
Abstract
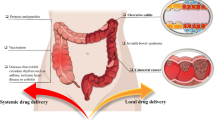
Colon-specific drug delivery systems (CDDS) are desirable for the treatment of a range of local diseases such as ulcerative colitis, Crohn’s disease, irritable bowel syndrome, chronic pancreatitis, and colonic cancer. In addition, the colon can be a potential site for the systemic absorption of several drugs to treat non-colonic conditions. Drugs such as proteins and peptides that are known to degrade in the extreme gastric pH, if delivered to the colon intact, can be systemically absorbed by colonic mucosa. In order to achieve effective therapeutic outcomes, it is imperative that the designed delivery system specifically targets the drugs into the colon. Several formulation approaches have been explored in the development colon-targeted drug delivery systems. These approaches involve the use of formulation components that interact with one or more aspects of gastrointestinal (GI) physiology, such as the difference in the pH along the GI tract, the presence of colonic microflora, and enzymes, to achieve colon targeting. This article highlights the factors influencing colon-specific drug delivery and colonic bioavailability, and the limitations associated with CDDS. Further, the review provides a systematic discussion of various conventional, as well as relatively newer formulation approaches/technologies currently being utilized for the development of CDDS.
Similar content being viewed by others


INTRODUCTION
Colon-targeted drug delivery has been the focus of numerous studies in recent years due to its potential to improve treatment of local diseases affecting the colon, while minimizing systemic side effects. Some examples of disease states which impact the colon include Crohn’s disease (CD), ulcerative colitis (UC), and irritable bowel syndrome (IBS) (1). Some of the frequently used drugs for the treatment of these ailments include sulfasalazine, dexamethasone, hydrocortisone, metronidazole, prednisolone, and others (2).
The delivery of these drugs specifically to the colon without being absorbed first in the upper gastrointestinal (GI) tract allows for a higher concentration of the drug to reach the colon with minimal systemic absorption (3). The colonic contents have a longer retention time (up to 5 days), and the colonic mucosa is known to facilitate the absorption of several drugs, making this organ an ideal site for drug delivery (3,4). A drug can be delivered to the colon via the oral, or the rectal route. Oral dosage forms are the most preferred delivery route for colon-specific delivery due to their convenience (4). Oral dosage forms also allow for a greater degree of flexibility in their manufacturing, design, improved patient adherence, relatively safe administration, and they do not require sterile preparation (2). Direct rectal delivery of drugs is challenging with respect to targeting a drug to specific sites within the colon (2,4). Additionally, the extent of drug distribution varies for different rectal dosage forms depending on their spreading capacity and retention time.
The success of a colon-specific drug delivery system (CDDS) depends on the drug’s physico-chemical properties, the type of delivery system, all other factors which may influence the GI transit time, as well as the degree of interaction between the drug and the GI tract (1). It is essential for oral CDDS to protect the drug from being released in the stomach and small intestine (4). Thus, the approaches used in developing a CDDS are aimed at delaying the drug release until the system reaches the colon, with some strategies demonstrating better success than others. Several marketed formulations report the use of a combination of conventional and newer approaches discussed above (Table I).
DRUG CRITERIA FOR A COLON-SPECIFIC DELIVERY
Drugs which are meant to be incorporated into a colon-specific delivery system should fulfill one or more of the following physico-chemical/therapeutic criteria (1,4). First, these drugs should exhibit local effects in the colon to treat intestinal diseases. Peptide drugs like amylin and non-peptide drugs such as oxyprenolol are some examples of agents with these effects. Secondly, these drugs may demonstrate a sub-optimal absorption in the upper gastrointestinal tract. This includes antianginal drugs such as isosorbide dinitrate. Agents used in the treatment of colon or rectal cancers (e.g., 5-flurouracil and capecitabine) are also ideal candidates for CDDS. The remaining criteria include a high likelihood of the drug’s degradation in the stomach by the acidic environment or enzymes (e.g., peptide drugs like insulin and gonadorelin), or a high risk for first-pass metabolism (e.g., corticosteroids).
LIMITATIONS OF COLONIC DRUG DELIVERY
The development of a colon-specific drug delivery system is associated with specific limitations and challenges. A predominant and an obvious challenge is the fact that the colon is located in the distal part of the gastrointestinal tract (GIT). An orally administered dosage form has to traverse the entire alimentary canal in order to reach the target site. The GIT physiology is complex and has a wide range of pH values, fluid volumes, and transit times. Moreover, the presence of food and metabolic enzymes also increases the physiological complexity. These factors are an obstacle to the reliable and efficient delivery of drugs to the colon. Another factor is the drug solubility. Due to a low colonic luminal fluid volume, higher viscosity, and a neutral pH, the solubilization of the drug could be a rate-limiting factor for colonic absorption. Finally, maintaining the stability of the drug in the colon can be a matter of concern. The non-specific interactions of the drug with the colonic content e.g., dietary residues, intestinal secretions, mucus, or fecal matter can have a negative influence on the stability of the drug (5). In addition, the colonic bacterial enzymes may also degrade the drug, rendering it ineffective.
FACTORS INFLUENCING COLON-SPECIFIC DRUG DELIVERY AND COLONIC BIOAVAILABILITY
Several factors may influence the formulation/development of a colon-specific drug delivery system (CDDS) and the colonic bioavailability of the drugs (2,6). Some of these factors are briefly discussed below.
Anatomical/Physiological Factors
The human large intestine is approximately 1.5-m long and forms the colon (ascending, transverse, and descending), with a small distal part forming the rectum. The colon is 2–3 in. in diameter, and its lumen is lined with mucus. The physiology of the colon differs significantly from other segments of the gastrointestinal tract (GIT) (Table II). Moreover, the physiology and the physical properties of the colonic contents also differ between the ascending, transverse, descending, and sigmoidal colon. In addition, there exists variability in movement of food and dosage forms across the colon, which may present a challenge in the development of colonic drug delivery systems (7). Another physiological factor that affects colonic drug delivery and bioavailability is the variation in pH of the GIT. Significant intra- and inter-subject variability in the pH of the GIT have been observed between disease states, fasted/fed states, sexes, and ages in humans (8–10). Factors such as the viscosity and volume of colonic fluids, the presence of microbial enzymes, and the resulting colonic metabolism are other important factors influencing CDDS performance which is discussed further.
