
Abstract
Antibody drug conjugates (ADCs) are a multi-component modality comprising of an antibody targeting a cell-specific antigen, a potent drug/payload, and a linker that can be processed within cellular compartments to release payload upon internalization. Numerous ADCs are being evaluated in both research and clinical settings within the academic and pharmaceutical industry due to their ability to selectively deliver potent payloads. Hence, there is a clear need to incorporate quantitative approaches during early stages of drug development for effective modality design and target selection. In this review, we describe a quantitative approach and framework for evaluation of the interplay between drug- and systems-dependent properties (i.e., target expression, density, localization, turnover, and affinity) in order to deliver a sufficient amount of a potent payload into the relevant target cells. As discussed, theoretical approaches with particular considerations given to various key properties for the target and modality suggest that delivery of the payload into particular effect cells to be more sensitive to antigen concentrations for targets with slow turnover rates as compared to those with faster internalization rates. Further assessments also suggest that increasing doses beyond the threshold of the target capacity (a function of target internalization and expression) may not impact the maximum amount of payload delivered to the intended effect cells. This article will explore the important application of quantitative sciences in selection of the target and design of ADC modalities.
Similar content being viewed by others


INTRODUCTION
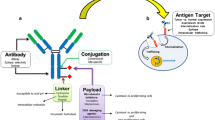
Antibody drug conjugates (ADCs) combine the selectivity of antibodies with the potent therapeutic activity of small molecule drugs (payload) (1–3). The three components of a typical ADC are (a) the antibody carrier with a high affinity for a well-expressed antigen, (b) a potent payload, and (c) a linker that is preferably cleaved site-specifically upon cellular internalization of the ADC (1–3). Hence, it is of critical importance to evaluate and optimize all these three components for the design of an effective ADC. By selectively targeting cell-specific antigens, the targeting antibody is intended to direct the payload to relevant effect cells (biophase) while minimizing exposure to non-target tissues, thus expanding the therapeutic index (TI) for the potent payload (4–7). In particular instances, the ADC may be required to undergo intracellular uptake via receptor-mediated endocytosis to traffic through endosomal compartments, and to release the free payload via changes in lysosomal pH and/or enzymatic conditions in order to exert its action in the cytoplasm or nucleus (8). Alternatively, a class of non-internalizing ADCs, referred to as extracellular drug conjugates (EDCs), is being developed. In the case of EDCs, drug release is not required as the payload acts on the cell surface while still attached to the antibody and interacts with a specific cell-surface antigen (9).
ADCs are emerging as a prominent therapeutic platform in oncology, where the approach has allowed minimizing the non-specific toxicities of potent chemotherapeutics by exploiting the exquisite specificity of antibodies (10–12). The two currently marketed ADCs are Adcetris® (SGN-35) for hematological malignancies and Kadcyla® (T-DM1) for metastatic breast cancer (3). Currently, there are more than 30 ADCs in clinical development for various hematological and solid-tumor malignancies (13). Although ADCs have been exploited to trigger specific tumor cell eradication, this therapeutic venue is not the only area where application of the targeted delivery of potent payloads may be advantageous. Additionally, improvements in the linker-payload technologies are broadening the potential utility of these agents outside of oncology (14).
Application of quantitative pharmacology is increasingly sought in order to accelerate the drug development process (15–17). Similarly for the development of ADCs, application of quantitative approaches with considerations given to the cellular and physiological properties should facilitate design and selection of effective drug candidates. In this review, we describe how an early quantitative assessment can provide insights applicable to the design of ADCs and to evaluate both drug- and system-dependent properties. A quantitative understanding of the key properties presented here should enable both rational target selection and successful modality design.
