
Abstract
Neutropenia and febrile neutropenia (FN) are serious side effects of cytotoxic chemotherapy which may be alleviated with the administration of recombinant granulocyte colony-stimulating factor (GCSF) derivatives, such as pegfilgrastim (PG) which increases absolute neutrophil count (ANC). In this work, a population pharmacokinetic-pharmacodynamic (PKPD) model was developed based on data obtained from healthy volunteers receiving multiple administrations of PG. The developed model was a bidirectional PKPD model, where PG stimulated the proliferation, maturation, and margination of neutrophils and where circulating neutrophils in turn increased the elimination of PG. Simulations from the developed model show disproportionate changes in response with changes in dose. A dose increase of 10% from the 6 mg therapeutic dose taken as a reference leads to area under the curve (AUC) increases of ~50 and ~5% for PK and PD, respectively. A full random effects covariate model showed that little of the parameter variability could be explained by sex, age, body size, and race. As a consequence, little of the secondary parameter variability (Cmax and AUC of PG and ANC) could be explained by these covariates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Neutropenia and febrile neutropenia (FN) are serious complications that can be induced by cytotoxic chemotherapy in the treatment of cancer (1,2). During FN, patients are more susceptible to opportunistic infections and sepsis, which may result in hospitalization and, in some severe cases, death (3). Additionally, if the neutropenia is severe enough or FN occurs, the chemotherapy doses need to be lowered or the treatment paused in order for the absolute neutrophil count (ANC) to recover, reducing the effectiveness of the treatment and thus potentially, overall survival (4).
Treatment guidelines for chemotherapy-induced neutropenia recommend the administration of granulocyte colony-stimulating factors (GCSFs) if a patient’s risk for severe neutropenia during chemotherapy is greater than 20% (3). GCSFs are proteins which bind to receptors on hematopoietic stem cells and neutrophil precursor cells, stimulating the differentiation, survival, and proliferation of neutrophil precursor cells (5,6). After administration of GCSF, ANC production is increased, transit time of granulocytes through the bone marrow is reduced, and the neutrophils are more readily expelled from bone marrow. Inspired by these properties, filgrastim (Neupogen®, Amgen), a recombinant methionylated and non-glycosylated human GCSF, was developed and approved by the US Food and Drug Administration (FDA) in 1991 (7). Frequent administrations of filgrastim due to the relatively short half-life (t1/2 ∼ 3.5 h) of the protein results in a high patient burden added on the already large burden of chemotherapy. During a 2-week cycle, as many as 14 injections may be administered to a patient (8). In order to reduce this burden, a pegylated filgrastim, pegfilgrastim (PG), was developed and marketed as Neulasta® (Amgen) (9). Pegylation (adding a polyethylene glycol [PEG] molecule) of therapeutic proteins can lead to increased resistance to enzymatic degradation and for smaller proteins, it can increase the molar mass of the protein beyond that which can be eliminated via the renal route (10). When added to filgrastim (molar weight ∼20 kDa), pegylation increased its hydrodynamic size by ∼2-fold, preventing the drug from being filtered by the glomeruli, reducing its elimination (11).
In order to understand the relationship between PG and ANC, a pharmacokinetic-pharmacodynamic (PKPD) model is of value. Various semi-mechanistic models for filgrastim have been published and adequately describe the disposition of filgrastim (12,13). Many of the principles explored by these models, such as dose-dependent bioavailability and neutrophil-mediated elimination, may also be applicable to PG and a model built on mechanistic principles, has previously been published for PG. (14) However, for addressing a number of questions related to PG therapy, it is valuable to have a model where interindividual and intraindividual aspects are captured. Also for a quantitative assessment of the bidirectional nature of the PG-ANC system, such a model may more precisely capture the interaction. In this work, individual PG concentration and ANC measurements were modeled using a population approach, thereby characterizing the bidirectionality in the system on an individual level. Further, the extent to which the variability in parameters of the system could be explained by demographic covariates was investigated.