Intestinal-Colonic Transit Time
The intestinal-colonic transit time plays an important role in the performance of CDDS and the colonic bioavailability of drugs. The transit times are also influenced by colonic disease states such as UC and CD. Patients with UC are known to have shorter colonic times (~24 h) compared to healthy subjects (~52 h) (11). Similarly, Rana et al. showed that in patients with IBD, the orocecal transit time was delayed (12). The transit of dosage forms generally depends on the time of administration, presence/absence of food, and the type of dosage form. Stubbs et al. studied the effect of dawn and dusk on the motility of dosage forms in the colon. The results showed that colonic transit was delayed during sleep, and larger dosage forms, e.g., capsules transited faster compared to smaller dosage forms, e.g., dispersed particles (13).
Colonic Fluid Volume
The average human food intake is approximately 1.5 kg/day and mainly consists of undigested proteins, carbohydrates, and fats. These food components may serve as substrates for the microbial enzymes in the colon (14). The colon has a high water-absorbing capacity and can absorb ~90% of the water entering the colon (15). The colonic fluid volume is calculated to be in the range of 1–44 ml with an average volume of approximately 13 ml (16). Due to this low volume of colonic fluids, the dissolution of drugs from the dosage forms becomes challenging and may affect the local drug bioavailability.
Viscosity of Colonic Luminal Contents
Due to a higher water-absorbing capacity, the viscosity of the colonic luminal contents is higher than upper GIT contents and presents a challenge for the dissolution of CDDS. Moreover, the viscosity of the contents progressively increases as it transits from the ascending colon towards the descending colon, resulting in a reduced drug dissolution and mucosal absorption (17). Viscosity also influences the penetration of the drug into the disease-causing bacteria in the colon. The mobility of bacteria in the colon has been shown to be dependent on the viscosity of colonic contents (18).
Colonic pH
The pH varies significantly between different regions of the GIT. For example, the pH of gastrointestinal contents can be as low as 1 to 2 in the stomach and rise to 7.5 in the distal small intestine (19,20). The pH then declines from the end of the small intestine to the colon and gradually increases once again in the colon (19,20). The pH of the colon may be influenced by a carbohydrate rich diet. This is due to the fermentation of polysaccharides by colonic bacteria and subsequent formation of short chain fatty acids (21). Similarly, polysaccharide-based drugs may also alter colonic pH. Laxative drugs like lactulose are known to be fermented by colonic bacteria to produce lactic acid and reduce colonic pH (22). Gastrointestinal disease states such as UC have also been found to influence the colonic pH (23). The pH of the colon affects the pharmacokinetic and pharmacodynamic behavior of a CDDS by influencing the solubility of drugs in the colonic fluid. Moreover, if one or more components of the dosage form are pH-sensitive, for example, a pH-sensitive coating membrane, the effect of colonic pH is even more pronounced on the drug release.
Colonic Enzymes and Metabolism
The colon is known to consist of over 400 different species of aerobic and anaerobic microorganisms like Escherichia coli and Clostridium species, respectively (24). These bacteria contain several hydrolytic and reductive metabolizing enzymes (25). The colonic enzymes catalyze a range of reactions, including the metabolism of xenobiotics (e.g., drugs) and other biomolecules (e.g., bile acid), deactivation of harmful metabolites as well as carbohydrate and protein fermentation (26). Polysaccharides such as chitosan, guar gum, pectin, etc., are commonly employed as release rate-controlling components in colon-targeted dosage forms. These polysaccharides are known to be resistant to gastric and intestinal enzymes, but are metabolized by anaerobic bacteria in the colon (27–29). Drugs are also known to be susceptible to biotransformation by colonic enzymes. The metabolism of drugs by the colonic enzymes may result in the formation of metabolites that are pharmacologically active, inactive, or sometimes even harmful (30,31). Formation of a pharmacologically active metabolite by the colonic metabolism of drugs is a commonly used “prodrug” approach for the colon-specific drug delivery systems (32).
Formulation Factors
The formulation factors that influence colonic drug delivery and bioavailability includes physicochemical properties of the drugs, the dose, and the dosage form factors. Due to the lower amount (1–44 ml) of colonic fluid available for dissolution, the solubility and the dose of a drug become important factors for its colonic bioavailability. Although the highly potent drug budesonide (dose, 9 mg) has a lower aqueous solubility, it is absorbed well in the colon and is used successfully in the treatment of UC (33). Mesalamine has a significantly higher solubility (3.64 mg/ml) compared to budesonide (0.24 mg/ml); however, it also has a significantly higher dose (4.8 g daily) which becomes a rate-limiting factor for its colonic absorption (34). Finally, the technology used in the dosage form development can also influence the colonic bioavailability of drugs. Useris® and Entocort EC® are currently approved budesonide products for the treatment of UC and CD, respectively (35). Useris® is a multi-matrix (MMX)-based delayed-release tablets, which ensures the drug release in the colon, while Entocort EC® is a capsule which releases the drug in the ileum to treat CD.
CONVENTIONAL APPROACHES FOR ACHIEVING COLONIC DELIVERY
Prodrugs
Prodrugs are inactive derivatives of a drug molecule which release the active ingredient once they are hydrolyzed by enzymes such as those in the colon (2,36). In order to optimize drug delivery specific to the colon, the extent of this hydrolysis should be minimal in the upper portions of the gastrointestinal tract and much more extensive in the colon. Azo conjugates are one of the most researched groups of compounds that fall into this category (37,38). However, this is not a very flexible method because it relies on the functional groups of the drug molecule (4). Kim et al. synthesized a prodrug of metronidazole, which was metabolized to the active drug, metronidazole when placed in rats’ cecal contents (39). Unlike metronidazole, this prodrug did not metabolize in the small intestine, and the systemic absorption of this prodrug was also found to be much lower compared to that of oral metronidazole (39). In another study, Kim et al. prepared a prodrug of metronidazole, using a sulfate group, and showed that that this formulation remained intact in the upper intestine, but was cleaved in the presence of rat cecal contents and active metronidazole was released. Similar to the first prodrug, much less of the conjugated prodrug was degraded and absorbed in the small intestine compared to the active drug after oral administration. Therefore, a minimal amount was absorbed into systemic circulation (40). Vaidya et al. utilized the prodrug approach by conjugating metronidazole with pectin and compared the drug release from this formulation to that from pectin microspheres which physically entrapped the drug (41). The pectin-metronidazole (PT-ME) prodrug showed significantly reduced drug release in the upper GIT compared to pectin microspheres containing metronidazole. In vitro and in vivo studies revealed that conjugating the drug to pectin can successfully target its delivery to the colon since no drug release occurred at an acidic pH from the PT-ME prodrug while nearly 100% of the metronidazole physically entrapped in pectin microspheres was released in this same environment. A significantly higher fraction of the drug was released from the PT-ME prodrug in the colon (41).