CONSIDERATIONS FOR TARGET SELECTION AND MODALITY DESIGN
The most critical driver underlying the efficacy profile for an ADC is the successful delivery of the payload (drug) into the intended effect cells. Delivery of potent payloads to the biophase is a result of the interplay between target- and modality-related properties (Fig. 1). Critical target-related properties are (a) expression profile, and density (receptor copy number), (b) target internalization and turnover rate, and (c) functional relevance to human disease (18–20). An ideal target should demonstrate a specific expression profile at the relevant site of action (typically tumor cells) with little to no expression in other healthy tissues (18, 19). Target expression and density should also be in sufficient concentrations to facilitate delivery of a potent payload into the effect compartment (“Impact of Target Density and Internalization Rate on Payload Delivery” section). In most instances, application of ADCs would necessitate an internalization step in order to facilitate intracellular uptake and payload release (18, 19). Rate of internalization or target turnover rate is a critical parameter that controls the efficiency of the payload delivery into the biophase (“Impact of Target Density and Internalization Rate on Payload Delivery” section).

Antibody-drug conjugates: interplay between target and modality-related properties
Modality-related parameters critical for selection of the appropriate antibody are (a) antibody isotype, (b) affinity to target and epitope selectivity, (c) pharmacological mode of action, and (d) species cross-reactivity (21). The selection of isotype will determine whether the antibody can elicit effector function [i.e., recruit effector cells for antibody-directed cellular cytotoxicity (ADCC) and/or elicit complement (CDC)], an additional mechanistic advantage for cell-killing endpoints in oncology applications (22, 23). For example, a human antibody of IgG1 isotype is known to recruit effector cells more efficiently and potentially elicit complement activation, thus demonstrating enhanced effector function relative to an IgG4 isotype (22, 23). For an ADC modality, the pharmacological effect can be elicited by both the payload and the antibody interacting with the antigen target, in conjunction with the recruitment of effector cells and/or complement (24). It is therefore critical to fine-tune the cytotoxic effect for both the antibody and payload within the desired therapeutic window (7). Currently, the ADC candidates evaluated in clinic settings are of both of IgG1 and IgG4 isotypes (13). Binding to membrane-associated internalizing antigens can greatly enhance the antibody clearance through a target-mediated specific process (commonly known as target-mediated drug disposition or TMDD) (25). TMDD contributes to an additional target-mediated saturable clearance process for the antibody, thus influencing dose selection (“Determination of In Vivo Target Capacity” section) (21).
Selection of a payload that is conjugated to the antibody is governed by properties such as (a) its ADME properties, (b) mechanism of action (intercellular vs. cellular uptake), and most importantly (c) the payload potency. Cytotoxic payloads currently employed in clinical settings have been mostly DNA-alkylators—acting in the nucleus such as calicheamicin and duocarmycins—and tubulin-binders—acting in the cytoplasm such as auristatins and maytansinoids (26). Non-cytotoxic payloads have not yet been evaluated in the clinic. Payloads for non-cytotoxic applications are required to continuously engage the relevant receptors in order to exert their pharmacological action. Hence for non-cell-killing payloads, the kinetics and extent of accumulation of the payload in the cell is critical for its mechanism of action.
One of the key parameters governing the payload accumulation at the site of action is its elimination rate from the effect compartment(s) (“Effect of Drug PK on Its Delivery to Intracellular Space” section). Additionally, apart from the overall payload ADME properties, it is critical to understand the relationships between drug exposure and the intended pharmacodynamic endpoints. Treatment with drugs known to have direct pharmacodynamic effects (Cmax-driven effects) will benefit from higher drug concentrations at the site of action (27, 28). However, drugs that show indirect pharmacodynamic effect properties (AUC-driven effects) will benefit from prolonged exposure at the biophase (27, 28).
ADC stability and the payload’s release rate can be tailored with the choice of linker (refer to article in the theme issue: Antibody Drug Conjugates: Design and selection of linker, payload and conjugation chemistry) (29). Non-specific release of the drug will alter the free drug as well as the intact ADC concentration in plasma, altering pharmacokinetics and toxicity (30, 31). The site-specific release rate will determine the free drug concentrations within the intracellular effect compartment (26, 32–37). Target, antibody, drug, and linker-related properties define the fate of the ADC in vivo via a multi-step kinetic process (Fig. 2). As with any multi-step kinetic system, the rate-limiting step will most significantly influence the intracellular drug exposure and the expected PD outcome (key parameters integrated in “Integration of Key Parameters for Target Selection and Modality Design” section).

