
Abstract
The aim of the study was to investigate the effect of novel polymer/lipid formulations on the dissolution rates of the water insoluble indomethacin (INM), co-processed by hot melt extrusion (HME). Formulations consisted of the hydrophilic hydroxypropyl methyl cellulose polymer (HPMCAS) and stearoyl macrogol-32 glycerides—Gelucire 50/13 (GLC) were processed with a twin screw extruder to produce solid dispersions. The extrudates characterized by X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC) and hot stage microscopy (HSM) indicated the presence of amorphous INM within the polymer/lipid matrices. In-line monitoring via near-infrared (NIR) spectroscopy revealed significant peak shifts indicating possible interactions and H-bonding formation between the drug and the polymer/lipid carriers. Furthermore, in vitro dissolution studies showed a synergistic effect of the polymer/lipid carrier with 2-h lag time in acidic media followed by enhanced INM dissolution rates at pH > 5.5.
Similar content being viewed by others

INTRODUCTION
The advances of high throughput screening and combinational chemistry resulted in an increased number of pharmaceutical active compounds which require further development to produce finish products (1). More than 40% of newly synthesized compounds are highly hydrophobic and water insoluble and thus it remains a key challenge for pharmaceutical scientists to improve the aqueous solubility of these poorly water-soluble drugs (1,2). Therefore, advanced formulation optimisation and pharmaceutical processing technologies are required to overcome solubility issues and thus enabling the oral delivery of poorly water-soluble drugs. A number of approaches and techniques have been investigated for dissolution enhancement including particle size reduction, salt formation, lipid-based formulation and solubilization (3), co-evaporation (4), hot spin mixing (5), roll-mixing or co-milling (6), freeze-drying (7), spray drying (8,9), supercritical fluid processing (SFP) (10) and hot melt extrusion (HME) (1,2,11). HME offers several advantages over the aforementioned technologies as it is cost efficient, solvent free and easy to scale up processes and it can be developed in a continuous manufacturing manner (12–16).
HME has been used extensively to enhance the dissolution rate of various poorly water-soluble drug candidates quite often leading to the formation of amorphous or molecularly dispersed drug forms (1,2) either in polymeric or lipidic carriers. In a traditional HME processing, mainly binary mixtures of powdered formulations (e.g. drug with polymers or lipids) are used. New novel processes of melt extrusion techniques involving two or more carriers (matrices) such as lipid and polymers, along with the active substance, can be proven advantageous over the traditional HME processing. This could be attributed to the simultaneous optimization and development a multi-layered extrudates at the end of the process (17). This emerging co-extrusion of polymer/lipid carriers could potentially enhance dissolution rates of poorly water-soluble drugs aiming to provide a synergistic effect (17). Hydrophilic polymers have shown significant increase of drug dissolution rates while several lipids are used to increase solubility of insoluble drugs. Recently, in order to ensure the product quality and in-line measurement of critical product parameters, Food and Drug Administration (FDA) launched a new initiative called ‘process analytical technology (PAT)’ to control and understand the manufacturing process (CDER PAT subcommittee, 2005). Near-infrared (NIR) spectroscopy is to date considered one of the most common techniques suitable for varied PAT applications, which have resulted in several studies in the pharmaceutical and nutritional fields. It has been reported that the NIR spectroscopy can be used to facilitate both quantitative and qualitative analysis such as drug crystallinity and polymer-drug or polymer-polymer interactions and the drug dissolution rates during an HME process (18–22).
Delayed release formulations are suitable vehicles for modification of the release of drug at specific target areas within the gastrointestinal tract. These formulations are usually designed to protect the drug against a gastric environment and prevent the release of the drug before reaching the target site. Thus, the delayed release formulations may have increased importance due to their distinctive advantages in both technological and therapeutic aspects such as modification of drug release, free dispersion in the gastro intestinal tract (23,24).