Another way to assist a drug formulation to remain intact as it passes through the stomach and small intestine is for it to be covalently linked to a carrier. A drug can bind to carrier molecules such as cyclodextrin, glucuronide, dextran, and amino acids. It can also be linked to a carrier through an azo bond. All of these bonds are broken down by colonic bacteria or enzymes (3). Modasiya et al. studied the use of sodium alginate (Na-Alg) and hydroxypropyl methylcellulose (HPMC) as carriers for the successful delivery of matrix, enteric-coated, and compression-coated tablets of curcumin to the colon (42). The in vitro results showed that the drug release was rapid from the matrix, and from the enteric-coated tablets in conditions representing the stomach and small intestine. It was also observed that increasing HPMC levels significantly restricted the release of curcumin in the upper GIT and assisted in the delivery of curcumin specifically to the colon.
Colon-Specific Biodegradable Delivery Systems
The colon contains many species of anaerobic bacteria which obtain their energy by fermenting substrates such as polymers which have not yet been digested. Bacteroides, eubacteria, clostridia, enterococci, and enterobacteria are some examples of these colon-specific species, and they produce numerous enzymes such as glucuronidase, xylosidase, nitroreductase, and azoreductase to ferment these polymers (10,43,44). Since these enzymes are localized to the colon, this appears to be a more promising approach for colon-specific delivery (4,45). Polymers used in the development of CDDS can be chemically modified, and these modifications can influence the extent of enzymatic degradation. For example, Roos et al. synthesized the acetyl derivative of guar gum (AcGGM) and used this polymer to make a hydrogel of bovine serum albumin (BSA) (46). They observed that the rate of hydrolysis for modified AcGGM by β-mannase was affected by its degree of substitution (DS). As DS increased, the hydrolysis rate actually decreased, indicating that the side chain hindered the enzyme. On the other hand, the addition of β-mannase significantly enhanced BSA release, with 95% of the BSA released after 8 h in the presence of this enzyme, and only 60% was released in its absence. Azo-aromatic polymers, which are among the most researched groups of compounds used as prodrugs, are susceptible to degradation by azoreductases (18,19). Therefore, they can be used to coat the drug molecules such as peptides to protect them from degradation by peptidases in the stomach and small intestine while still permitting drug release in the colon. Hita et al. carried out a study in which metronidazole capsules were coated with a film of azo-aromatic polymers, and polymers sensitive to pH. The in vitro and in vivo results revealed that the microbiota specific to the colon degraded these polymers and released the metronidazole locally in the colon (47).
Matrix-Based Systems
Another approach towards colon-targeted drug delivery includes embedding the drug in polymer matrices to trap it and release it in the colon. These matrices can be pH-sensitive or biodegradable. Ahmad et al. developed matrix tablets containing metronidazole using a natural polymer called Assam Bora rice starch (48). The prepared tablets were evaluated by in vitro drug release studies using 0.1 N HCl, phosphate buffer with a pH of 7.4, and goat cecal content. The results showed that the tablets exhibited a sustained release of the drug in the alkaline environment, which was believed to be due to erosion and dissolution of the polymer during its prolonged exposure to this environment (48). The release of the drug, however, was observed throughout the GIT. This indicated that these matrix tablets were not colon-specific delivery systems, but rather controlled-release systems.
Timed-Release Systems
Timed-released formulations are based on the drug being released in the colon after a specified amount of time (3). This approach is dependent on the transit time through the small intestine, which is and known to vary between 3 and 4 h (2,49). Gastric emptying time is inconsistent between individuals and also fluctuates depending on food intake (50). Additionally, diseases associated with the colon, such as irritable bowel syndrome and ulcerative colitis can influence transit time through the colon (4). Gazzaniga et al. used a combination of pH-sensitive polymers and a timed-release approach to achieve colon-specific delivery (51). A formulation consisting of a drug-containing core enclosed within three polymeric layers (a hydrophilic layer sandwiched between two pH-sensitive layers) was developed. The in vitro evaluation results revealed a sustained drug release due to pH protection and hydrogel formation.
Bioadhesive Systems
Bioadhesive systems allow a formulation to remain in contact within an organ, in this case the colon, for a long period of time to assist poorly absorbable drugs to be absorbed (48,52). Some of the polymers which have been explored as bioadhesive components for these systems include polycarbophils, polyurethanes, and polyethylene oxide (3). Ahmad et al. used Assam Bora rice starch to develop a bioadhesive microsphere (BAM) for targeting the delivery of metronidazole to the colon (48). These BAMs were found to have higher retention time in the colon, and helped increase absorption of the drug in the colon. The in vitro drug release studies showed that only 10–12.5% of the metronidazole was released on conditions representing the stomach and less than 25% was released in a simulated small intestine. However, over 90% of the drug was rapidly released in cecal content. Additional in vivo studies showed that the drug was only released when the BAM reached the colon and was equally pharmacologically effective compared to marketed formulations.
Multiparticulate Systems
Multiparticulate systems have a smaller particle size compared to single-unit systems, and studies have shown that they can reach the colon more quickly since they pass through the GI tract more easily (1,53). Microspheres are one example of a multiparticulate system that can be loaded with a drug for colonic delivery. Microspheres that are prepared using biodegradable components can be taken up by macrophages (1). Chourasia et al. carried out in vitro drug-release studies on a metronidazole multiparticulate system consisting of cross-linked chitosan microspheres coated with pH-sensitive Eudragit® polymers (52). Metronidazole was released only after Eudragit® polymers dissolved in the alkaline pH of the small intestine. Since there was increased drug release in the presence of rat cecal contents, chitosan was thought to be susceptible to degradation by colonic enzymes. Vaidya et al. developed a multiparticulate system in which microspheres of the polysaccharide pectin were coated with a pH-sensitive polymer, Eudragit® S 100 (41,54). The in vitro drug release studies showed that no metronidazole was released at the acidic pH of the stomach. However, once the system was in a more alkaline environment of the colon, metronidazole was released continuously. Metronidazole release was found to be even more significant in the presence of rat cecal contents indicating that this system was biodegradable, in addition to being pH-sensitive. Furthermore, the metronidazole concentration in various parts of the GIT as shown through in vivo studies also demonstrated the ability of this system to target this drug specifically to the colon. Perera et al. synthesized and evaluated microparticles based on a pectin-4-aminothiophenol (Pec-ATP) conjugate and observed that these particles were much more stable in vivo, than unmodified pectin microparticles (55). These particles thus appeared to be a better option for colon-targeted delivery based on this study. In a recent study, Liu et al. developed guar gum base microspheres for the colonic delivery of budesonide (56). The in vitro release studies showed that these microspheres extended the release of budesonide over 24 h. The in vivo evaluation of the colon targeting and pharmacokinetic studies showed that the prepared budesonide microspheres effectively delivered budesonide to the colon in high concentrations.