Compartmental model for ADC pharmacokinetics, target interaction, intracellular uptake, and drug release (Ab ADC, R receptor, AbR ADC-receptor complex, K D affinity for AbR formation, k syn zero-order synthesis rate of the receptor, k int complex/receptor internalization rate, CL int clearance of complex/receptor, SM small molecule/drug/payload, CL SM elimination clearance of small molecule). The balance of ksyn and kint determine the receptor-recycling rate (41). In this case, the ksyn was estimated from target capacity as described previously (39, 40). ksyn highlights the synthesis rate of the antigen and was estimated based on equation: kint x Css (concentration of the target at steady state). CLRES reflect the linear clearance reported for antibodies (38); CLSM reflects the clearance of the payload selected based on internal historical data; Vd_SM reflect volume of distribution for the payload; KD reflects the affinity of the antibody for the target antigen; Vd_Ab reflects volume of the central compartment selected based on historical data (38). Each mole of antibody is assumed to deliver two moles of the payload to the effect compartment. PK of the payload assumed to follow a 1-compartment model
QUANTITATIVE PHARMACOLOGY FRAMEWORK
Topics addressed in this review are focused on the key design parameters relevant for selection of an effective ADC modality using a quantitative approach framework (Fig. 2). The compartmental model shown in Fig. 2 accounts for the target-mediated disposition of an ADC along with a linear residual clearance from plasma (CLRES) (38). The target-related clearance is assumed to be independent of drug loading on the ADC and similar for different species of an ADC (39). Furthermore, the target receptor (R) is eliminated/catabolized upon internalization of the ADC-receptor complex (AbR) and the ADC-receptor complex internalizes at the same rate as the receptor alone (kint). The CLint was determined from the internalization rate (kint) and volume of distribution of the antibody (Vd_Ab) (39, 40). The antibody-receptor complex assumed to have a similar volume of distribution as of the antibody. The number of receptors per cell, receptor concentration in blood, and receptor internalization rate (kint) were assigned from in vitro measurements of a well-perfused target in blood. The receptor is assumed to be synthesized at a constant zero-order synthesis rate (ksyn) (41). The quantitative approach presented here further assumes a fast distribution of ADC to the putative effect compartment(s) as driven by the receptor (R) and ADC (Ab) concentration and antibody affinity (KD). These assumptions are generally applicable for therapeutic application of ADCs in hematological malignancies or well-perfused organs such as the liver (partition coefficient Kp = 1) (42). For ADC applications in solid tumor therapy, where these assumptions may not be applicable (i.e., Kp < 1), further adjustments can be incorporated in the proposed compartmental model (43, 44). It is important to note that the compartmental models have inherent limitations with respect to antibody biodistribution. Antibody distribution to various organs, including the tumor, is thought to be predominantly due to convection rather than diffusion as presumed in the compartmental models (21). The model also assumes instantaneous lysosomal release of the payload. Drug release rates, tailored by choice of the linker, may not always be rapid. Hence, an additional rate constant can be incorporated to account for enzymatic release of the drug from the ADC modality. Upon release, the free drug is assumed to undergo fast diffusion into the interstitial space in the effect compartment which then is followed by a fast diffusion into systemic circulation. For non-oncology applications, the residence time of the payload is a critical consideration for effective application of an ADC modality (see “Effect of Drug PK on Its Delivery to Intracellular Space” section). It is assumed that upon drug release, volume of distribution of the released payload be similar to that observed for the free payload. All parameters have been specified in the figure legends.