The aim of this study was to investigate for the first time the effect of a novel polymer/lipid combinations as carrier matrices during the extrusion processing and evaluate the synergistic effects on the dissolution rates of the water insoluble drug, indomethacin (INM). In addition, an in-line NIR probe was used to monitor critical product quality attributes (e.g. amorphicity or drug/polymer/lipid interactions)
MATERIALS AND METHODS
Materials
Indomethacin (INM) was purchased from Sigma Aldrich (Gillingham, Dorset, UK). Hydrophilic polymer hydroxypropyl methyl cellulose grade AQOAT AS-LF (HPMC) and Stearoyl macrogol-32 glycerides grade Gelucire 50/13 (GLC) were provided by ShinEtsu (Japan) and Gattefosse (Lyon, France), respectively. The HPLC solvents were of analytical grade and purchased from Fisher Chemicals (UK). All materials were used as received.
Hot Melt Extrusion Processing
Various compositions of INM/GLC/HPMCAS physical blends were mixed properly in 100 g batches as shown in Table I. A Turbula TF2 mixer (Basel, Switzerland) was used to blend drug/polymer/lipid physical mixtures properly for 10 min to achieve uniformity of the powder. Formulations were prepared at various INM/HPMCAS/GLC (%, w/w) ratios with 20–40% drug loadings (%, w/w ratios). The extrudates were processed with a twin screw extruder (Eurolab 16, ThermoFisher, Germany) in order to obtain drug/polymer/lipid extrudates with a diameter of 2 mm at 50–150 rpm using 1-kg/h feed rate. The screw configuration (L/D ratio 40) used has thee kneading zones with 10 kneading blocks each. The first kneading zone was configured as 30°, 40° and 60° angle while the second kneading zone was configured at 60° followed by a third kneading zone just before the die end at 90° angles. The collected extrudates were cut into small pellets (of 1 mm length) by using ThermoFisher (Germany) pelletizer. Extrusion process was optimized and performed at 120–140°C (as shown in Table I). The manufactured pellets were further micronized to obtain granules by using a cutter mill (Retsch, Germany) with 250 μm mesh.
The Hansen solubility parameters (δ) of both the drug and the polymer were calculated by using the Hoftyzer and van Krevelen method (25,26) according to the following equation:
Where
i = structural groups within the molecule, δ = the total solubility parameter, F di = molar attraction constant due to molar dispersion forces, F 2 pi = molar attraction constant due to molar polarization forces, E hi = hydrogen bonding energy, Vi = group contribution to molar volume.
Scanning Electron Microscopy Analysis
The spatial distribution of discreet chemical phases was evaluated using a cold-cathode field-emission gun scanning electron microscope (Hitachi SU8030 FEG-SEM, Japan). The samples were placed on double-sided carbon adhesive tabs and coated with carbon (Edwards 306 high vacuum carbon evaporation) before scanning electron microscopy (SEM) analysis. The surface analysis to characterize the morphology of the pellets was evaluated using SEM at a nominal magnification of 1000× (area = 0.127 mm × 0.095 mm). The accelerating voltage of the incident electron beam was set at 8 kV. This value was selected in order to minimize beam damage to the sample while maintaining adequate excitation.
Differential Scanning Calorimetry Analysis
The physical state of pure drug, pure lipid, physical mixtures and extrudates was examined by using Mettler-Toledo 823e (Greifensee, Switzerland). Samples were prepared with an initial weight of approximately 4–5 mg in sealed aluminium pan with lid. The samples heated from −30 to 200°C at 10°C/min scanning rate under nitrogen (N2) atmosphere. The GLC was heated from 0 to 80°C at the same scanning rate due to the low melting point.
Hot Stage Microscopy Analysis
Characterization of INM in the molten lipid/polymeric carrier was assessed using hot stage microscopy. During testing, an Olympus BX60 microscope (Olympus Corp., Center Valley, PA) with Insight QE camera (Diagnostic Instruments, Inc., Sterling Heights, MI) was used to visually observe samples, while a FP82HT hot stage controlled by a FP 90 central processor (Mettler Toledo, Columbus, OH) was used to maintain temperatures between 20 and 150°C. Images were captured under visible and polarized light using Spot Advance Software (Diagnostic Instruments, Inc.).
Powder X-Ray Diffraction
X-ray diffraction was used to assess the solid state of the extrudates. X-ray powder diffraction (XRPD) was performed for bulk drug, lipid, physical mixtures and extruded formulations. A Bruker D8 Advance in theta-theta mode was used with Cu anode at 40 kV and 40 mA, parallel beam Goebel mirror, Lynxeye Position Sensitive Detector with 3° opening and LynxIris at 6.5 mm, 0.2 mm exit slit and sample rotation was 15 rpm. The samples were scanned from 2° to 40° 2 theta with a step size of 0.02° 2 theta using 0.2 s per step counting time; 176 channels actives on PSD making a total counting time of 35.2 per step.