Beckert et al. synthesized and evaluated a multiparticulate system in which the drug in the core of the formulation consisted of two forms of pellets (57). The first type of pellet, pellet A, contained an inner polymer coating which allowed the drug to be released continuously and an outer polymer which broke down only at a pH greater than 5.5. The polymer coating on pellet B limited the drug release to less than 20% after 6 h at pH 6.8. However, more than 50% of the drug was released within the same time period at pH 7.2. Therefore, the combination of these two forms of pellets with their polymer coatings was found to be promising for a targeted drug delivery to the colon. Agarwal et al. in a recent study prepared and characterized calcium alginate-carboxymethyl cellulose (CA-CMC) beads for colon-specific oral drug delivery (58). The researchers explored the combined properties of CA-CMC, i.e., pH sensitivity, degradation by colonic microflora, and preferential colonic mucoadhesivity in designing colon-specific delivery of 5-flurouracil, an anticancer drug. The in vitro drug release results showed that the CA-CMC beads were able to significantly extend the release of the drug beyond 24 h. Additionally, the CA-CMC beads were demonstrated to have a significantly high mucoadhesiveness at colonic pHand degrade in the presence of colonic microflora.
The use of nanoparticles as carriers for orally administered drugs targeted to the colon has also been reported previously (59–61). Studies have shown that these nanoparticulate systems improve the bioavailability of these drugs due to their increased surface area and thus increased contact with biological surfaces. Nanoparticles are also taken up by macrophages at inflamed regions of the colon, which allows the system to remain at the target site for a prolonged period of time. However, the nanoparticulate systems must be protected from being taken up by Payer’s patches or degraded by enzymes before reaching the colon in order for this approach to be successful. Calabrese et al. prepared nanotechnology-based hybrid formulations of metronidazole and K10-montmorillonite (MMT-K10) clay, and carried out kinetic and equilibrium studies to determine the release of MNE from this clay (60). The results showed that these newer nanoparticle formulations allowed for longer action in the colon. It was also found that an enteric coating is unnecessary due to low drug release in the simulated gastric fluid.
Polysaccharide-Based Delivery Systems
Polysaccharide-based delivery systems have several advantages and are therefore becoming a popular option for colon-specific delivery of drugs. Some of the advantages with polysaccharide use include availability, easy modifications, stability, safety, and biodegradability (4). Mundargi et al. compared several polysaccharides for their usefulness in colon-targeted metronidazole delivery (62). The results showed that the release rate of metronidazole does depend on the nature and the concentration of the polysaccharide used in the formulation. Gauri et al. used various amounts of xanthan gum and guar gum to prepare matrix tablets of metronidazole (63). The in vitro evaluation of tablets in 0.1 N HCl, pH 7.4 phosphate buffer, and pH 6.8 phosphate buffer with 4% w/v rat cecal content showed that the amount of drug released from the matrix tablets during the first 5 h, which represented time spent in the stomach and small intestine, ranged from 12 to 33%. It was also observed that the increasing xanthan gum content in the matrix tablets delayed the drug release and caused them to be more susceptible to colonic enzymes.
The use of a combination of polysaccharides in CDDS has been found to be more effective for achieving colon-specific delivery compared to the use of a single polysaccharide. Cellulose derivatives are frequently used in combination to develop these delivery systems because cellulose is not absorbed systemically when administered orally. There are two groups of cellulose esters which can be used in drug formulations. Non-enteric cellulose esters such as cellulose acetate are insoluble in water and their solubility is independent of pH. These can be used in insoluble, permeable coatings. The enteric cellulose esters such as cellulose acetate phthalate (CAP) and hydroxypropyl methylcellulose phthalate (HPMCP) have solubilities which are pH-dependent. They are insoluble in highly acidic conditions, but when the pH reaches a certain range they dissolve. The pH at which the polymer dissolves varies depending on the extent of esterification. Some examples of carbohydrate mixtures which have been studied include pectin-HPMC, chitosan-HPMC, chitosan-pectin, guar gum-chitosan, and dextran-chitosan (64–66).
As mentioned in the previous sections, polysaccharides such as pectin, chitosan, chondroitin sulfate, galactomannan, and amylose are ideal materials for achieving colon-specific delivery because they can be degraded by the colonic enzymes and are harmless to the organisms. The use of these polysaccharides in thin film coatings is believed to have the potential to allow for increased drug delivery to the target regions at a faster rate, compared to other formulations which utilize these materials within matrix systems or as compression coatings. Pectin is a hydrophilic polysaccharide which can modify drug release due to its gelling ability. An insoluble polymer such as ethyl cellulose (EC) is often mixed with the pectin in the coating layer to help reduce water permeability and protect the drug core (67). Wakerly et al. mixed an aqueous dispersion of EC (Surelease®) with pectin to coat tablets of paracetamol (68). These film coatings had various pectin/EC ratios, and after evaluating the coated tablets in vitro, the results showed that the rate of drug release increased as the amount of pectin in the film increased. The drug diffused through the EC as well as through pores which formed in the film coating after pectinolytic enzymes degraded the pectin (68).
Colon Targeting by Coatings
Incorporating the drug in the pH-sensitive polymers allow for delayed release by protecting the active ingredient from the acidic pH of the stomach and proximal small intestine. These polymers then break down in the more basic pH of the terminal ileum, thus providing a targeted drug delivery to the colon (1,3). Although the solubility of these polymers increases as pH rises, there are some disadvantages to this approach. The pH fluctuations along the GIT can cause the formulation to dissolve early in the small intestine and the lag time can be too long at the ileo-cecal junction and ascending colon (4). Some examples of commonly used pH-sensitive polymers in the design of colon-targeted drug delivery systems include methacrylic-acid based polymers, also known as Eudragit® (2,36).