Impact of Target Density and Internalization Rate on Payload Delivery
The two key target-related parameters affecting drug concentration at the intracellular site are expression density of the target receptor and its internalization or turnover rate which define the target capacity (20). Understanding the target capacity—the interplay between these parameters—provides a rational approach for target selection. In simulations shown in Fig. 3, drug concentration profiles in the intracellular space were predicted by varying the target receptor concentrations and internalization rates (Fig. 3). For relatively fast target turnover rates (half-life of internalization <0.5 h), a change in receptor expression did not result in a significant difference in accumulation of the payload at the intracellular effect compartment (Fig. 3a). However, when target turnover rates were assumed to be slower (half-life of internalization approximates 5 h), changes in the receptor density had a greater impact on drug accumulation in the biophase (Fig. 3b). As highlighted, using this quantitative framework, it is feasible to establish a potential range for combinations of the target density profile and the effective internalization rates required for effective payload delivery into a particular effect compartment. This paper describes a compartmental model that integrates in vivo PK of antibody with target engagement and drug delivery. However, the model can be simplified to an in vitro cellular model where the interplay of target selection and modality-design parameters can be evaluated. This quantitative pharmacology approach can then be utilized in early discovery for in vitro screening of targets and ADC constructs.

Impact of receptor concentration and rate of internalization on small molecule delivery to intracellular space; (left panel a). Intracellular drug concentration profile for a well-expressed (80,000 receptor copies per cell), fast internalizing target (t1/2 = 0.5 h) at target receptor concentrations 0.5, 1.0, and 2.0 nM; (right panel b). Intracellular drug concentration profile for a well-expressed (80,000 receptor copies per cell), slow internalizing target (t1/2 = 5 h) at target receptor concentrations 0.5, 1.0, and 2.0 nM. Note: All other parameters besides receptor internalization rate and receptor concentrations (as shown in the figures) are kept constant, i.e., CLRES = 5 mL/day/Kg; CLSM = 2.7 L/day/Kg; Vd_SM = 0.6 L/Kg; KD = 0.8 nM; ksyn = 64 μg/mL/day; Vd_Ab = 40 mL/Kg. In both a, b, kint was kept constant (in a: kint = 1.4 h−1 and in b: kint = 0.14 h−1) and ksyn was adjusted to achieve different scenarios with respect to receptor concentrations
Determination of In Vivo Target Capacity
Target-mediated drug disposition (TMDD) is a result of the capacity-limited interaction of the antibody with the target (25, 45). In general, a simple dose-ranging pharmacokinetic study can generate highly useful information with respect to the magnitude of the target capacity and its impact on the antibody clearance and potential payload delivery (46). In practical terms, delivery of the payload via an ADC to the effect cells will be limited by the target capacity (a composite of both target density and the internalization rate). To determine target capacity, it is acceptable to use either TMDD or Michaelis-Menten (MM) modeling approaches (40). A recent literature report has compared these modeling approaches (i.e., a TMDD and the MM (40)). As described, an accurate estimation of the target capacity was reported with the MM-approximation approach (and comparable to the TMDD estimation method) when target binding, target internalization, and target recycling were fast and not rate-limiting (40). Additionally, the presence of multiple ADC species that potentially could be generated in an in vivo setting, as a result of partial deconjugation of the payload, will need to be considered when these approaches are employed for evaluation of target capacity (39). This point is a critical consideration as different ADC species, with varying degrees of drug loading, may potentially have different biodistribution, elimination, and efficacy profiles under in vivo conditions (47–49).