MATERIAL AND METHODS
Data
Data from a double-blind three-way cross-over study where healthy subjects (n = 192) were treated with single 6 mg doses of potentially biosimilar pegfilgrastim (BIOS_PG), US-sourced Neulasta® (US_PG), and EU-sourced Neulasta® (EU_PG) were used in this analysis. The study was conducted in accordance with the ethical principles stated by the Declaration of Helsinki, International Conference on Harmonization, Technical Requirements for the Registration of Pharmaceuticals for Human Use of Good Clinical Practice and applicable drug and data protection laws in the USA. All subjects were required to sign a subject information sheet before any study-related procedures were performed. All documents pertaining to the ethical conduct of the study were signed by an Institutional Review Board. The study included a washout of at least 5 weeks was observed between periods of single administration of each PG formulation. Subjects were sampled for PG concentrations and ANC. The primary aim of the study was to compare the pharmacokinetics (PK) and pharmacodynamics (PD) of the proposed biosimilar, BIOS_PG, to the reference products US_PG and EU_PG. The secondary aim of the study was to compare the safety, tolerability, and immunogenicity of the three formulations. If an individual tested positive for either anti-PEG antibodies or anti-drug antibodies (ADA) within an occasion, all records for that individual for that particular occasion were excluded in the analysis data set.
Pharmacokinetic/Pharmacodynamic Model Development
A one-compartment model with sequential absorption processes was used as a starting point for the PK model development. In this model, a zero-order input into the absorption compartment was followed by first-order absorption dictating absorption into the central compartment. The PK of pegfilgrastim has previously been described using a one-compartment model with dual absorption processes (14). The typical value of Frel in the model was fixed to one as no intravenous data was available. Elimination of PG has previously been described as consisting of a saturable neutrophil-mediated and a linear non-specific pathway which may indicate proteolysis or other elimination (15). Several models for elimination were tested, including linear non-specific elimination, saturable non-specific elimination, linear ANC-dependent elimination, saturable ANC-dependent elimination, and combinations of the aforementioned mechanisms (resulting in parallel elimination routes). Saturable processes were described using a Michaelis-Menten equation:
where V(t) is the amount of drug being eliminated at time = t, Vmax is the maximal elimination rate in amount per hour, CPG(t) is the PG concentration at time = t, and KM is the concentration of PG that elicits half the PG effect on the elimination rate. To test whether saturable ANC-dependent elimination was supported by the data, Vmax was multiplied by ANC to scale the parameter.
The starting point for the PD model of ANC was a semi-mechanistic indirect effect model with transit compartments adapted from Quartino et al. (16) and Friberg et al. (17). The PD model was linked to the PK model by testing linear and maximum effect (Emax) drug effects on different parameters in the PD model by multiplying the rates with (1 + Effect), indicating stimulation of the rates by PG.
Model development was sequential; first, a PK model was developed where linearly interpolated ANC observations were used to investigate ANC-mediated effects, then a PD model was developed where the linearly interpolated PG observations were used to investigate the drug effects, and thereafter, the models were combined without any interpolation, refined, and finalized.
The stochastic portion of the model was divided into interindividual variability (IIV), interoccasion variability (IOV), and residual error. The distribution of parameters associated with IIV and IOV was assumed to be log-normally distributed. Other distributions were investigated as indicated. Residual errors in the PK and PD models were additive on the log scale, resulting in proportional residual errors on a normal scale. Other residual models were investigated as indicated.
Covariate relationships were evaluated in the model using the full random effects model (FREM) approach (18,19). FREM is a covariate modeling methodology which reduces covariate selection bias by estimating all covariate relationships simultaneously. In this approach, all covariates are modeled as random effects (observations with a fixed mean and estimated IIV) and are coupled to the parameters through a full covariance matrix, including both the random effects for the parameters and the covariates. The basic workflow of the usage of FREM can be summarized as:
-
1.
Pre-specify all potential covariates of interest for explaining parameter variability.
-
2.
Add covariates to a base model as observations and estimate IIV and covariances between all parameters and covariates.
-
3.
Post-process the parameter-covariate covariance matrix to obtain information of interest: correlation coefficients, covariate-parameter relations for a traditional covariate model, etc.
The time constant covariates may explain parameter IIV, while the time-varying covariates here may explain parameter IOV in the model. The FREM was developed by first considering only time-constant covariates (race, sex, age, lean body weight (LBW) calculated according to Janmahasatian et al. (20), body mass index (BMI), and weight) and secondly including also time-varying covariates (formulation and period).