Enteric-soluble polymers are resistant to dissolution in the acidic environment of the stomach, but can dissolve at the higher pH values of the intestine. These polymers have been studied extensively for their use as coatings in formulations intended to deliver active pharmaceutical ingredients specifically to the colon (Fig. 1). Polymers based on poly-methacrylate such as Eudragit® L and Eudragit® S have frequently been used for this purpose, and each one has its own unique pH value at which it dissolves. These two polymers have been mixed in different ratios to form a coating with an optimized dissolution rate. Additionally, coatings with these polymers are designed to be relatively thick to prolong their dissolution, and provide a controlled or an extended drug release (69). Obitte et al. investigated the capability of the hydrophobic polymers Landolphia owariensis latex (LOL) and Eudragit® L-100 to control the release of metronidazole aimed at colon targeting (70). The in vitro dissolution studies showed that drug release increased with increasing pH. The Eudragit® L 100 and LOL also demonstrated an additive effect on delaying and then increasing drug release at pH 7.4 over an extended period of time.

Schematic representation of the cross-section of the enteric-coated colon-targeted drug delivery system
In addition to enteric-soluble polymers, acid-soluble polymers have also been investigated as potential agents to be used in colon-targeted formulations. The pH of the proximal large intestine decreases in those with IBS. For example, the colonic pH is typically 6.4–7.0 for a healthy person, but can drop to 2.3–4.7 with someone who has UC (71). Leopold et al. developed dexamethasone minitablets coated with the acid-soluble polymer Eudragit® E and found that the Eudragit® E coating rapidly dissolved in the buffers at pH 2.0–5.0 allowing the drug to be released within 10–50 min. However, the extent to which the drug released was delayed in the pH 6.8 buffer depended on the composition of the drug core as well as the coating thickness. (69,71).
Compression-coating (tablet-in-a-tablet), also known as “dry coating” is a tablet coating technique where the core tablet (containing the drug) is coated with a coating-excipient (powder) on a tablet press. This technique has gained interest in the formulation development in recent years due to the dry nature of dosage form development, i.e., avoiding the process complexities and stability challenges associated with spray coatings (wet, hot). Several researchers have explored this technique for the development of colon-specific oral solid dosage formulations.
Yassin et al. applied a granulated chitosan coating to a colon-targeted tablet formulation of 5-fluorouracil using compression-coating with a goal of targeting this drug specifically to the colon for a more effective treatment of colon cancer with less toxic side effects (72). The in vitro evaluation of the formulation showed that increasing the thickness of the coating resulted in a progressive decrease in the drug release at acidic pH. Additionally, the in vivo studies showed that this formulation did not break down until it reached the large bowel. Recently, Kadiyam et al. developed an almond-gum, matrix-based colonic drug delivery system of tramadol HCl, compression-coated with Eudragit® S100 (73). The study results showed that compression-coated tablets successfully delayed the release of tramadol HCl over 24 h. The in vivo X-ray imaging studies in rats revealed that the compression-coated tablets efficiently delivered the drug to the colon without being disintegrated in the upper GIT.
Pulsatile drug delivery systems are time-dependent formulations that are designed to release the drug after a predictable period known as the “lag phase” (74). The pulsatile systems which are currently being studied do not depend on the different environmental conditions of the GIT for the drug release. Most of these orally administered systems consist of a drug-containing core, coated with a polymer (74). Film coatings used for pulsatile delivery include rupturable, permeable, and semipermeable coatings.
Rupturable film coatings allow a drug to be released after undergoing a timed disruption caused by hydrostatic pressure within the core (69). Since these polymeric films are permeable, an influx of water and subsequent swelling of the hydrophilic polymers can initiate the disruption. Permeable film coatings allow water to pass through and dissolve the drug-containing core, but the polymeric coating itself is insoluble. These coatings do not rupture after exposure to an aqueous medium because they are permeable and resistant to dissolution. Additionally, the materials within these coatings do not expand after an influx of water. Since it takes time for the drug to diffuse out from the core after dissolving, this results in a lag phase before drug release occurs (69).
Another type of time-dependent coating is a semipermeable film coating which is similar to permeable coatings in that they are permeable to water (75). However, these coatings are impermeable to solutes. Water moves into the tablet core of the formulation due to osmotic pressure, and when the hydrostatic pressure within the system exceeds the osmotic pressure after a programmed lag phase, small orifices in the outer membrane allow the drug which has dissolved in the aqueous medium to be pumped out (76).
INTEGRATED APPROACHES FOR ACHIEVING COLONIC DELIVERY
In recent years, several integrated approaches have been explored to achieve colon-specific drug delivery. These approaches utilize physiological factors such as luminal pressure, or physical phenomena such as osmotic pressure in the design of the delivery systems.
Pressure Controlled Delivery
Peristaltic motion causes the luminal pressure of the large intestine to increase more than that of the small intestine because its contents are more viscous due to the reabsorption of water (77). Several studies have been carried out to utilize the colonic luminal pressure to develop colon-specific drug delivery systems. Takaya et al. developed capsules that deliver a drug to the colon based on luminal pressure (78). Although these systems allow for drugs to be delivered to the colon rather than the small intestine due to higher colonic pressure, reabsorption of water from the colon causes its content to be highly viscous which may become an obstacle for site-specific delivery (4,79).
Osmotic Controlled Delivery
Although the concept of osmotic controlled drug delivery has been around for several years, the applications of this technology in the design of colon-specific oral dosage forms have gained popularity only in the 10–15 years. The OROS-CT is an example of a system regulated by osmotic pressure. It consists of a hard gelatin capsule which dissolves in the pH of the small intestine and allows water to enter the unit. This then causes it to swell and the drug is forced out (3). Within each capsule there can be as many as 5–6 units, and each unit is surrounded by a drug impermeable enteric coating which prevents water from entering in the acidic environment of the stomach (Fig. 2). However, this coating dissolves and the water enters once the capsule enters the higher pH of the small intestine. Within the enteric coating there is a semipermeable membrane which encompasses an osmotic push compartment as well as a drug compartment. The water causes the push compartment to swell and forms a gel in the drug compartment that is forced out of an orifice through the membrane next to the drug compartment. The rate at which the drug flows out depends on the rate at which water enters. To prevent drug release in the small intestine, these systems can also be designed such that there is a lag time between when the enteric coating dissolves and the drug is released (80).