Additionally, evaluation of the target capacity is a critical tool for establishing affinity requirements for an ADC modality. For targets with high capacity, the payload delivery is less likely to depend on the antibody affinity under steady-state conditions (“Integration of Key Parameters for Target Selection and Modality Design” section). Any change in affinity will be offset by the high target capacity. For targets with lower capacity, improvements in affinity are more likely to impact receptor occupancy and payload delivery (Fig. 5). However, under non-steady-state conditions, high-affinity antibodies have been shown to penetrate tumors less efficiently (the binding-site barrier hypothesis) and hence tumor penetration is also a critical consideration for effective targeting of ADCs to solid tumors (50–54). Antibodies that bind to target antigen with lower affinity, however, may have a higher penetration index into the tumor core and potentially be more effective in tumor killing (50–54). Furthermore, target accessibility is another key criterion for successful ADC design and payload delivery. Effective dose selection, within the intended therapeutic window, may be challenging if the target cells are localized in multiple tissues with different biodistributional barriers. For a given dose, a toxic concentration may be attained in the more accessible tissue while a sub-efficacious concentration may be reached in a less accessible tissue (50–54).
This article describes a strategic approach in integrating the relevant data for the design of a successful modality and appropriateness of the target properties. The parameters used for simulations, outlined in various sections, are selected based on historical data and are within the acceptable physiological framework described in the literature (Figs. 2, 3, 4, and 5). However, the approach described here should be tailored on a case-by-case basis with addition of complexities that may be relevant to each particular situation as discussed in the literature (47–49, 55, 56).

Effect of elimination rate of released small molecule on its delivery to intracellular space at varying doses. Note: All other parameters besides small molecule elimination rate are kept constant, i.e., CLRES = 5 mL/day/Kg; Vd_SM = 0.6 L/Kg; KD = 0.8 nM; ksyn = 64 μg/mL/day; Vd_Ab = 40 mL/Kg; kint = 2.77 h−1; number of receptor copies = 80,000 per cell; 1000 T cells per mm3 of blood

Interplay of target affinity, target expression, and rate of internalization on delivering efficacious drug concentration to effect site. Note: Antibody affinity for receptor, receptor internalization rate and receptor concentration are varied. All other parameters are kept constant: CLRES = 5 mL/day/Kg; CLSM = 2.7 L/day/Kg; Vd_SM = 0.6 L/Kg (t1/2_SM ∼ 4 h); Vd_Ab = 40 mL/Kg
Effect of Drug PK on Its Delivery to Intracellular Space
The pharmacokinetics of the free payload, upon release from the antibody carrier, is an additional consideration impacting payload accumulation at the effect site particularly for non-cell-killing applications. Simulations of free payload concentrations in the intracellular compartment at various elimination half-lives of the payload reflect that its accumulation increases with the increase in half-life or decrease in elimination rate of the drug (Fig. 4). Under the conditions employed, payload accumulation cannot be increased beyond a dose of 1 mg/kg due to target-capacity limitations as discussed in “Determination of In Vivo Target Capacity” section. The efficacy driver for antibody-drug conjugates is typically related to the effective concentrations that are achieved at the effect cells (27, 28). An increase in payload potency relaxes the requirement on the amount of payload that needs to be delivered in order to exert the desired PD effect, thus compensating for other potential limitations with respect to target capacity, payload release rate, and payload residence time within the biophase. Payload efflux from intracellular site can additionally result in a bystander effect, where it is available to diffuse across neighboring cells and elicit cytotoxicity (57). This bystander effect must be accounted for while assessing the pharmacodynamic potential of ADCs. Concentration of the target, payload release rates, dissociation rates from low- or high-affinity binders, and payload efflux rates are determinants of the payload residence time within the intracellular compartments and the bystander effect. For non-cell-killing applications, payload actions can be governed by its residence time. The balance of the payload efflux rate may be tailored by using more hydrophilic, cell-impermeable payloads that will limit their diffusion to the extracellular space upon release, thus increasing local intracellular payload concentrations (58). Additionally, payload binding to intracellular proteins can affect its pharmacokinetics and likely improve accumulation at the site of action (as defined by koff of binding to intracellular target). DNA intercalators and tubulin-binders are the most popular class of cytotoxics utilized in ADCs, and they are known to bind to nuclear or mitochondrial matter, respectively. This potentially may contribute to a longer residence time in effect cells, thus improving the ADC’s PD profile. Payload binding to intracellular targets and payload efflux post intracellular delivery may be viewed as competing kinetic processes with impact on therapeutic index in particular for non-oncology targets.