Model Evaluation
Model evaluation was based on the inspection of graphical diagnostics such as predictions, residuals, and visual predictive checks (VPCs) (21), as well as parameter estimates and parameter precision. Discrimination between models was mainly based on approximations of -2·log(likelihood), referred to here as the objective function value (OFV). The reported OFV was obtained using an importance sampling assisted by mode a posteriori (IMPMAP) estimation method. For nested models, the likelihood ratio test was used.
Simulations
The R-package ncappc was used to generate and plot non-compartmental analysis (NCA) statistics based on both the measured data and on model simulations using the same design structure as the measured data (22). NCA statistics calculated based on the model simulations were compared with observed NCA statistics to measure general model performance. Simulated NCA statistics with different administered doses using the base model before covariate inclusion served as an indication of the sensitivity of the PK and PD to changes in the administered dose. A 6 mg dose and increases from it of 2, 4, 6, 8, and 10% were simulated. For each dose, the geometric mean computed from 1000 simulated individuals’ area under the curve (AUC) and maximum concentration (Cmax) were compared. Simulations with subsequent NCA calculations (445,000 profiles each of PG and ANC) were also performed from the final model with all covariate relations to assess the correlation between NCA metrics and parameters and covariates.
Data Analysis and Software Details
Data were analyzed using NONMEM version 7.3.0 (23) installed on an Intel Xeon-based server. Perl-speaks-NONMEM (PsN) version 4.7.0 (24,25) was used for NONMEM run control. Data management, further processing of NONMEM output, and goodness-of-fit evaluations were performed using R version 3.2.3 (26). Parameter estimation in NONMEM was performed using an expectation maximization (EM) algorithm, IMPMAP, and the model was MU-referenced. Standard errors of the parameter estimates in the final model were computed by the $COVARIANCE step in NONMEM.
RESULTS
Data
Profiles corresponding to dosing occasions were ADA toward an identifiable part of the molecule (PEG and/or filgrastim) have been detected where excluded from the analysis. Although ADA are frequently identified as having an effect on drug elimination, this could not be evaluated due to the low frequency of ADA. Further, individuals (n = 15) with measureable baseline concentrations of PG were excluded since being drug naive was an inclusion criteria for participation in the study. The total number of individuals included in the analysis was 174, of which 43% were females and 86% were Caucasians. The administered doses deviated from the nominal doses of 6 mg. Hence, the median doses of remaining syringes analyzed per batch of drug administered to the subject were used as individual dose amounts. This constituted amounts of 6.6 and 6.7 mg for two different batches of BIOS_PG, 6.3 mg for US_PG and 6.2 mg for EU_PG. Observed concentrations of PG and ANC are presented in Fig. 1.

Observed PG concentrations and ANC versus time after dose in study PG-01-003. Black dots and lines show concentration-time profiles in individuals at each dosing occasion (one line per individual administration)
Pharmacokinetic/Pharmacodynamic Model Development
The starting point for the PK model was a one-compartment model with a depot compartment and sequential zero- and first-order absorption. Simpler absorption models did not describe data as well as the starting model and more complex models, for example incorporating ANC-dependent elimination from a presystemic depot compartment offered no better description of data. Therefore, the starting absorption model was retained in the final model. An elimination model using a linear ANC-dependent process and a saturable non-specific process described the data as well as having both processes as saturable. Also, any simplification of the two-process elimination model made description of data significantly (p < 0.05) worse.
The PD model included a transit chain of four transit compartments followed by a compartment representing ANC in circulation. The feedback mechanism that made the original models from Quartino et al. (16) and Friberg et al. (17) self-regulating was not found to be significant (p > 0.05) during model development. Given our data, where concentrations of an exogenous GCSF analogue are expected to be in a very large excess compared to endogenous GCSF (moreover expected to be at low concentrations due to suppression by high neutrophil counts), this mechanism, which is based on endogenous levels, may not be identifiable, and thus the self-regulating component was removed. A baseline ANC (BASE) representing the baseline neutrophil count was estimated. Other structural components of the PD model were fixed to literature values as the estimation of their values in the absence of exogenous GCSF was not sufficiently informed by the present data. The typical value of circulating neutrophil elimination was fixed to the frequently reported half-life (∼7 h) (27). The baseline value of mean maturation time (MMT) was fixed to 120 h, since 5 days has been reported as the time it takes for the cells to mature and migrate from the bone marrow to the blood stream (27). The effect of fixing MMT to a certain value with this relatively low number of transit compartments effectively reduced the model to a turnover model with a maturation component and an indirect drug response model (28,29).