In-Line Near-Infrared Spectroscopy
Near-infrared spectrometry was performed during extrusion using an Antaris II NIR spectrometer (Thermo Scientific, UK) equipped with a halogen NIR source and InGaAs detector. The instrument used a fibre optic probe connected to the NIR spectrometer and was used for in-line monitoring. NIR spectra were collected in real-time during the entire extrusion process via a fibre optic diffuse reflectance probe. All NIR in-line spectra were continuously collected using the RESULT Integration software (Version 3.0 Thermo Scientific, UK). Each spectrum was acquired by averaging 32 scans with a resolution of 16 cm−1 over the 4000–10,000 cm−1 wavenumber range. The acquisition of a full spectrum took approximately 16 s. The spectral pre-processing was performed using the TQ Analyst software (Version 8.6.12 Thermo Scientific, UK).
In vitro Drug Release Studies
In vitro dissolution study was carried out by using a Varian 705 DS dissolution paddle apparatus (Varian Inc. North Carolina, USA) at 100 rpm and 37 ± 0.5°C. One hundred milligrammes equivalent of INM was used in each dissolution vessels. Seven hundred fifty millilitre of 0.1 M HCl solution was used as dissolution media to keep the pH 1.2 for the first 2 h, and then the pH was adjusted to 6.8 using a solution of 0.2 M of dihydrogen sodium orthophosphate (adjusted with NaOH). About 2–3 ml of solutions were withdrawn in predetermine time intervals for determining the release of INM. All dissolutions were performed in triplicates.
HPLC Analysis
The release of INM was determined by HPLC. An Agilent Technologies system equipped with a HYCROME 4889, 5 μm × 150 mm × 4 mm column at 276 nm was used for the INM HPLC assay. The mobile phase consisted of methanol/water/acetic acid (60:40:1, v/v/v). The flow rate was 1.5 ml/min and the retention time INM was about 4 min. The INM calibration curve (R 2 = 0.9998), at concentrations varying from 10 to 50 μg/ml, were used to evaluate all the samples with 20 μl injection volume.
RESULTS AND DISCUSSION
HME Processing
Extrusion processing was optimized to obtain the extrudates as yellowish pellets of 1 mm length of granules by feeding the pellets in a cutter mill (Fig. 1). The temperature profiles, screw speed and the feed rate were found to be the critical processing parameters (CPP) for the development of the extruded pellets or granules. During the optimisation of the process to manufacture INM controlled release formulations, a range of temperature profiles from 120 to 140°C were evaluated and subsequently processing conditions and outputs such as shear force, torque, feed rate and screw speed were optimized.

Manufacturing of INM pellets through HME processing
By keeping the final processing condition and the throughput of the drug/polymer/lipid powder blends, final batches of formulations were extruded as shown in Table I with a feed rate of 1 kg/h and a screw speed of 50–150 rpm. All conveyed extruded strands via a compressed air conveyor belt facilitated the feeding of the extrudates to the pelletizer attached at the end of the conveyor. The pelletizer resulted in the production of the pellets with 1 mm length. The length of the pellets was homogenously cut into pieces by pelletizer as the speed and processing conditions were adjusted.
All formulations were easily extruded to produce strands with INM loadings varying from 20 to 40% (w/w ratio). All materials were found to be miscible according to the Hansen solubility parameters (δ2) (25,26). The estimated solubility values of INM (22.8 MPa1/2), HPMCAS (25.30 MPa1/2) and GLC (19.8 MPa1/2) showed a difference of Δδ < 7.0 MPa1/2 suggesting complete miscibility with each other (Supp. Table 1). Although INM presents a high melting point, the polymer/lipid formulations were extruded at relative low temperatures due to the addition of the lipid, which acted as a plasticizer. However, as it can be seen in Table I, due to the requirements for increased drug loadings (30–40%) and high throughput (1 kg/h), the extrusion temperature profiles were adjusted to higher temperatures. The reason for these changes was to improve formulation processability and prevent the creation high torque in the extrusion barrels.
