sample stability is mandated by all pharmaceutical regulatory agencies. These agencies require that sample stability be demonstrated from the time a sample is collected through to instrumental analysis. Biomarkers are becoming more important for regulatory decisions, so there is a greater expectation that biomarker stability is demonstrated in a fashion similar to therapeutic compounds.
Sample stability has been an essential part of every BMV guidance since the first was published in 2001. The most current examples are the 2012 EMA BMV Guidance (30), the 2018 BMV FDA Guidance (18), and the 2013 BMV MHLW Guideline (84). Combined, these offer a comprehensive description of experiments and acceptance criteria for ensuring the integrity of bioanalytical samples and offer a good starting point for creating a biomarker stability plan. Tables VI and VII list recommended stability experiments along with biomarker-specific considerations, which support fit-for-purpose approaches (see also the “Sample Type” section for discussion on pre-analytical considerations) and Table VIII lists recommendations for sample collection. Additionally, prior to implementation of a biomarker assay in a clinical study, several factors such as biomarker preservation and intra-subject variability owing to food effects and diurnal variations need to be considered to minimize pre-analytical variability of blood specimens, especially serum, and plasma (Table VIII) (85, 93).
FUTURE OPPORTUNITIES
The preceding was undertaken to provide a framework for both the practitioner and the regulator. It is a collection of the current state presented as best practices that will encourage the continued creation and use of high-quality LC-MS assays for drug discovery and development activities that go beyond exogenous analytes to include biomarkers. We include a summary of recommendations for BAV acceptance criteria (Table IX). As a group, we view this work as a starting point, not an end. Biomarkers present unique challenges, but they also present new opportunities. Biomarkers present a new and evolving interface between drug development and clinical chemistry. Translational science is the term most often applied where knowledge from research is taken into clinical evaluation of safety and efficacy. Where biomarkers are concerned, that interface might be extended into clinical testing used in the treatment of patients. While the aim of pharmaceutical research is not to develop diagnostic or prognostic tests, the assays developed for novel biomarkers should and likely will become useful tools in a clinical setting. Nowhere is this more evident than in cancer research.
The need to provide the foundation for harmonization of novel biomarkers exists. The key aspects of harmonization lie in reference materials and reference methods, both of which are developed early on as part of pharmaceutical research and development. The fundamental objective is to provide a means of generating comparable results among and between different suppliers of clinical test results, be they laboratory-developed tests or any health authority-approved commercial tests.
Standard reference materials are those that are certified by a metrology organization such as the National Institute of Standards and Technology. The demand for such materials has thus far outpaced the capacity of such organizations, leading to a long list of requested reference standards. One means to address the temporary gap in certified reference materials is to establish reference methods. Methods that can be used to value assign reference materials for calibrators and quality controls. Many of the fully validated assays developed to support pharmaceutical clinical trials likely meet the requirements for a reference method. The Joint Committee for Traceability in Laboratory Medicine (JCTLM) is an organization that works toward the goal of harmonization in clinical laboratory testing. Their program outlines the requirements and the process for worldwide adoption of a method as a reference method (https://www.jctlm.org/). While it is unlikely that most companies focused on pharmaceutical development would provide services such as reference material value assignment, it is foreseeable that methods developed and validated to support clinical trials could be reproduced and made available through organizations like JCTLM as reference methods for the value assignment of reference materials.
Involvement of pharmaceutical and contract research companies in the harmonization of biomarker assay results would fill a gap that is difficult to overcome; it would improve biomarker assay results and potentially increase access to the biomarkers as clinical tests. Use of the assays developed for pharmaceutical clinical trials as reference methods also has the potential to add value to the company that develops them. (e.g., Roche, Merck, and BMS companion diagnostics). Translation of biomarker assays used as drug development tools to reference methods also provides a means for the bioanalytical community to play a bigger role in improving health care by connecting pharmaceutical development and clinical laboratory medicine.
References
FDA, Food and Drug Administration (2001) Guidance for Industry: bioanalytical method validation. U.S. Department of Health and Human Services, 34 p.
Lee JW, Weiner RS, Sailstad JM, Bowsher RR, Knuth DW, O’Brien PJ, et al. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: a conference report. Pharm Res. 2005;22(4):499–511.
Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, et al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res. 2006;23(2):312–28.