Schematic representation of the cross-section of the OROS-CT colon-targeted drug delivery system
Pulsincap Systems
Time-dependent systems are not always ideal for delivering drugs to the colon due to variability in the gastric emptying time and the changes in gastrointestinal transit due to peristalsis or disorders such as IBS. Therefore, the integration of a timed-release system with pH-sensitive properties can be beneficial in achieving colon-targeted delivery. A pulsincap system is one example of a formulation that utilizes both these techniques (81,82). The system consists of a water insoluble capsule body containing the drug, a hydrogel plug which seals the opened end of this capsule body and a water soluble cap which covers the hydrogel plug (Fig. 3). Additionally, the capsule is coated with an acid insoluble film coating which prevents the drug from being released in the stomach. The hydrogel plug begins swelling when this enteric coating dissolves in the small intestine. The swelling of the plug allows for a lag time before the drug is released and the amount of lag time depends on the length of the plug and the extent at which it is inserted (81). Abraham et al. developed a pulsincap system in which they tested several polymers as the plug material. The formulations were tested at pH 1.2 for 2 h to simulate gastric fluid, pH 7.4 for 3 h to simulate intestinal fluid, and pH 6.8 for 7 h to simulate the colon. The study found that no significant drug release occurred within 5 h from the start of the experiment, and it was concluded that this modified pulsincap system can successfully target metronidazole to the colon (81).

Schematic representation of the mechanism of the pulsincap colon-targeted drug delivery system
CONCLUSIONS
The development of colon-targeted oral drug delivery systems has gained increasing interest among formulation scientists in recent years. As discussed above, colon-specific drug delivery systems provide significant therapeutic benefits to the patients in terms of safety, efficacy, and patient compliance. Factors including the physicochemical characteristics of the drug, formulation and process variables, as well as the GI physiological factors influence, and may present a challenge to the successful formulation of a colon-specific drug delivery system. The formulation approaches utilized to overcome these challenges mainly focus on an individual mechanism of drug delivery, including bypassing the complex pH environment of the upper GIT by the dosage form, preventing the drug release and drug-absorption in the upper GIT, and releasing the drug in the colon for absorption. The metabolizing capacity of the colon enzymes is also being explored as an approach to target drug delivery specifically in the colonic region. To ensure a balance between efficiency, target-specificity, cost, and patient compliance, it appears that a combination of conventional and newer approaches is the key to the development of colon-specific drug delivery systems. In addition to the combined approaches, the exploration of nanotechnology seems to be an area of future research for colon targeting of drugs (59–61,83).
REFERENCES
Das S, Deshmukh R, Jha A. Role of natural polymers in the development of multiparticulate systems for colon drug targeting. Syst Rev Pharmacy. 2010;1(1):79–85.
Leuva VR, Patel BG, Chaudhary DJ, Patel JN, Modasiya MMK. Oral colon-specific drug delivery system. J Pharm Res. 2012;5(4):2293–7.
Kumar M, Ali A, Kaldhone P, Shirode A, Kadam VJ. Report on pharmaceutical approaches to colon targeted drug delivery systems. J Pharm Res. 2010;3(3).
Philip AK, Philip B. Colon targeted drug delivery systems: a review on primary and novel approaches. Oman Med J. 2010;25(2):79–87.
Kumar P, Mishra B. Colon targeted drug delivery systems—an overview. Curr Drug Deliv. 2008;5(3):186–98.
Malayandi R, Kondamudi P, Ruby PK, Aggarwal D. Biopharmaceutical considerations and characterizations in development of colon targeted dosage forms for inflammatory bowel disease. Drug Deliv Transl Res. 2014;4(2):187–202.
Coupe AJ, Davis SS, Wilding IR. Variation in gastrointestinal transit of pharmaceutical dosage forms in healthy subjects. Pharm Res. 1991;8(3):360–4.
Dressman JB, Berardi RR, Dermentzoglou LC, Russell TL, Schmaltz SP, Barnett JL, et al. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm Res. 1990;7(7):756–61.
Ibekwe VC, Fadda HM, McConnell EL, Khela MK, Evans DF, Basit AW. Interplay between intestinal pH, transit time and feed status on the in vivo performance of pH responsive ileo-colonic release systems. Pharm Res. 2008;25(8):1828–35.
Rubinstein A. Microbially controlled drug delivery to the colon. Biopharm Drug Dispos. 1990;11(6):465–75.
Hebden JM, Blackshaw PE, Perkins AC, Wilson CG, Spiller RC. Limited exposure of the healthy distal colon to orally-dosed formulation is further exaggerated in active left-sided ulcerative colitis. Aliment Pharmacol Ther. 2000;14(2):155–61.
Rana SV, Sharma S, Malik A, Kaur J, Prasad KK, Sinha SK, et al. Small intestinal bacterial overgrowth and orocecal transit time in patients of inflammatory bowel disease. Dig Dis Sci. 2013;58(9):2594–8.
Stubbs JB, Valenzuela GA, Stubbs CC, Croft BY, Teates CD, Plankey MW, et al. A noninvasive scintigraphic assessment of the colonic transit of nondigestible solids in man. J Nucl Med. 1991;32(7):1375–81.
Christl SU, Scheppach W. Metabolic consequences of total colectomy. Scand J Gastroenterol Suppl. 1997;222:20–4.
Sandle GI. Salt and water absorption in the human colon: a modern appraisal. Gut. 1998;43(2):294–9.
Schiller C, Frohlich CP, Giessmann T, Siegmund W, Monnikes H, Hosten N, et al. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment Pharmacol Ther. 2005;22(10):971–9.
Shameem M, Katori N, Aoyagi N, Kojima S. Oral solid controlled release dosage forms: role of GI-mechanical destructive forces and colonic release in drug absorption under fasted and fed conditions in humans. Pharm Res. 1995;12(7):1049–54.
Pijper A, Discombe G. Shape and motility of bacteria. J Pathol Bacteriol. 1946;58(3):325–42.
Evans DF, Pye G, Bramley R, Clark AG, Dyson TJ, Hardcastle JD. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut. 1988;29(8):1035–41.