SEM Analysis
Surface morphology was examined by SEM for both the drug/polymer/lipid physical mixtures and the extrudates (EXT). As can be seen in Fig. 2a–c, the drug crystals appeared to be irregular in shape (needle type) and size. The physical mixture of the drug and carrier (lipid/polymer) showed the presence of drug in the crystalline form, which is distinguishable from the HME granules. In case of extrudates (Fig. 2d–f), it was difficult to distinguish the presence of drug crystals. This could be due to the presence of amorphous INM onto the lipid/polymeric matrices during the extrusion processing (more details in the DSC and XRD sections). Furthermore, the sieving analysis (data not shown) after the optimized micronisation of the pellets presented particle sizes lower than 500 μm for all extruded formulations ranging from 40 to 250 μm.

SEM images of (a–c) INM/HMPCAS/GLC physical mixture and (d–f) INM/HMPCAS/GLC extruded formulation (F1)
Thermal Analysis
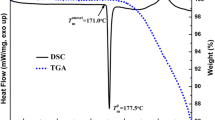
Differential scanning calorimetry (DSC) was used to determine the solid state of the drug in the extruded matrices as well as to outline any possible intermolecular drug-lipid-polymer interactions. Figure 3 shows the thermal transitions of pure INM, GLC and HMPCAS. The bulk INM showed an endothermic thermal transition at 161.18°C (ΔH = 115.33 j/g), which corresponds to its melting peak while GLC exhibited an endothermic sharp melting peak at 45.32°C (ΔH = 143.67°C). Similarly, the bulk HMPCAS pure showed a step change due to glass transition temperature at 128.21°C. The DSC analysis (Fig. 3) of the extruded F1-F2 formulations showed a shift of the HMPC glass transition at lower temperatures at 86.14 and 88.31°C, respectively, while the INM melting endotherm disappeared from the 161°C compare to the bulk substance. These results indicate the existence of INM amorphous state within the extruded matrices (1), and it is attributed to the possible drug-lipid-polymer intermolecular interactions during the extrusions. The same composition (F3) was processed at high screw rates and extrusion temperatures, and IND was also found in amorphous state (data not shown). However, different thermal events were recorded for the other extruded formulations (F4-F6) as shown in Table II. These formulations revealed melting endotherms due to the presence of crystalline drug in the extruded formulations (data not shown). In addition, the binary physical mixture (F6) showed a melting endotherm due to the presence of crystalline INM at 135.84°C (ΔH = 17.85°C). In contrast, the extrudates revealed two endothermic thermal events at 71.46°C corresponding to the melting of crystalline drug and another one at relatively higher temperature at 145.91°C due to the Tg of the amorphous polymer (Supp. Fig.1) indicating the existence of crystalline INM in the system.

DSC thermal transitions of bulk IND, HPMCAS, GLC and the extrudates
Hot stage microscopy (HSM) studies were conducted to visually determine the thermal transitions and the extent of drug melting within the polymer/lipid matrices at different stages of heating. Images taken using HSM under optical light are shown in Fig. 4. The bulk INM showed no changes up to 150–160°C, which is in agreement with the DSC results as thermal transition due to the melting of the drug occurred at 161°C. Similarly to the DSC thermograms, the INM/HMPC/GLC extrudates (F2) showed nominal API solubilization into the hydrophilic molten polymer/lipidic matrices up to 100–120°C and thereafter showed complete solubilization of the drug (Fig. 4). This could potentially be attributed to the intermolecular interactions or possibly the solubilization of the drug into polymer/lipid matrices during the extrusion processing.