Heterogeneous conjugation of ADCs results in a distribution of species with a range of drug loading and positional variants of the conjugated payload. The heterogeneous conjugation process can introduce additional challenges with respect to ADC production and batch-to-batch variability for the final drug product (59). To overcome these challenges, site-specific conjugation technologies are increasingly employed to control the extent and position of drug loading on ADCs (60, 61). Conjugation methods may impact drug loading and potential release rates, thus impacting the ability to deliver an efficacious amount of payload to the site of action (4, 62–64). The choice of linker impacts stability of the ADC in circulation and site-specific release rates in the intracellular compartment (32–35). ADCs typically employ lysosomal enzyme-based linker cleavage strategies such as cathepsin B and phosphatases (26, 29). Some ADCs have non-cleavable linkers that rely on antibody catabolism for payload release (65). The balance of the release rates and the payload elimination will determine the overall payload exposure. For slow release and fast payload elimination, free payload concentration profile may be governed by the formation rate-limited kinetics (47). Instability of linker in circulation increases non-specific exposure of free payload (30).
Integration of Key Parameters for Target Selection and Modality Design
The amount of payload delivered to the site of action is a complex interplay of drug and system-dependent parameters. Simulations in Fig. 5 demonstrate the interplay of target affinity, receptor density, and rate of internalization for delivering efficacious concentrations of payload to the cell (payload potency of 0.3 nM is assumed in this case study). The ratio of ADC internalization rate and the payload elimination rate is a critical index for payload accumulation in biophase. In simulations presented in Fig. 5, payload accumulation has been assessed over a range of receptor concentrations and affinities for the putative target. Simulation lines represent a potential parameter range required to deliver an average payload concentration equal to the required payload concentrations (0.3 nM).
These simulations provide the following critical insights:
-
1.
Figure 5a highlights the bottom region in the graph where target internalization rate—and turnover rate—is slowest with respect to payload elimination rate. These simulations reflect that below a critical ratio of ADC internalization rate to small molecule elimination rate, delivery of the necessary payload concentrations may not be feasible despite improvements in binding affinity of ADC to target (Fig. 5a).
-
2.
Figure 5b demonstrates that for targets that internalize at 1/10th the rate of the payload elimination, a higher binding affinity of 1 nM is required to achieve efficacious payload concentration. On the other hand, a faster internalizing target (∼10 times faster than payload elimination) can uptake ADC more efficiently and can deliver equivalent efficacious amounts at a much lower target binding affinity of 100 nM.
-
3.
Figure 5c shows that in order to deliver equivalent amounts of payload and for a given internalization and elimination clearance, targets that show lower levels of receptor expression require a higher binding affinity than targets that are highly expressed. Increased binding affinity (KD) improves the probability of internalization as a result of the slower dissociation rate (koff) thus improving the ADC uptake.
-
4.
Figure 5d highlights two regions in the graph: case A where increase in receptor density increases intracellular payload exposure (AUC) and case B where increase in receptor density only increases the attained maximum concentration (Cmax) but not the overall payload exposure (AUC). Cell-killing dynamics of cytotoxics is known to be driven by the overall drug exposure (AUC-driven effects) (27, 28). When ADC target binding and payload elimination kinetics are similar to the proposed scenario as described in case study A, therapeutic outcome may benefit from selection of targets with higher expression density.
-
5.
Simulations compiled in a three-dimensional scale define a surface representing the system properties required to deliver sufficient amount of payload into the effect compartment (Fig. 5e). Similarly, an analogous analysis can be carried out in order to evaluate payload delivery into non-target site to evaluate the existence and extent of undesired effects. Selection of target and design of drug-related properties must take into account the design space described above where the combination of parameters suffices to deliver efficacious amounts of payload into the effect compartments while reducing undesired effects.