An early decrease in ANC was observed after PG administration, indicating the necessity of a margination effect in the model. Margination describes the rapid expansion of the volume of distribution in the ANC compartment, and was described using an Emax effect. Additionally, drug effect was included as an Emax effect on changes in the production rate (proliferation effect) and the transit rate (maturation effect) from baseline (the inverse of the MMT). Correlations between the Emax and EC50 parameters were high and it was found that estimating a single EC50 parameter for these two effects did not worsen model fit as indicated by the OFV. Further, high correlations between both Emax,prol and EC50 parameters and the parameters describing the margination effect were also identified. The margination effect was therefore described by scaling both Emax,prol and EC50 via multiplication by separate fixed effect parameters. The final PD model included the following drug effects:
where Emax,mat is the maximal (proportional) effect of PG on maturation rate (1/MMT) change, Emax,prol is the maximal (proportional) effect of PG on proliferation rate change, and Emax,Scale and EC50Scale are scaling parameters for the margination effect.
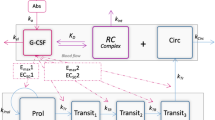
The final structure of the PKPD model structure is presented in Fig. 2. IIV was included as log-normal distributions on all parameters in the PK model apart from Vmax (unsupported) with a full variance-covariance block (Ω). IOV was included between parameters associated with absorption, first-order absorption rate constant (Ka), Frel, and the input rate. IIV in the PD model was included on BASE, neutrophil half-life, EC50, Emax,mat, and Emax,prol, the latter three in an Ω block. IOV in the PD model was only included on neutrophil half-life. Final estimates of the combined model before covariate inclusion can be seen in Table I.

Pharmacokinetic-pharmacodynamic (PKPD) model structure of the final model. The system is bidirectional where pegfilgrastim (PG) influences the absolute neutrophil count (ANC) by stimulating proliferation, maturation, and margination. The margination effect increased the apparent volume of distribution of the ANC compartment
Model Evaluation
The predictive performance of the model without covariates is presented in VPCs in Fig. 3. The median of the PG concentration and ANC data (solid lines) falls within or close to the 95% prediction interval of the median (dark shaded area) resulting from 500 simulations from the model indicating adequate model performance. However, the model somewhat overpredicts the variability at the outer percentiles of the ANC data.

Visual predictive check for the combined model stratified by observation type. Pegfilgrastim (PG) concentration and absolute neutrophil count (ANC) are presented in the left and right panels, respectively. Then, 500 simulated dataset were generated. The black lines are the 2.5th, 50th (solid) and 97.5th percentiles based on the observed data. The shaded areas are 95% confidence intervals for the 2.5th, 50th (dark gray), and 97.5th percentile prediction intervals based on the simulated data
Simulations
Model performance was also evaluated by comparing observed NCA statistics to simulated NCA statistics across all dosing occasions. Figure 4 demonstrates that the simulated AUC and Cmax mimic the observed percentiles of the data. The observed geometric means of PK and PD AUCs were 4359 ng h/mL and 6214·109cells h/L, respectively, and the corresponding simulated geometric means were 4523 and 6035. The model slightly overpredicts Cmax for both PG and ANC.

Simulated individual area under the curve (AUC) and maximum concentration (Cmax) (shown as histograms, solid gray lines for geometric mean and dashed gray lines for 2.5th and 97.5th quantiles) compared with the same observed non-compartmental analysis metrics (solid black lines for geometric mean and dashed black lines for 2.5th and 97.5th quantiles) for pegfilgrastim (PG) and absolute neutrophil count (ANC). One hundred simulations of the reference data set were performed. AUCs were calculated from time = 0 to the last observed time point in each individual and occasion
The sensitivity of PK and PD to small changes in administered dose from the reference (6 mg therapeutic dose) can be seen in Fig. 5. In general, PK AUC increased > 5 times more than the increase of dose while PD AUC increased by ∼0.5 times the dose increase. A similar trend was observed for Cmax where PK Cmax increased > 4 times more than the increase in dose while PD Cmax increased by ∼0.4 times the dose increase.