Lee JW, Hall M. Method validation of protein biomarkers in support of drug development or clinical diagnosis/prognosis. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(13):1259–71.
Houghton R, Horro Pita C, Ward IMR. Generic approach to validation of small-molecule LC-MS/MS biomarker assays. Bioanalysis. 2009;1(8):1365–74.
Hougton R, Gouty D, Allinson J, Green R, Losauro M, Lowes S, et al. Recommendations on biomarker bioanalytical method validation by GCC. Bioanalysis. 2012;4(20):2439–46.
Amaravadi L, Song A, Myler H, Thway T, Kirshner S, Devanarayan V, Ni GY, et al. WP in bioanalysis part3: focus on new technologies and biomarkers. Bioanalysis. 2015;7(24):3107–24.
Richards S, Amaravadi L, Pillutla R, Birnboeck H, Torri A, Cowan KJ, Papadimitriou A, et al. WP in bioanalysis part 3: focus on biomarker assay validation (BAV). Bioanalysis. 2016;8(23):2475–96.
Gupta S, Richards S, Amaravadi L, Piccoli S, Desilva B, Renuka P, Stevenson L, Mehta D, Carrasco-Triguero M, et al. WP in bioanalysis part 3: a global perspective on immunogenicity guidelines & biomarker assay performance. Bioanalysis. 2017;9(24):1967–96.
Stevenson L, Richards S, Pillutla R, Torri A, Kamerud J, Mehta D, Keller S, Purushothama S, et al. WP in bioanalysis part 3: focus on flow cytometry, gene therapy, cut points and key clarifications on BAV. Bioanalysis. 2018;10(24):1973–2001.
Neubert H, Olah T, Lee A, Fraser S, Dodge R, Laterza O, Szapacs M, Alley S, et al. WP in bioanalysis part 2: focus on immunogenicity assays by hybrid LBA / LCMS and regulatory feedback. Bioanalysis. 2018;10(23):1897–917.
Piccoli S, Metha D, Vitaliti A, Allinson J, Amur S, Eck S, Green C, Hedrick M, et al. WP in bioanalysis part 3: FDA immunogenicity guidance, gene therapy, critical reagents, biomarkers and flow cytometry validation. Bioanalysis. 2019;11(24):2207–44.
Neubert H, Alley S, Lee A, Jian W, Buonarati M, Edmison A, Garofolo F, Gorovits B, et al. WP in bioanalysis part 1: BMV of hybrid assays, acoustic MS, HRMS, data integrity, endogenous compounds, microsampling and microbiome. Bioanalysis. 2021;13(4):203–38.
Spitz S, Zhang Y, Fischer S, McGuire K, Sommer U, Amaravadi L, Bandukwala A, Eck S, et al. WP in bioanalysis part 2: BAV guidance, CLSI H62, biotherapeutics stability, parallelism testing. CyTOF and regulatory feedback Bioanalysis. 2021;13(5):295–361.
Draft guidance for industry on bioanalytical method validation - a notice by the Food and Drug Administration on 09/13/2013 - https://www.federalregister.gov/documents/2013/09/13/2013-22309/draft-guidance-for-industry-on-bioanalytical-method-validation-availability
Booth B, Arnold ME, DeSilva B, Amaravadi L, Dudal S, Fluhler E, Gorovits B, Haidar SH, Kadavil J, Lowes S, Nicholson R, Rock M, Skelly M, Stevenson L, Subramaniam S, Russell Weiner EW. Workshop report: Crystal City V—quantitative bioanalytical method validation and implementation: the 2013 revised FDA guidance. AAPS J. 2015;17(2):277–88.
Arnold ME, Booth B, King L, Ray C. Workshop report: Crystal City VI—bioanalytical method validation for biomarkers. AAPS J 2016;18(6):1366–72. Available from: http://link.springer.com/10.1208/s12248-016-9946-6.
FDA, Food and Drug Administration (2018) Guidance for industry bioanalytical method validation, (May):1–22. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
ICH M10, Draft Bioanalytical Method Validation (2019). https://www.ich.org/fileadmin/PublicWeb Site/ICH products/Guidelines/Multidisciplinary/M10/M10EWGStep2DraftGuideline 2019 0226.pdf.
FDA (2014) Guidance for industry and FDA staff qualification process for drug development tools. Food Drug Adm Cent Drug Eval Res (January).