SUMMARY/CONCLUSION
A quantitative framework has been discussed to integrate critical ADC design and target selection attributes. Intracellular delivery of potent payloads depends on the following: interplay between drug and system-dependent properties such as ADC PK, biodistribution of ADC, target-mediated drug disposition, antibody affinity, internalization rate, payload release, and elimination from target site as well as payload mechanism of action and potency. Such quantitative assessments in early stages of drug development are critical in informing ADC design and target selection, thus facilitating timely go/no-go decisions.
References
Zolot RS, Basu S, Million RP. Antibody-drug conjugates. Nat Rev Drug Discov. 2013;12(4):259–60.
Adair JR et al. Antibody-drug conjugates-a perfect synergy. Expert Opin Biol Ther. 2012;12(9):1191–206.
Perez HL et al. Antibody-drug conjugates: current status and future directions. Drug Discov Today. 2014;19(7):869–81.
Junutula JR et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925–32.
McDonagh CF et al. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol Cancer Ther. 2008;7(9):2913–23.
Hughes B. Antibody-drug conjugates for cancer: poised to deliver? Nat Rev Drug Discov. 2010;9:665–7.
Junutula JR et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin Cancer Res. 2010;16(19):4769–78.
Kovtun YV, Goldmacher VS. Cell killing by antibody-drug conjugates. Cancer Lett. 2007;255(2):232–40.
Sweeny L et al. A novel extracellular drug conjugate significantly inhibits head and neck squamous cell carcinoma. Oral Oncol. 2013;49(10):991–7.
Schrama D, Reisfeld RA, Becker Jr C. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5(2):147–59.
Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13(3):235–44.
Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29.
Mullard A. Maturing antibody-drug conjugate pipeline hits 30. Nat Rev Drug Discov. 2013;12(5):329–32.
Graversen JH et al. Targeting the hemoglobin scavenger receptor CD163 in macrophages highly increases the anti-inflammatory potency of dexamethasone. Mol Ther. 2012;20(8):1550–8.
Tabrizi MA, Bornstein GG, Klakamp SL. Development of antibody-based therapeutics: translational considerations. New York: Springer; 2012.
Luu KT et al. Modeling, simulation, and translation framework for the preclinical development of monoclonal antibodies. AAPS J. 2013;15(2):551–8.
Sheiner LB, Steimer JL. Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol. 2000;40:67–95.
Mathur R, Weiner GJ. Picking the optimal target for antibody-drug conjugates. Proc Am Soc Clin Oncol. 2013;e103–7.
Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets. 2009;9(8):982–1004.
Tabrizi M, Funelas C, Suria H. Application of quantitative pharmacology in development of therapeutic monoclonal antibodies. AAPS J. 2010;12(4):592–601.
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58.
Jiang XR et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10(2):101–11.
Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25(12):1369–72.
Girish S et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69(5):1229–40.
Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32.
Drachman JG, Senter PD. Antibody-drug conjugates: the chemistry behind empowering antibodies to fight cancer. Hematol Am Soc Hematol Educ Program. 2013; 306–310.
Jusko WJ. Pharmacodynamics of chemotherapeutic effects: dose-time-response relationships for phase-nonspecific agents. J Pharm Sci. 1971;60(6):892–5.
Levasseur LM et al. Modeling of the time-dependency of in vitro drug cytotoxicity and resistance. Cancer Res. 1998;58(24):5749–61.
Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21(1):5–13.
Lin K, Tibbitts J. Pharmacokinetic considerations for antibody drug conjugates. Pharm Res. 2012;29(9):2354–66.
Sapra P, Betts A, Boni J. Preclinical and clinical pharmacokinetic/pharmacodynamic considerations for antibody-drug conjugates. Expert Rev Clin Pharmacol. 2013;6(5):541–55.
Alley SC et al. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008;19(3):759–65.
Doronina SO et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug Chem. 2006;17(1):114–24.
Erickson HK et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66(8):4426–33.
Erickson HK et al. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem. 2009;21(1):84–92.