The percent increase in area under the curve (AUC) and maximum concentration (Cmax) of simulated pegfilgrastim (PG) concentrations (PK) and absolute neutrophil count (ANC) (PD) versus percent increase in administered dose. A nominal dose of 6 mg was used as the reference baseline. The dotted line is the identity line
Full Random Effects Modeling
The interindividual and interoccasion variability in parameters are illustrated in Figs. 6 and 7, together with the fraction of the total variability explained by individual covariates or by all covariates jointly. The ability of covariates to describe the parameter variability is clearly small; as a consequence, no individual covariate could not explain more than 2% of the variability in the derived metrics AUC and Cmax (see Appendix 2). Forest plots showing the size of the effects (including uncertainty) of the covariates on the different parameters in the model are presented in Appendix 1.

Parameter standard deviation after inclusion of covariates in the FREM interindividual variability (IIV) component (colored bars), with the explained part of the variation visible at the top (gray bands)

Parameter standard deviation after inclusion of formulation and period in the FREM interoccasion variability (IOV) component (colored bars), with the explained part of the variation visible at the top (gray bands)
DISCUSSION
The disposition of non-pegylated and pegylated exogenous GCSF products has been described using PKPD models previously (12,13,14). These models, in general, describe the impact of GCSF administration on ANC, and the effects of ANC on the elimination of the administered product in turn. This self-regulating mechanism is important to characterize in order to perform realistic simulations using the models. For PG, the previously published PKPD analysis was based on mean data ignoring the bidirectional feedback mechanisms that operate at the individual level (14). The present model is developed recognizing the individual, and occasion-specific, bidirectional feedback. This is not only important for respecting the nature of the system but also to allow us to inspect the quality of the model using simulations as well as performing simulations to learn about properties of the therapy.
There are many similarities between the presently developed model and the previously developed model for PG concentrations (14). Both models have a dual processes PG absorption model, one-compartment PG distribution, parallel ANC-dependent, and non-specific PG elimination pathways, and in both models PG has a triple effect on ANC: proliferation, maturation rate, and margination. There are also differences between the models as further discussed below. These differences may have their origin in the data, where the previous model was developed based on data from fewer individuals (n = 32 vs n = 174) and fewer occasions (n = 32 vs n = 445), but with four dose levels compared to the single dose level in the present study. Also, the previous model incorporated data on immature precursors to neutrophils which were not available in the present analysis.
The absorption of PG was described by a sequential zero- and first-order absorption, whereas the previous model used a first-order absorption model with lag time equivalent to an alternative parametrization of a sequential first-order transit compartment model. For the model described in this work, the mean absorption time was 3.7 days when estimated across the different absorption processes compared to 1.84 days estimated in the previous model. However, this difference could be attributed to differences in the administration site, method, or other unspecified reasons. Similar in both models was that the first-order absorption contributed to a majority of the mean absorption time. The observed time of maximum concentration Tmax of PG profiles in the previous study was dose dependent with increasing Tmax for increased dose. The observed Tmax, calculated with NCA, for similar doses (0.1 mg/kg and 6 mg) were comparable for the two studies (16 and 20 h). The present model predicts a dose-dependent increase in Tmax similar to the one previously observed. The dose-dependency in Tmax can be understood both from the zero-order absorption process as well as the saturable elimination. Mechanistic models accounting for distribution of drug to lymph before reaching plasma can be used to describe absorption of subcutaneously administered large molecules but require extensive data in the absorption phase for parameter estimation (30).
The distribution volume of PG was estimated to be small, 1.8 L, in this analysis. In the previous model, the volume was estimated to be approximately 5 L. The value of 1.8 L is lower than the expected plasma volume of approximately 3 L. This discrepancy may indicate that some aspect of the model is misspecified; however, the distribution volume of PG was similar to a reported distribution volume for filgrastim (~2 L) (12). As absorption process parameters and distribution volume often have high correlation, any misspecification is most likely in the nature of the absorption process. For this reason, different absorption models were investigated, but none better than the present one was identified.
The elimination of PG was, in the present model, dominated by the saturable non-specific process, and the ANC-dependent elimination played more than a marginal role only at the highest PG concentrations where it could account for up to around 50% of the elimination rate. In the model by Roskos et al. (14), the dominating route of elimination varied over time and depended on the concentration of PG and ANC. In these determinations of elimination pathways, there is a benefit with multiple dose levels, as in Roskos et al., but also individual-based analysis, as in the present work. Presently, the issue of which elimination pathway is dominating, and when, is difficult to fully resolve. Additional data obtained at low ANC could assist such an analysis via an increased fraction of non-ANC-dependent elimination.