Piccoli S., Sauer JM., Ackermann B., Allinson J., Arnold M., Amur S., Aubrecht J., Baker A., Becker R., Buckman-Garner S. et al (2019) Points to consider document: scientific and regulatory considerations for the analytical validation of assays used in the qualification of biomarkers in biological matrices. Critical Path Institute (C-Path). https://c-path.org/wp-content/uploads/2019/06/EvidConsid-WhitePaper-AnalyticalSectionV20190621.pdf
Chau CH, Rixe O, McLeod H, Figg WD. Validation of analytic methods for biomarkers used in drug development. Clin Cancer Res. 2008;14(19):5967–76.
Wu Y, Lee JW, Uy L, Abosaleem B, Gunn H, Ma M, et al. (2009) Tartrate-resistant acid phosphatase (TRACP 5b): a biomarker of bone resorption rate in support of drug development: modification, validation and application of the BoneTRAP® kit assay. J Pharm Biomed Anal 49(5):1203–12. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0731708509001629.
Cummings, J., Raynaud, F., Jones, L., Sugar, R., Dive, C. (2010) Fit-for-purpose biomarker method validation for application in clinical trials of anticancer drugs. Br J Cancer, 103:1313. Available from: https://doi.org/10.1038/sj.bjc.6605910.
Valentin, M.-A., Ma, S., Zhao, A., Legay, F., Avrameas, A. (2011) Validation of immunoassay for protein biomarkers: bioanalytical study plan implementation to support pre-clinical and clinical studies. J Pharm Biomed Anal 55(5):869–77. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0731708511001853.
Lowes S, Ackermann BL. AAPS and US FDA Crystal City VI workshop on bioanalytical method validation for biomarkers. Bioanalysis. 2016;8(3):163–7.
Tu and Bennett. Parallelism experiments to evaluate matrix effects, selectivity and sensitivity in ligand binding assay method development: pros and cons. Bioanalysis. 2017;9(14):1107–22.
Smith, L.M., Kelleher, N.L., et al. (2013) Proteoform: a single term describing protein complexity. Nat methods 10(3):186-7. Available from: https://doi.org/10.1038/nmeth.2369.
Sturgeon, C., Hill, R., Hortin, G.L., Thompson, D. (2010) Taking a new biomarker into routine use – a perspective from the routine clinical biochemistry laboratory. PROTEOMICS – Clin Appl 4(12):892–903. Available from: https://doi.org/10.1002/prca.201000073.
Zhao, X., Qureshi, F., Eastman, P.S., Manning, W.C., Alexander, C., Robinson, W.H., et al. (2012) Pre-analytical effects of blood sampling and handling in quantitative immunoassays for rheumatoid arthritis. J Immunol Methods 378(1–2):72–80. Available from: https://www.sciencedirect.com/science/article/pii/S0022175912000427.
EMA (2012) Guideline on bioananalytical method validation. Committee for Medicinal Products for Human Use (CHMP).
Duggan, J.X., Vazvaei, F., Jenkins, R. (2015) Bioanalytical method validation considerations for LC–MS/MS assays of therapeutic proteins. Bioanalysis 7(11):1389–95. Available from: https://doi.org/10.4155/bio.15.69.
Cowan KJ, Amaravadi L, Cameron MJ, Fink D, Jani D, Kamat M, et al. Recommendations for selection and characterization of protein biomarker assay calibrator material. AAPS J. 2017;19(6):1550–63.
Kunz U, Goodman J, Loevgren U, Piironen T, Elsby K, Robinson P, et al. Addressing the challenges of biomarker calibration standards in ligand-binding assays: a European bioanalysis forum perspective. Bioanalysis. 2017;9(19):1493–508.
Jiang H, Zeng J, Titsch C, Voronin K, Akinsanya B, Luo L, et al. Fully validated LC-MS/MS assay for the simultaneous quantitation of coadministered therapeutic antibodies in cynomolgus monkey serum. Anal Chem. 2013;85(20):9859–67.
Stevenson L, Garofolo F, DeSilva B, Dumont I, Martinez S, Rocci M, Amaravadi L, Brudny KM, Musuku A, Booth B, et al. WP in bioanalysis: ‘hybrid’ – the best of LBA and LCMS. Bioanalysis. 2013;5(23):2903–18.