HSM thermograms of INM/HMPCAS/GLC extrudates
X-Ray Powder Diffraction Analysis
The drug-polymer-lipid extrudates, including the bulk drug and physical mixtures of the same composition, were studied by X-ray analysis, and the diffractograms were recorded to examine the crystalline state of INM. As can be seen from Fig. 5a, the diffractogram of pure INM (average particle size below 100 μm) showed distinct intensity peaks at 10.17°, 11.62°, 17.02°, 19.60°, 21.82°, 23.99°, 26.61°, 29.37°, 30.32°, 33.55° degree 2θ. The physical mixtures of INM formulation showed identical peaks at lower intensities suggesting that the drug retained its crystallinity. In contrast, no distinct intense peaks due to INM were observed in the diffractograms of the extruded F1-F3 formulations. The absence of INM intensity peaks indicates the presence of amorphous INM into the polymer/lipid matrix complementing the findings from DSC. However, INM/HMPCAS (binary) extruded formulation (F6) revealed distinct intensity peaks of the drug at 12–17° 2θ and 22–27° 2θ values (Fig. 5b), simply indicates the presence of crystalline drug in the extruded formulation. Some additional XRD results of F4-F6 extruded formulations are provided in supplementary documents (Supp. Fig. 1a–d). All additional formulations showed characteristic peaks of the crystalline INM at relatively lower intensity which indicates the presence of crystalline drug in the extruded formulations. This could be due the averse or incomplete mixing during the extrusion processing while the presence of the lipid as third excipient has facilitated the homogenous mixing and thus the interactions. Hence, the presence of the lipidic carrier facilitated the formation of amorphous solid dispersions with high drug loading.

a X-ray diffractograms of INM pure and INM/HMPCAS/GLC physical mix (PM) and INM/HMPCAS/GLC extrudates (EXT). b X-ray diffractograms of INM/HMPCAS extrudates (EXT)
In-Line Near-Infrared Spectroscopy
In a continuous manufacturing system of any dosage forms, it is paramount to control and monitor quality attributes of the processed materials for enhanced product performance. In order to monitor critical quality attributes of the drug within the hydrophilic polymeric/lipidic matrices, a near-infrared (NIR) fibre probe was used as process analytical technology (PAT) tool and all the in-line data generated during the process were collected using the appropriate software package. NIR spectra of INM, HMPCAS, GLC pure were measured off-line to determine the characteristic vibrational bands attributable to the chemical structures of the bulk substance (Fig. 6a). Subsequently, in-line NIR spectra of the INM/HMPCAS/GLC extruded formulations were collected from different mixing zones during extrusion processing. The second derivative spectra, in Fig. 6b, shows a significant band shift between the NIR spectrum of bulk INM and the extruded formulations. The peak shifts at 4664, 5789, 6006 cm−1 are mainly due to the interactions between INM and the carriers. In addition, the increased peak intensity suggests the formation of strong intermolecular interactions. Interestingly, a new peak at 2549 cm-1, which does not exist in the bulk materials and the physical mixture, appeared in the extruded formulations. This peak corresponds to second overtone of the C = O stretching mode of the C–H···O = C hydrogen bonding between INM and probably HPMCAS. It is obvious that the second derivative peaks exhibited a frequency shift towards higher wave numbers compared to that of bulk substances. These changes in the spectrum could be attributed to the interaction (e.g. hydrogen bonding) (27) involved in the solid dispersion formation between the drug and carriers.

a NIR spectra of INM, GLC, HMPCAS and the extruded formulation (raw data and second derivatives) from 4000 to 1000 cm−1 region. b Second derivatives NIR spectra of INM, GLC, HMPCAS and the extruded formulation
In vitro Dissolution Study
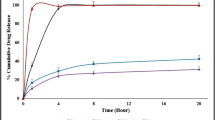
The dissolution behaviour of the processed formulations was assessed for extruded granules (average particle size 250 μm) and pellets (1 mm) in comparison to the bulk INM powder. As shown if Fig. 7 (and Supp. Fig. 3), a lag time with no drug release was observed for 2 h in acidic dissolution media for all of the extruded formulations. It appears that the polymer has a predominant effect and prevents drug release at low pH values. At higher pH values, INM was rapidly released within 1 h. The lag time was attributed to the pH dependency of HMPCAS, which dissolves at higher pH 5.5 (28).

In vitro release profiles of F1 (granules), F2 (pellets and granules) and F3 (granules), extruded formulations (n = 3, 37°C, paddle speed 100 rpm)
Interestingly, HMPCAS/GLC showed a synergistic effect resulting INM release in faster rates compared to those of INM/HMPC extrudates. It can be clearly seen that extruded polymer/lipid formulations affected significantly INM dissolution rates (Fig. 7). The synergistic effect of GLC/HMPCAS revealed a release of about 90% in 30 min while for HMPCAS/INM (F6), only 20% was released after 3 h in basic media. There was no significant difference observed between the extruded pellets and granules of F1-F3 investigated by calculating the similarity factor (f2). According to the FDA guidelines, release curves are considered similar when the calculated f2 is 50–100 (29).