The impact of PG on ANC was predominantly an increase in ANC levels. However, for the first hours after the PG administration, there was a clear decrease in ANC levels. A similar decrease was observed by Roskos et al. (14) in both cases; this was modeled as a margination effect where the apparent distribution of the ANC compartment increased resulting in a sudden drop in ANC. This “paradoxical” drop has been reported before and the authors speculate that this may be due to increased adhesion to tissue after GCSF administration (31). The second noticeable effect of PG on ANC is the increase in estimated proliferation rate whereby the number of neutrophils entering the maturation chain increased. The third effect of PG on ANC, included in both the present model and that by Roskos et al. (14), is the decrease in maturation time with administration of PG. Such a decrease has been well documented by for example flash labeling studies (32). There is good agreement between the two models in the potency of PG, where the estimated values of EC50 were 9.86 and 9.24 ng/mL. In this analysis, a high correlation was identified between the drug effect on proliferation (Emax,prol) and a margination effect. Therefore, to achieve a parsimonious model, a scaling parameter was estimated to scale both Emax,prol and EC50.
In this work, MMT in the absence of exogenous GCSF was fixed to a physiologically plausible value (120 h) which should be appropriate given that neutrophil kinetics in health should be relatively stable and that variability in neutrophil kinetics may be due to GCSF influence (29). Further, since the rates in the system are derived from MMT, this allowed for the estimation of separate drug effects on the maturation and proliferation features. Thus, the only fixed effect parameter estimated for the PD model, disregarding drug effects, was the ANC baseline.
The present PKPD model described and predicted the observed concentration data well. Although the central trend was well captured in both PG and ANC, there was a slight overprediction of ANC variability. Due to the bidirectional nature of the system where PK influences PD and PD influences the elimination of the drug, it is difficult to pinpoint whether misspecifications in the PK or PD models were responsible.
The simulated AUC and Cmax for both the PG concentrations and ANC demonstrated good model performance, although there was a slight overprediction in Cmax of both the PG and ANC. For complex systems, such as the present one (due to its bidirectionality), simulations can also be a way to learn about its properties. Below we illustrate this with examples of how AUC and Cmax depend on model parameters, covariates, and PG dose. Other aspects not explored here, but possible to assess based on simulations from the model include for example how the ANC response to PG would depend on the baseline ANC value, as in neutropenic patients, and/or with more frequent dosing, as may be therapeutically required.
The potential for demographic covariates to explain inter- and intraindividual variability in parameters was explored using a FREM approach. This makes covariate model building superfluous and avoids selection bias in the final model covariate relations by simultaneous estimation of all parameter relations with covariates of interest. Time constant covariates can only explain interindividual variability, whereas time-varying covariates can explain intraindividual variability in parameter values. In the present analysis, covariates explained little of either type of parameter variability. Sex, followed by lean body weight, was the most influential covariates, but even these did not explain more than a minority of the variability in any parameter. Given the complexity of the system, it is also of interest to assess how much covariates can influence secondary metrics such as AUC and Cmax. A simulation-based assessment revealed that only about 2% of the variability could, at most, be explained by covariates.
It may not be surprising that little variability is explained by demographic covariates in healthy volunteers, but it is also possible that the interactive nature of the system further makes impact of individual parameter deviations (due to covariate effects) less pronounced when assessed on AUC and Cmax. Through simulations, the relationship between parameter values in the model and AUC and Cmax were assessed. ANC baseline was, not surprisingly, the most influential parameter on AUC and Cmax of ANC, explaining 46 and 39% of the interindividual variability. For PG, it was Ka that explained most variability in both peak concentration (Cmax) and AUC, although the explained portion was only ca 10%.
Simulations where small changes in dose of PG (from the 6 mg reference dose) were made illustrate the sensitivity of the system. Exposure to PG increased five times more than the dose. This high sensitivity does not depend on the bidirectional nature of the system as increased concentrations of PG will drive an increase in ANC, which in turn will decrease PG concentrations. Instead, it is the saturable nature of the non-specific elimination that is responsible. For saturable systems, e.g., characterized by a Michaelis-Menten elimination, the exposure will increase more than proportionally with dose. An additional consideration here is that slow absorption contributes to the magnitude of disproportionality. For a drug with Michaelis-Menten elimination, and one-compartment distribution kinetics, the AUC changes maximally with the squared dose size if it is administered as an intravenous bolus dose. It is the relatively slow input to the system that is responsible for the even more disproportionate change in AUC observed in this situation. Also, this link between rate of input and AUC can also explain why Ka was the most influential parameter not only for Cmax but also AUC of PG.