King LE. Parallelism experiments in biomarker ligand-binding assays to assess immunological similarity. Bioanalysis. 2016;8(23):2387–91.
Guideline on Bioanalytical Method Validation in Pharmaceutical Development (25 July 2013, MHLW, Japan). Available from: http://www.nihs.go.jp/drug/BMV/250913_BMV-GL_E.pdf
Gerszten, R.E., Accurso, F., Bernard, G.R., Caprioli, R.M., Klee, E.W., Klee, G.G., et al. (2008) Challenges in translating plasma proteomics from bench to bedside: update from the NHLBI clinical proteomics programs. Am J Physiol Lung Cell Mol Physiol 295(1):L16–22. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18456800.
Tuck, M.K., Chan, D.W., Chia, D., Godwin, A.K., Grizzle. W.E., Krueger, K.E., et al. (2009) Standard operating procedures for serum and plasma collection: early detection research network consensus statement standard operating procedure integration working group. J Proteome Res 8(1):113–7. Available from: https://doi.org/10.1021/pr800545q.
Hoofnagle AN, et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry-based assays. Clin Chem. 2016;62(1):48–69.
Agrawal, L., Engel, K.B., Greytak, S.R., Moore, H.M. (2018) Understanding preanalytical variables and their effects on clinical biomarkers of oncology and immunotherapy. Semin Cancer Biol 52:26–38. Available from: https://www.sciencedirect.com/science/article/pii/S1044579X17302481.
Yi, J., Warunek, D., Craft, D. (2015) Degradation and stabilization of peptide hormones in human blood specimens. PLoS One 10(7):e0134427. Available from: https://doi.org/10.1371/journal.pone.0134427.
Gupta, V., Davancaze, T., Good, J., Kalia, N., Anderson, M., Wallin, J.J., et al. (2016) Bioanalytical qualification of clinical biomarker assays in plasma using a novel multi-analyte simple PlexTM platform. Bioanalysis 8(23):2415–28. Available from: https://doi.org/10.4155/bio-2016-0196.
Ayache, S., Panelli, M., Marincola, F.M., Stroncek, D.F. (2006) Effects of storage time and exogenous protease inhibitors on plasma protein levels. Am J Clin Pathol 126(2):174–84. Available from: https://doi.org/10.1309/3WM7XJ7RD8BCLNKX.
Oe, T., Ackermann, B.L., Inoue, K., Berna, M.J., Garner, C.O., Gelfanova, V., et al. (2006) Quantitative analysis of amyloid β peptides in cerebrospinal fluid of Alzheimer’s disease patients by immunoaffinity purification and stable isotope dilution liquid chromatography/negative electrospray ionization tandem mass spectrometry. Rapid Commun mass Spectrom 20(24):3723–35. Available from: https://doi.org/10.1002/rcm.2787.
Shuford, C.M., Walters, J.J., Holland, P.M., Sreenivasan, U., Askari, N., Ray K, et al. (2017) Absolute protein quantification by mass spectrometry: not as simple as advertised. Anal Chem 89(14):7406–15. Available from: https://doi.org/10.1021/acs.analchem.7b00858.
van den Broek I, Smit NPM, Romijn FPHTM, van der Laarse A, Deelder AM, van der Burgt YEM, Cobbaert CM. Evaluation of interspecimen trypsin digestion efficiency prior to multiple reaction monitoring-based absolute protein quantification with native protein calibrators. J Proteome Res. 2013;12(12):5760–74. https://doi.org/10.1021/pr400763d.
Lowenthal MS, Liang Y, Phinney KW, Stein SE. Quantitative bottom-up proteomics depends on digestion conditions. Anal Chem. 2014;86(1):551–8. https://doi.org/10.1021/ac4027274.
Nouri-Nigjeh E, Zhang M, Ji T, Yu H, An B, Duan X, Balthasar J, Johnson RW, Qu J. Effects of calibration approaches on the accuracy for LC−MS targeted quantification of therapeutic protein. Anal Chem. 2014;86(7):3575–84. https://doi.org/10.1021/ac5001477.
Scott KB, Turko IV, Phinney KW. Quantitative performance of internal standard platforms for absolute protein quantification using multiple reaction monitoring mass spectrometry. Anal Chem. 2015;87(8):4429–35. https://doi.org/10.1021/acs.analchem.5b00331.