Fallingborg J, Christensen LA, Ingeman-Nielsen M, Jacobsen BA, Abildgaard K, Rasmussen HH. pH-profile and regional transit times of the normal gut measured by a radiotelemetry device. Aliment Pharmacol Ther. 1989;3(6):605–13.
Macfarlane GT, Gibson GR, Cummings JH. Comparison of fermentation reactions in different regions of the human colon. J Appl Bacteriol. 1992;72(1):57–64.
Bown RL, Gibson JA, Sladen GE, Hicks B, Dawson AM. Effects of lactulose and other laxatives on ileal and colonic pH as measured by a radiotelemetry device. Gut. 1974;15(12):999–1004.
Nugent SG, Kumar D, Rampton DS, Evans DF. Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut. 2001;48(4):571–7.
Gibson SA, McFarlan C, Hay S, MacFarlane GT. Significance of microflora in proteolysis in the colon. Appl Environ Microbiol. 1989;55(3):679–83.
Rowland IR. Factors affecting metabolic activity of the intestinal microflora. Drug Metab Rev. 1988;19(3–4):243–61.
Chung KT, Fulk GE, Egan M. Reduction of azo dyes by intestinal anaerobes. Appl Environ Microbiol. 1978;35(3):558–62.
Hejazi R, Amiji M. Chitosan-based gastrointestinal delivery systems. J Control Release. 2003;89(2):151–65.
Ratner BD, Gladhill KW, Horbett TA. Analysis of in vitro enzymatic and oxidative degradation of polyurethanes. J Biomed Mater Res. 1988;22(6):509–27.
Sinha VR, Kumria R. Polysaccharides in colon-specific drug delivery. Int J Pharm. 2001;224(1–2):19–38.
Kang M, Khanal T, Kim H, Lee D, Yeo H, Lee Y, et al. Role of metabolism by human intestinal microflora in geniposide-induced toxicity in HepG2 cells. Arch Pharm Res. 2012;35(4):733–8.
Nutt JG, Fellman JH. Pharmacokinetics of levodopa. Clin Neuropharmacol. 1984;7(1):35–49.
Azadkhan AK, Truelove SC, Aronson JK. The disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br J Clin Pharmacol. 1982;13(4):523–8.
Hamedani R, Feldman RD, Feagan BG. Drug development in inflammatory bowel disease: budesonide-a model of targeted therapy. Aliment Pharmacol Ther. 1997;3:98–107.
French DL, Mauger JW. Evaluation of the physicochemical properties and dissolution characteristics of mesalamine: relevance to controlled intestinal drug delivery. Pharm Res. 1993;10(9):1285–90.
Nicholls A, Harris-Collazo R, Huang M, Hardiman Y, Jones R, Moro L. Bioavailability profile of Uceris MMX extended-release tablets compared with Entocort EC capsules in healthy volunteers. J Int Med Res. 2013;41(2):386–94.
Rabito MF, Reis AV, Freitas Ados R, Tambourgi EB, Cavalcanti OA. A pH/enzyme-responsive polymer film consisting of Eudragit® FS 30 D and arabinoxylane as a potential material formulation for colon-specific drug delivery system. Pharm Dev Technol. 2012;17(4):429–36.
Friend DR, Chang GW. A colon-specific drug-delivery system based on drug glycosides and the glycosidases of colonic bacteria. J Med Chem. 1984;27(3):261–6.
Friend DR, Chang GW. Drug glycosides: potential prodrugs for colon-specific drug delivery. J Med Chem. 1985;28(1):51–7.
Kim D, Hong S, Jung S, Jung Y, Kim YM. Synthesis and evaluation of N-nicotinoyl-2-{2-(2-methyl-5-nitroimidazol-1-yl)ethyloxy}-D,L-glycine as a colon-specific prodrug of metronidazole. J Pharm Sci. 2009;98(11):4161–9.
Kim H, Lee Y, Yoo H, Kim J, Kong H, Yoon JH, et al. Synthesis and evaluation of sulfate conjugated metronidazole as a colon-specific prodrug of metronidazole. J Drug Target. 2012;20(3):255–63.
Vaidya A, Jain S, Agrawal RK, Jain SK. Pectin-metronidazole prodrug bearing microspheres for colon targeting. J Saudi Chem Soc. 2012
Modasiya MK, Patel VM. Design of colon specific drug delivery using sodium alginate and HPMC. J Pharm Res. 2012;5(4):2253–8.
Cummings JH, Englyst HN. Fermentation in the human large intestine and the available substrates. Am J Clin Nutr. 1987;45(5 Suppl):1243–55.
Scheline RR. Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol Rev. 1973;25(4):451–523.
Basit A, Bloor J. Perspectives on colonic drug delivery business briefing. Pharmatech. 2003:185–9.
Roos AA, Edlund U, Sjoberg J, Albertsson AC, Stalbrand H. Protein release from galactoglucomannan hydrogels: influence of substitutions and enzymatic hydrolysis by beta-mannanase. Biomacromolecules. 2008;9(8):2104–10.
Hita V, Singh R, Jain SK. Colonic targeting of metronidazole using azo aromatic polymers: development and characterization. Drug Deliv. 1997;4(1):19–22.
Ahmad MZ, Akhter S, Ahmad I, Singh A, Anwar M, Shamim M, et al. In vitro and in vivo evaluation of Assam Bora rice starch-based bioadhesive microsphere as a drug carrier for colon targeting. Expert Opin Drug Deliv. 2012;9(2):141–9.
Alvarez-Fuentes J, Fernández-Arévalo M, González-Rodríguez ML, Cirri M, Mura P. Development of enteric-coated timed-release matrix tablets for colon targeting. J Drug Target. 2004;12(9/10):607–12.
Fukui E, Miyamura N, Uemura K, Kobayashi M. Preparation of enteric coated timed-release press-coated tablets and evaluation of their function by in vitro and in vivo tests for colon targeting. Int J Pharm. 2000;204(1–2):7–15.
Gazzaniga A, Iamartino P, Maffione G, Sangalli ME. Oral delayed-release system for colonic specific delivery. Int J Pharm. 1994;108(1):77–83.
Chourasia MK, Jain SK. Design and development of multiparticulate system for targeted drug delivery to colon. Drug Deliv. 2004;11(3):201–7.
Hardy JG, Wilson CG, Wood E. Drug delivery to the proximal colon. J Pharm Pharmacol. 1985;37(12):874–7.