However, the other extruded formulations F4-F6 showed relatively low drug release due to the presence of higher INM crystalline content in the extrudates (Supp. Fig 3). A careful look in the release profiles of these formulations indicates that high drug loadings screw speeds are critical parameters and facilitate increased IND crystalline content in the extrudates.
CONCLUSIONS
In this study, novel polymer/lipid formulations were processed via HME to increase the dissolution rates of the poorly water-soluble INM. The optimized process resulted in high INM loaded formulations in the form of pellets or granules. The solid state characterization revealed the amorphous state of INM in the extruded formulations while the in-line NIR monitoring showed significant peaks shifts (region 4600–6000 cm-1) indicating possible drug/polymer/lipid interactions. At low pH values (acidic media), the HMPC grade polymer presented a predominant effect and no drug release was observed while in alkaline media the polymer/lipid formulations showed a synergistic effect with rapid dissolution rates.
References
Maniruzzaman M, Rana M, Boateng JS, Douroumis D. Dissolution enhancement of indomethacin and famotidine processed by hot-melt extrusion. Drug dev Ind Pharm. 2013;39(2):218–27.
Grycze A, Schminke GS, Maniruzzaman M, Beck J, Douroumis D. Development and evaluation of orally disintegrating tablets (ODTs) containing ibuprofen granules prepared by hot melt extrusion. Coll Surf B Bio. 2011;86:275–84.
Laukamp EJ, Vynckier AK, Voorspoels J, Thommes M, Breitkreutz J. Development of sustained and dual drug release co-extrusion formulations for individual dosing. Eur J Pharm Biopharm. 2015;89:357–64.
Hong SW, Lee BS, Park SJ, Jeon HR, Moon KY, Kang MH, et al. Solid dispersion formulations of megestrol acetate with copovidone for enhanced dissolution and oral bioavailability. Arch Pharm Res. 2011;34(1):127–35.
Codoni D, Belton P, Qi S. Nanostructural analysis of water distribution in hydrated multicomponent gels using thermal analysis and NMR relaxometry. Mol Pharm. 2015.
Repka MA, Shah S, Lu J, Maddineni S, Morott J, Patwardhan K, et al. Melt extrusion: process to product. Expert Opin Drug Deliv. 2012;9(1):105–25.
Sekikawa H, Fukyda W, Takada M, Ohtani K, Arita T, Nakano M. Dissolution behavior and gastrointestinal absorption of dicumarol from solid dispersion systems of dicumarol-polyvinylpyrrolidone and dicumarol-beta-cyclodextrin. Chem Pharm Bull. 1983;31(4):1350–6.
Caron V, Tajber L, Corrigan OI, Healy AM. A comparison of spray drying and milling in the production of amorphous dispersions of sulfathiazole/polyvinylpyrrolidone and sulfadimidine/polyvinylpyrrolidone. Mol Pharm. 2011;8(2):532–42.
Wu K, Li J, Wang W, Winstead DA. Formation and characterization of solid dispersions of piroxicam and polyvinylpyrrolidone using spray drying and precipitation with compressed antisolvent. J Pharm Sci. 2009;98(7):2422–31.
Patil A, Chirmade UN, Trivedi V, Lamprou DA, Urquhart A, Douroumis D. Encapsulation of water insoluble drugs in mesoporous silica nanoparticles using supercritical carbon dioxide. J. Nanomed. Nanotech. 2011; A211.
Maniruzzaman M, Boateng JS, Snowden MJ, Douroumis D. A review of hot-melt extrusion: process technology to pharmaceutical products. ISRN Pharm. 2012;2012:436763.
Witzleb R, Kanikanti VR, Hamann HJ, Kleinebudde P. Solid lipid extrusion with small die diameters—electrostatic charging, taste masking and continuous production. Eur J Pharm Biopharm. 2011;77(1):170–7.
Crowley MM, Zhang F, Repka MA, Thumma S, Upadhye SB, Battu SK, et al. Pharmaceutical applications of hot-melt extrusion: part I. Drug Dev Ind Pharm. 2007;33(9):909–26.