Benesova E, Vidova V, Spacil Z. A comparative study of synthetic winged peptides for absolute protein quantification. Sci Rep. 2021;11(1):10880. https://doi.org/10.1038/s41598-021-90087-9.
Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat Methods. 2005;2(8):587–9. https://doi.org/10.1038/nmeth774.
Brun V, Dupuis A, Adrait A, Marcellin M, Thomas D, Court M, Vandenesch F, Garin J. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol Cell Proteomics. 2007;6(12):2139–49. https://doi.org/10.1074/mcp.M700163-MCP200.
Brun V, Masselon C, Garin J, Dupuis A. Isotope dilution strategies for absolute quantitative proteomics. J Proteome. 2009;72(5):740–9. https://doi.org/10.1016/j.jprot.2009.03.007.
Reddy PT, Jaruga P, Nelson BC, Lowenthal MS, Jemth A-S, Loseva O, Coskun E, Helleday T, Dizdaroglu M. Production, purification, and characterization of 15N-labeled DNA repair proteins as internal standards for mass spectrometric measurements. Methods Enzymol. 2016;566:305–32. https://doi.org/10.1016/bs.mie.2015.06.044.
Bronsema KJ, Bischoff R, van de Merbel NC. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;893-894:1–14. https://doi.org/10.1016/j.jchromb.2012.02.021.
Grant RP, Hoofnagle AN. From lost in translation to paradise found: enabling protein biomarker method transfer by mass spectrometry. Clin Chem. 2014;60(7):941–4.
Zhao, Y., Liu, G., Kwok, S., Jones, B.R., Liu, J., Marchisin, D., et al. (2017) Highly selective and sensitive measurement of active forms of FGF21 using novel immunocapture enrichment with LC–MS/MS. Bioanalysis 10(1):23–33. Available from: https://doi.org/10.4155/bio-2017-0208.
Jones, B.R., Schultz, G.A., Eckstein, J.A., Ackermann, B.L. (2012) Surrogate matrix and surrogate analyte approaches for definitive quantitation of endogenous biomolecules. Bioanalysis 4(19):2343–56. Available from: https://doi.org/10.4155/bio.12.200.
Song A, Lee A, Garofolo F, Kaur S, Duggan J, Evans C, Palandra J, Di Donato L, Xu K, Bauer R, et al. WP in bioanalysis part 2: focus on biomarker assay validation (BAV) hybrid LBA/LCMS and input from regulatory agencies. Bioanalysis. 2016;8(23):2457–74.
Cox JM, Butler JP, Lutzke BS, Jones BA, Buckholz JE, Biondolillo R, et al. (2015) A validated LC–MS/MS method for neurotransmitter metabolite analysis in human cerebrospinal fluid using benzoyl chloride derivatization. Bioanalysis 7(19):2461–75. Available from: https://doi.org/10.4155/bio.15.170.
Lame, M.E., Chambers, E.E., Blatnik, M. (2011) Quantitation of amyloid beta peptides Aβ1–38, Aβ1–40, and Aβ1–42 in human cerebrospinal fluid by ultra-performance liquid chromatography–tandem mass spectrometry. Anal Biochem 419(2):133–9. Available from: https://www.sciencedirect.com/science/article/pii/S0003269711005306.
Jian, W., Edom, R., Weng, N., Zannikos, P., Zhang, Z., Wang, H. (2010) Validation and application of an LC–MS/MS method for quantitation of three fatty acid ethanolamides as biomarkers for fatty acid hydrolase inhibition in human plasma. J Chromatogr B. [cited 2018 Dec 11];878(20):1687–99. Available from: https://www.sciencedirect.com/science/article/pii/S1570023210002734.