The less than proportional increase in ANC metrics with increasing doses can be understood from the saturable relation with concentration of PG of the ANC proliferation and maturation. With an EC50 of 9 ng/mL and peak concentration approximately ten times higher, increased PG exposure will result in much less than proportional increase in ANC. Further, the bidirectionality of the system may limit the effect of increasing dose by limiting the prolongation in the period of maximum neutrophil production induced by increasing doses due to increased PG clearance by the quicker initial increase in ANC.
CONCLUSIONS
In conclusion, a population PKPD model for PG and ANC following PG administration that well describes PG and ANC profiles has been developed. The mechanistic components of the model are similar to the model of Roskos et al. (14), but also with some differences. Primarily, by including individual and occasion parameter variability, the new model is able to quantify the influence of PK on PD and vice-versa on an individual level. Thereby, it can serve as a tool to understand the system, identify covariates, and through simulations investigate the consequences of for example dosing strategies, patient population characteristics, and trial designs. A sensitive dependence of PG AUC and Cmax on PG dose was identified through such simulations.
References
Leonard RC, Miles D, Thomas R, Nussey F. Impact of neutropenia on delivering planned adjuvant chemotherapy: UK audit of primary breast cancer patients. Br J Cancer. 2003;89:2062–8.
Zielinski CC, Awada A, Cameron DA, Cufer T, Martin M, Aapro M. The impact of new European Organisation for Research and Treatment of Cancer guidelines on the use of granulocyte colony-stimulating factor on the management of breast cancer patients. Eur J Cancer. 2008;44:353–65.
Klastersky J, de Naurois J, Rolston K, Rapoport B, Maschmeyer G, Aapro M, et al. Management of febrile neutropaenia: ESMO Clinical Practice Guidelines. Ann Oncol. 2016;27:v111–8.
Lustberg MB. Management of neutropenia in cancer patients. Clin Adv Hematol Oncol. 2012;10:825–6.
Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008;42:277–88.
Bendall LJ, Bradstock KF. G-CSF: from granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev. 2014;25:355–67.
US Food and Drug Administration / Center for Drug Evaluation and Research, 1991, Summary Basis of Approval. Application Number: 103353/000. URL: https://www.accessdata.fda.gov/drugsatfda_docs/bla/pre96/103353Orig1s000.pdf, Accessed on: 2017–11-28.
Georgala A, Klastersky JA. Prophylaxis of febrile neutropenia in adults receiving chemotherapy needs to be adapted to the risk. Expert Rev Hematol. 2015;8:115–21.
Yang BB, Kido A, Salfi M, Swan S, Sullivan JT. Pharmacokinetics and pharmacodynamics of pegfilgrastim in subjects with various degrees of renal function. J Clin Pharmacol. 2008;48:1025–31.
Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J Pharm Sci. 2010;99:2557–75.
Molineux G. The design and development of pegfilgrastim (PEG-rmetHuG-CSF, Neulasta). Curr Pharm Des. 2004;10:1235–44.
Wiczling P, Lowe P, Pigeolet E, Lüdicke F, Balser S, Krzyanski W. Population pharmacokinetic modelling of filgrastim in healthy adults following intravenous and subcutaneous administrations. Clin Pharmacokinet. 2009;48:817–26.
Wang B, Ludden TM, Cheung EN, Schwab GG, Roskos LK. Population pharmacokinetic-pharmacodynamic modeling of filgrastim (r-metHuG-CSF) in healthy volunteers. J Pharmacokinet Pharmacodyn. 2001;28:321–42.
Roskos LK, Lum P, Lockbaum P, Schwab G, Yang BB. Pharmacokinetic/pharmacodynamic modeling of pegfilgrastim in healthy subjects. J Clin Pharmacol. 2006;46:747–57.
Molineux G, Foote M, Arvedson T. Twenty years of G-CSF: clinical and nonclinical discoveries. Milestones in Drug Therapy. Basel: Springer; 2012.