Vaidya A, Jain A, Khare P, Agrawal RK, Jain SK. Metronidazole loaded pectin microspheres for colon targeting. J Pharm Sci. 2009;98(11):4229–36.
Perera G, Barthelmes J, Bernkop-Schnurch A. Novel pectin-4-aminothiophenole conjugate microparticles for colon-specific drug delivery. J Control Release. 2010;145(3):240–6.
Ye L, Hong Z. Budesonide-loaded guar gum microspheres for colon delivery: preparation, characterization and in vitro/in vivo evaluation. Int J Mol Sci. 2015;16(2):2693–704.
Beckert T, Petereit HU, Dressman J, Rudolph M. Multi-particulate form of medicament, comprising at least two differently coated forms of pellet. Google Patents; 2005.
Agarwal T, Narayana SNGH, Pal K, Pramanik K, Giri S, Banerjee I. Calcium alginate-carboxymethyl cellulose beads for colon-targeted drug delivery. Int J Biol Macromol. 2015;75:409–17.
Beloqui A, Coco R, Memvanga PB, Ucakar B, des Rieux A, Preat V. pH-sensitive nanoparticles for colonic delivery of curcumin in inflammatory bowel disease. Int J Pharm. 2014;473(1–2):203–12.
Calabrese I, Cavallaro G, Scialabba C, Licciardi M, Merli M, Sciascia L, et al. Montmorillonite nanodevices for the colon metronidazole delivery. Int J Pharm. 2013;457(1):224–36.
Spada G, Gavini E, Cossu M, Rassu G, Giunchedi P. Solid lipid nanoparticles with and without hydroxypropyl-beta-cyclodextrin: a comparative study of nanoparticles designed for colonic drug delivery. Nanotechnology. 2012;23(9).
Mundargi RC, Patil SA, Agnihotri SA, Aminabhavi TM. Development of polysaccharide-based colon targeted drug delivery systems for the treatment of amoebiasis. Drug Dev Ind Pharm. 2007;33(3):255–64.
Gauri B, Singh SK, Mishra D. Formulation and evaluation of colon targeted oral drug delivery systems for metronidazole in treatment of amoebiasis. Int J Drug Del. 2011;3(3).
Shukla RK, Tiwari A. Carbohydrate polymers: applications and recent advances in delivering drugs to the colon. Carbohydr Polym. 2012;88(2):399–416.
Bassi P, Kaur G. pH modulation: a mechanism to obtain pH-independent drug release. Expert Opin Drug Deliv. 2010;7(7):845–57.
Kosaraju SL. Colon targeted delivery systems: review of polysaccharides for encapsulation and delivery. Crit Rev Food Sci Nutr. 2005;45(4):251–8.
Liu L, Fishman ML, Kost J, Hicks KB. Pectin-based systems for colon-specific drug delivery via oral route. Biomaterials. 2003;24(19):3333–43.
Wakerly Z, Fell JT, Attwood D, Parkins D. Studies on drug release from pectin/ethylcellulose film-coated tablets: a potential colonic delivery system. Int J Pharm. 1997;153(2):219–24.
Maroni A, Zema L, Loreti G, Palugan L, Gazzaniga A. Film coatings for oral pulsatile release. Int J Pharm. 2013;457(2):362–71.
Obitte N, Chukwu A. The synergistic effect of Landolphia owariensis latex and Eudragit L-100-coated capsules on the in vitro controlled release of metronidazole for possible colon targeting. Asian J Pharm. 2011;5(2):75–83.
Leopold CS, Eikeler D. Eudragit E as coating material for the pH-controlled drug release in the topical treatment of inflammatory bowel disease (IBD). J Drug Target. 1998;6(2):85–94.
Yassin AE, Alsarra IA, Alanazi FK, Al-Mohizea AM, Al-Robayan AA, Al-Obeed OA. New targeted-colon delivery system: in vitro and in vivo evaluation using X-ray imaging. J Drug Target. 2010;18(1):59–66.
Kadiyam R, Muzib YI. Colon specific drug delivery of tramadol HCl for chronotherapeutics of arthritis. Int J Pharm Investig. 2015;5(1):43–9.
Maroni A, Del Curto MD, Cerea M, Zema L, Foppoli A, Gazzaniga A. Polymeric coatings for a multiple-unit pulsatile delivery system: preliminary study on free and applied films. Int J Pharm. 2013;440(2):256–63.
Santus G, Baker RW. Osmotic drug delivery: a review of the patent literature. J Control Release. 1995;35(1):1–21.
Verma RK, Krishna DM, Garg S. Formulation aspects in the development of osmotically controlled oral drug delivery systems. J Control Release. 2002;79(1–3):7–27.
Sethi S, Harikumar SL, Nirmala. Review on advances in colon targeted drug delivery system. Int J Pharm Sci Res. 2012;3(9):2989–3000.
Takaya T, Niwa K, Muraoka M, Ogita I, Nagai N, Yano R-i, et al. Importance of dissolution process on systemic availability of drugs delivered by colon delivery system. J Control Release. 1998;50(1–3):111–22.
Hay DJ, Sharma H, Irving MH. Spread of steroid-containing foam after intrarectal administration. Br Med J. 1979;1(6180):1751–3.
Philip AK, Pathak K. Osmotic flow through asymmetric membrane: a means for controlled delivery of drugs with varying solubility. AAPS PharmSciTech. 2006;7(3):56.
Abraham S, Srinath M. Development of modified pulsincap drug delivery system of metronidazole for drug targeting. Indian J Pharm Sci. 2007;69(1):24–7.
Krogel I, Bodmeier R. Pulsatile drug release from an insoluble capsule body controlled by an erodible plug. Pharm Res. 1998;15(3):474–81.
Huanbutta K, Sriamornsak P, Luangtana-Anan M, Limmatvapirat S, Puttipipatkhachorn S, Lim LY, et al. Application of multiple stepwise spinning disk processing for the synthesis of poly(methyl acrylates) coated chitosan-diclofenac sodium nanoparticles for colonic drug delivery. Eur J Pharm Sci. 2013;50(3–4):303–11.
Marieb EN, Hoehn K. Human anatomy and physiology. 7th ed. San Francisco: Pearson Benjamin Cummings; 2007.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Amidon, S., Brown, J.E. & Dave, V.S. Colon-Targeted Oral Drug Delivery Systems: Design Trends and Approaches. AAPS PharmSciTech 16, 731–741 (2015). https://doi.org/10.1208/s12249-015-0350-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0350-9