Ongay, S., Hendriks, G., Hermans, J., van den Berge, M., ten Hacken, N.H.T., van de Merbel, N.C., et al. (2014) Quantification of free and total desmosine and isodesmosine in human urine by liquid chromatography tandem mass spectrometry: a comparison of the surrogate-analyte and the surrogate-matrix approach for quantitation. J Chromatogr A Jan 24 [cited 2018];1326:13–9. Available from: https://www.sciencedirect.com/science/article/pii/S002196731301916X
Kinoshita, K., Jingu, S., Yamaguchi, J. (2013) A surrogate analyte method to determine d-serine in mouse brain using liquid chromatography–tandem mass spectrometry. Anal Biochem. [cited 2018 Dec 11];432(2):124–30. Available from: https://www.sciencedirect.com/science/article/pii/S0003269712004915
Liu, L., Cui, Z., Deng, Y., Dean, B., Hop, C.E.C.A., Liang, X. (2016) Surrogate analyte approach for quantitation of endogenous NAD+ in human acidified blood samples using liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J Chromatogr B [cited 2018];1011:69–76. Available from: https://www.sciencedirect.com/science/article/pii/S1570023215303652
Li, W., Cohen, L.H. (2003) Quantitation of endogenous analytes in biofluid without a true blank matrix. Anal Chem 75(21):5854–9. Available from: https://doi.org/10.1021/ac034505u.
Jemal, M., Schuster, A., Whigan, D.B. (2003) Liquid chromatography/tandem mass spectrometry methods for quantitation of mevalonic acid in human plasma and urine: method validation, demonstration of using a surrogate analyte, and demonstration of unacceptable matrix effect in spite of use of a stable. Rapid Commun mass Spectrom 17(15):1723–34. Available from: https://doi.org/10.1002/rcm.1112.
Yang E, Welink J, Cape S, Woolf E, Sydor J, James C, Goykhman D, Arnold M, Addock N, Bauer R, et al. WP in bioanalysis part 1: focus on biomarker assay validation (BAV). Bioanalysis. 2016;8(22):2363–78.
Metzler, G., King, R.C., Fernández-Metzler, C., Crathern, S. (2019) Reducing the need for surrogate matrix or surrogate analyte in biomarker assays, 67TH ASMS conference on mass spectrometry and allied topics, Atlanta, Georgia, June 2–6.
Jenkins R, Duggan JX, Aubry A-F, Zeng J, Lee JW, Cojocaru L, Dufield D., Garofolo F., Kaur S., A. G Schultz, et al. (2015) Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. AAPS J 17(1):1–16. Available from: http://link.springer.com/10.1208/s12248-014-9685-5
Huber-Lang, M., Sarma, J.V., Zetoune, F.S., Rittirsch, D., Neff, T.A., McGuire, S.R. et al. (2006) Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 12:682. Available from: https://doi.org/10.1038/nm1419.
Yu, Z., Kastenmüller, G., He, Y., Belcredi, P., Möller, G., Prehn, C., et al. (2011) Differences between human plasma and serum metabolite profiles. PLoS One 6(7):e21230. Available from: https://doi.org/10.1371/journal.pone.0021230.
Zhou W, Yang S, Wang PG. Matrix effects and application of matrix effect factor. Bioanalysis. 2017;9(23):1839–44.
Van Bruijnsvoort M, Meijer J, Van Den Beld C. The application of control charts in regulated bioanalysis for monitoring long-term reproducibility. Bioanalysis. 2017;9(24):1955–65.
Lowes S, LeLacheur R, Shoup R, Garofolo F, Dumont I, Martinez S, Zimmer J, Caturla MC, Couerbe P, Awaiye K, Fatmi S, et al. Recommendations on incurred sample stability (ISS) by GCC. Bioanalysis. 2014;6(18):2385–90.
DeSilva B, Garofolo F, Rocci M, Martinez S, Dumont I, Landry F, Dicaire C, Szekely-Klepser G, Weiner R, Arnold M, Bansal S, et al. WP in bioanalysis: alignment of multiple guidelines. Bioanalysis. 2012;4(18):2213–26.
Dijksman J, Timmerman P, Abbott R, Barroso B, Kloeppel MB, Companjen A, et al. Conference report: less is more, defining modern bioanalysis. Bioanalysis. 2012;4(6):633–42.
Yadav, M.S., Shrivastav, P.S., de Boer, T., Wieling, J., Singhal, P. (2013) Current understanding of bioanalytical assay reproducibility: incurred sample reanalysis, incurred sample stability, and incurred sample accuracy. In John Wiley & Sons, Inc.; p. 47–64.
Rocci M, Lowes S, Shoup R, Garofolo F, Farmen R, Zhang T, Allinson J, Gouty D, Hayes R, Nicholson R, Houghton R, et al. 7th GCC insights: incurred samples use; fit-for-purpose validation, solution stability, electronic laboratory notebook and hyperlipidemic matrix testing. Bioanalysis. 2014;6(20):2713–20.