Quartino AL, Karlsson MO, Lindman H, Friberg LE. Characterization of endogenous G-CSF and the inverse correlation to chemotherapy-induced neutropenia in patients with breast cancer using population modeling. Pharm Res. 2014;31:3390–403.
Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy- induced myelosuppression with parameter consistency across drugs. Journal of Clinical Oncology: Offi- cial Journal of the American Society of Clinical Oncology. 2002;20:4713–21.
Karlsson MO, 2012, A full model approach based on the covariance matrix of parameters and covariates, PAGE 21, Abstract 2455. URL:http://www.page-meeting.org/?abstract=2455, Accessed on: 2017–11-01.
Yngman G, Nyberg J, Jonsson NE and Karlsson MO, 2017, Practical considerations for using the full random effects modeling (FREM) approach to covariate modeling, PAGE 26, Abstract 7365. URL:https://www.page-meeting.org/default.asp?abstract=7365, Accessed on: 2017–11-01.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44:1051–65.
Karlsson M and Holford N, 2008, A tutorial on visual predictive checks, PAGE 17, Abstract 1434.URL:http://www.page-meeting.org/?abstract=1434, Accessed on: 2017–11-24.
Acharya C, Hooker AC, Turkylmaz GY, Jonsson S, Karlsson MO. A diagnostic tool for population models using non-compartmental analysis: the ncappc package for R. Comput Methods Prog Biomed. 2016;127:83–93.
Beal SL, Sheiner LB, Boeckmann AJ and Bauer RJ (2014) NONMEM User’s Guides. (1989–2014) Icon Development Solutions, Ellicott City.
Lindbom L, Pihlgren P, Jonsson EN. PsN-toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Prog Biomed. 2005;79:241–57.
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN)–a Perl module for NONMEM related programming. Comput Methods Prog Biomed. 2004;75:85–94.
R Development Core Team (2007), R: A language and environment for statistical computing. R Found- ation for Statistical Computing, Vienna, Austria. ISBN 3–900051–07-0, URL http://www.R-project.org
Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31:318–24.
De Souza DC, Craig M, Cassidy T, Li J, Nekka F, Bélair J, et al. Transit and lifespan in neutrophil production: implications for drug intervention. J Pharmacokinet Pharmacodyn. 2018;45:59–77.
Krzyanski W. Interpretation of transit compartments pharmacodynamic models as lifespan based indirect response models. J Pharmacokinet Pharmacodyn. 2011;38:179–204.
Fathallah AM, Balu-Iyer SV. Anatomical, physiological, and experimental factors affecting the bioavailability of sc-administered large biotherapeutics. J Pharm Sci. 2015;104:301–6.
Gordon BC, Revenis AM, Bonifacino AC, Sander WE, Metzger ME, Krouse AE, et al. Paradoxical drop in circulating neutrophil count following granulocyte-colony stimulating factor and stem cell factor administration in rhesus macaques. Exp Hematol. 2007;35:872–8.
Price TH, Chatta GS, Dale DC. Effect of recombinant granulocyte colony-stimulating factor on neutrophil kinetics in normal young and elderly humans. Blood. 1996;88:335–4.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix 1 Covariate-parameter effect sizes
Forest plots showing the size of the effect of the covariates on the parameters of the model are shown in Figs. 8 and 9.

Effect size of covariate-parameter interactions from the IIV FREM, with uncertainty. First and second line of continuous covariates show the effect of the covariate’s 5th and 95th percentile, respectively. For categorical covariates (sex and race), the effect of the presented category is compared to the other category

Effect size of covariate-parameter interactions from the IIV FREM, with uncertainty. First and second line of continuous covariates show the effect of the covariate’s 5th and 95th percentile, respectively. For categorical covariates (sex and race) the effect of the presented category is compared to the other category
Appendix 2 Correlation matrices
Correlations in the simulations implementing the covariate effects characterized with FREM are shown in Table II. In Tables III and IV, the correlation coefficients of the IIV and IOV components of FREM are displayed, respectively.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brekkan, A., Lopez-Lazaro, L., Yngman, G. et al. A Population Pharmacokinetic-Pharmacodynamic Model of Pegfilgrastim. AAPS J 20, 91 (2018). https://doi.org/10.1208/s12248-018-0249-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-018-0249-y