Maddineni S, Battu SK, Morott J, Majumdar S, Repka MA. Formulation optimization of hot melt extruded abuse deterrent pellet dosage form utilizing design of experiments (DOE). J Pharm Pharmacol. 2014;66(2):309–22.
Mohammed NN, Majumdar S, Singh A, Deng A, Murthy NS, Pinto E, et al. Klucel™ EF and ELF polymers for immediate-release oral dosage forms prepared by melt extrusion technology. AAPS PharmSciTech. 2012;13(4):1158–69.
Chen M, Lu J, Deng W, Singh A, Mohammed NN, Repka MA, et al. Influence of processing parameters and formulation factors on the bioadhesive, temperature stability and drug release properties of hot-melt extruded films containing miconazole. AAPS PharmSciTech. 2014;15(3):522–9.
Vynckier AK, Dierickx L, Voorspoels J, Gonnissen Y, Remon JP, Vervaet C. Hot-melt co-extrusion: requirements, challenges and opportunities for pharmaceutical applications. J Pharm Pharm. 2013;66:167–79.
Saerens L, Dierickx L, Lenain B, Vervaet C, Remon JP, Beer TD. Raman spectroscopy for the in-line polymer–drug quantification and solid state characterization during a pharmaceutical hot-melt extrusion process. Eur J Pharm Biopharm. 2011;77(1):158–63.
Krier F, Mantanus J, Sacré PY, Chavez PF, Thiry J, Pestieau A, et al. PAT tools for the control of co-extrusion implants manufacturing process. Int J Pharm. 2015;458(1):15–24.
De Beer T, Burggraeve A, Fonteyne M, Saerens L, Remon JP, Vervaet C. Near infrared and Raman spectroscopy for the in-process monitoring of pharmaceutical production processes. Int J Pharm. 2011;417(1–2):32–47.
Islam MT, Maniruzzaman M, Moradiya HG, Trivedi V, Snowden MJ, Halsey S, et al. Development of sustained-release formulations processed by hot-melt extrusion by using a quality-by-design approach. Drug Del Trans Res. 2014;4:377–87.
Wahl PR, Treffer D, Mohr S, Roblegg E, Koscher G, Khinast JG. Inline monitoring and a PAT strategy for pharmaceutical hot melt extrusion. Int J Pharm. 2013;455(1-2):159–68.
Abdul S, Chandewar AV, Jaiswal SB. A flexible technology for modified-release drugs: multiple-unit pellets system (MUPS). J Control Release. 2010;147:2–16.
Kim YI, Pradhan R, Paudel BK, Choi JY, Taek H, Kim JO. Preparation and evaluation of enteric-coated delayed-release pellets of duloxetine hydrochloride using a fluidized bed coater. Arch Pharm Res. 2015. doi:10.1007/s12272-015-0590.
Maniruzzaman M, Morgan DJ, Pang J, Douroumis D. Drug-polymer intermolecular interactions in hot-melt extruded solid dispersions. Int J Pharm. 2013;443(1-2):199–208.
Maniruzzaman M, Islam MT, Moradiya HG, Halsey SA, Slipper IJ, Chowdhry BZ, et al. Prediction of polymorphic transformations of paracetamol in solid dispersions. J Pharm Sci. 2014;103(6):1819–28.
Kelly, Gough T, Dhumal RS, Halsey SA, Paradkar A. Monitoring ibuprofen–nicotinamide cocrystal formation during solvent free continuous cocrystallization (SFCC) using near infrared spectroscopy as a PAT tool. Int J Pharm. 2012;426:15–20.
ShinEtsu Pharma Excipients, http://www.se-pfmd.com/en/pharma_excipients/Shin_etsu_aquoat/index.pmode. Last accssed August 2015.
Guidance for Industry. Dissolution testing of immediate release solid oral dosage forms U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER). 1997.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editors: Feng Zhang and Michael A. Repka
Rights and permissions
About this article

Cite this article
Maniruzzaman, M., Islam, M.T., Halsey, S. et al. Novel Controlled Release Polymer-Lipid Formulations Processed by Hot Melt Extrusion. AAPS PharmSciTech 17, 191–199 (2016). https://doi.org/10.1208/s12249-015-0470-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0470-2