Andisik M, DeStefano L, Stefan C, Gathani M, Laurino K, Lai VC, et al. Effects of blood processing and sample storage on the stability of biotherapeutics and anti-drug antibodies. Bioanalysis. 2015;7(11):1325–35.
Neubert H, Song A, Lee A, Wei C, Duggan J, Xu K, Woolf E, Evans C, Palandra J, Laterza O, et al. WP in bioanalysis part 2: rise of hybrid LBA/LCMS immunogenicity assays. Bioanalysis. 2017;9(23):1895–912.
Barelli, S., Crettaz, D., Thadikkaran, L., Rubin, O., Tissot, J-D (2007) Plasma/serum proteomics: pre-analytical issues. Expert Rev Proteomics 4(3):363–70. Available from: https://doi.org/10.1586/14789450.4.3.363.
Blair IA, Ciccimaro E. Stable-isotope dilution LC – MS for quantitative biomarker ana lysis. Bioanalysis. 2010;2:311–41.
Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75(13):3019–30. https://doi.org/10.1021/ac020361s.
Guideline on Bioanalytical method validation, European Medicines Agency, 21 July 2011, EMEA/CHMP/EWMP/EWP/192217/2009 Rev. 1 Corr. 2, Committee for Medicinal Products for Human Use (CHMP).
Panuwet, P., Hunter, R.E., D’Souza, P.E., Chen, X., Radford, S.A., Cohen, J.R., et al. (2016) Biological matrix effects in quantitative tandem mass spectrometry-based analytical methods: advancing biomonitoring. Crit Rev Anal Chem 46(2):93–105. Available from: https://doi.org/10.1080/10408347.2014.980775.
Welink J, Xu Y, Yang E, Wilson A, Henderson N, Luo L, Fraser S, Kavita U, Musuku A, et al. WP in bioanalysis part 1: a global bioanalytical community perspective on last decade of incurred samples reanalysis (ISR). Bioanalysis. 2018;10(22):1781–801.
Jenkins, R.G. (2016) Accuracy: a potential quandary in regulated bioanalysis of ‘endogenous’ analytes. Bioanalysis 8(23):2393–7. Available from: http://www.future-science.com/doi/10.4155/bio-2016-0247.
Stevenson, L.F., Purushothama, S. (2014) Parallelism: considerations for the development, validation and implementation of PK and biomarker ligand-binding assays. Bioanalysis 6(2):185–98. Available from: https://doi.org/10.4155/bio.13.292.
Xiao D, Hiing K, Ling J, Custodio J, Majeed SR, Tarnowski T. Quantitation of intracellular triphosphate metabolites of antiretroviral agents in peripheral blood mononuclear cells (PBMCs) and corresponding cell count determinations: review of current methods and challenges. Expert Opin Drug Metab Toxicol. 2018;14(8):781–802.
Weng N, Jian W. Targeted biomarker quantitation by LC-MS: John Wiley & Sons; 2017.
Wang, J., Lee, J., Burns, D., Doherty, D., Brunner, L., Peterson, M., et al. (2009) “Fit-for-purpose” method validation and application of a biomarker (C-terminal telopeptides of type 1 collagen) in denosumab clinical studies. AAPS J 11(2):385–94. Available from: http://www.springerlink.com/index/10.1208/s12248-009-9115-2.
Acknowledgements
The authors thank Drs. Vinita Gupta and Richard C. King for the helpful discussions and review of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing, review, and final approval of this manuscript.
Corresponding author
Ethics declarations
The views expressed in this article are those of the authors and do not reflect the official policy of the authors’ organizations.
Conflict of Interest
B.L.A. is employed by Ely Lilly and Company; M.E.A. is employed by Labcorp Drug Development; B.deS. and H.G. are employed by Bristol-Myers Squibb; C.F-M. is employed by PharmaCadence Analytical Services, LLC; F.G. is employed by BRI a Frontage Company; O.L. and F.V. are employed by Merck & Co.; M.J.R. is employed by Gossmer Bio, Inc.; and Y.M. is employed by Boehringer Ingelheim Pharmaceuticals, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernández-Metzler, C., Ackermann, B., Garofolo, F. et al. Biomarker Assay Validation by Mass Spectrometry. AAPS J 24, 66 (2022). https://doi.org/10.1208/s12248-022-00707-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-022-00707-z