Okeley NM et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16(3):888–97.
Sutherland MSK et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J Biol Chem. 2006;281(15):10540–7.
Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1–2):81–8.
Gibiansky L, Gibiansky E. Target-mediated drug disposition model and its approximations for antibody-drug conjugates. J Pharmacokinet Pharmacodyn. 2014;41(1):35–47.
Yan X, Mager DE, Krzyzanski W. Selection between Michaelis-Menten and target-mediated drug disposition pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2010;37(1):25–47.
Lauffenburger DA, Linderman JJ. Receptors: models for binding, trafficking, and signaling, vol. 365. New York: Oxford University Press; 1993.
Shah DK, Betts AM. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. mAbs. 2013;5(2):297–305.
Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990;50(3 Suppl):814s–9.
Rudnick SI et al. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71(6):2250–9.
Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol. 2006;72(1):1–10.
Bauer RJ et al. Population pharmacokinetics and pharmacodynamics of the anti-CD11a antibody hu1124 in human subjects with psoriasis. J Pharmacokinet Biopharm. 1999;27(4):397–420.
Bender B et al. A mechanistic pharmacokinetic model elucidating the disposition of trastuzumab emtansine (T-DM1), an antibody-drug conjugate (ADC) for treatment of metastatic breast cancer. AAPS J. 2014;16(5):994–1008.
Chudasama VL et al. Semi-mechanistic population pharmacokinetic model of multivalent trastuzumab emtansine in patients with metastatic breast cancer. Clin Pharmacol Ther. 2012;92(4):520–7.
Lu D et al. An integrated multiple-analyte pharmacokinetic model to characterize trastuzumab emtansine (T-DM1) clearance pathways and to evaluate reduced pharmacokinetic sampling in patients with HER2-positive metastatic breast cancer. Clin Pharmacokinet. 2013;52(8):657–72.
Fujimori K et al. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31(7):1191–8.
Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990;9(3):253–66.
Juweid M et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52(19):5144–53.
Saga T et al. Targeting cancer micrometastases with monoclonal antibodies: a binding-site barrier. PNAS. 1995;92(19):8999–9003.
van Osdol W, Fujimori K, Weinstein JN. An analysis of monoclonal antibody distribution in microscopic tumor nodules: consequences of a “binding site barrier”. Cancer Res. 1991;51(18):4776–84.
Shah DK, Haddish-Berhane N, Betts A. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab-vedotin. J Pharmacokinet Pharmacodyn. 2012;39(6):643–59.
Haddish-Berhane N et al. On translation of antibody drug conjugates efficacy from mouse experimental tumors to the clinic: a PK/PD approach. J Pharmacokinet Pharmacodyn. 2013;40(5):557–71.
Kovtun YV et al. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66(6):3214–21.
Jackson D et al. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008;68(22):9367–74.
Singh R, Erickson HK. Antibody-cytotoxic agent conjugates: preparation and characterization. Methods Mol Biol. 2009;525:445–67.
Axup JY et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. PNAS. 2012;109(40):16101–6.
Panowksi S et al. Site-specific antibody drug conjugates for cancer therapy. mAbs. 2014;6(1):34–45.
Jackson D et al. In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates. PLoS One. 2014;9(1), e83865.
Shen B-Q et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30(2):184–9.
Strop P et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20(2):161–7.
Shen BQ et al. Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine (T-DM1): an emphasis on preclinical and clinical catabolism. Curr Drug Metab. 2012;13(7):901–10.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editors: M. Tabrizi, I. Figueroa, and S. Sadekar
S. Sadekar and I. Figueroa contributed equally to this work.
Rights and permissions
About this article

Cite this article
Sadekar, S., Figueroa, I. & Tabrizi, M. Antibody Drug Conjugates: Application of Quantitative Pharmacology in Modality Design and Target Selection. AAPS J 17, 828–836 (2015). https://doi.org/10.1208/s12248-015-9766-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-015-9766-0