
Abstract
Drug-induced kidney injury is often observed in the clinics and can lead to long-term organ failure. In this work, we evaluated a novel in vitro system that aims at detecting whether compounds can cause renal proximal tubule damage in man. For this, we implemented organotypic cultures of human conditionally immortalized proximal tubule epithelial cells overexpressing the organic anion transporter 1 (ciPTEC-OAT1) in a three-channel OrganoPlate under microfluidic conditions. Cells were exposed to four known nephrotoxicants (cisplatin, tenofovir, cyclosporine A, and tobramycin). The effect on cell viability and NAG release into the medium was determined. A novel panel of four miRNAs (mir-21, mir-29a, mir-34a, and mir-192) was selected as potential biomarkers of proximal tubule damage. After nephrotoxicant treatment, miRNA levels in culture medium were earlier indicators than cell viability (WST-8 assay) and outperformed NAG for proximal tubule damage. In particular, mir-29a, mir-34a, and mir-192 were highly reproducible between experiments and across compounds, whereas mir-21 showed more variability. Moreover, similar data were obtained in two different laboratories, underlining the reproducibility and technical transferability of the results, a key requirement for the implementation of novel biomarkers. In conclusion, the selected miRNAs behaved like sensitive biomarkers of damage to tubular epithelial cells caused by several nephrotoxicity mechanisms. This biomarker panel, in combination with the 3D cultures of ciPTEC-OAT1 in the OrganoPlate, represents a novel tool for in vitro nephrotoxicity detection. These results pave the way for the application of miRNAs in longitudinal, time-course in vitro toxicity studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Drug-induced kidney injury (DIKI) is often observed in the clinics, can be a dose-limiting factor, and can lead to chronic kidney failure. The use of nephrotoxic drugs has been implicated in up to 25% of all cases of severe acute renal failure, in particular tubular necrosis, in critically ill patients (1). Despite its clinical relevance, detection of DIKI during the drug development process relies mainly on animal experimentation and histopathological examination (2,3). In the last years, advances have been made in qualifying novel biomarkers to detect kidney injury using body fluids and seven biomarkers have been successfully qualified for preclinical animal tests (4,5). Some biomarkers, for example kidney injury molecule 1 (KIM-1) and urinary N-acetyl-beta-D-glucosaminidase (NAG), have been implemented in the clinic as well as in vitro (6,7,8). In addition to the advancements in biomarkers of kidney injury in vivo, there is quite some interest in developing human in vitro test systems and biomarkers that can be used early in the drug screening process. Currently used systems apply either simple two-dimensional (2D) cell cultures or more complex, 3D cell cultures combined with microfluidics devices (3,9). Some published results on human proximal tubule epithelial cells (PTEC) grown on filter inserts or on microphysiologic systems (kidney-on-a-chip) used gene expression analysis for the detection of drug-induced cell injury (10,11). The results indicate that gene expression changes are associated with PTEC damage in vitro. The disadvantage of gene expression is that transcripts can only be measured at the end of an experiment (endpoint measurement), since the detection requires cell lysis. Extracellular biomarkers that can potentially be collected over time would be ideal. Enzymes, like NAG, or miRNAs that can be leaked or secreted from damaged cells would be better suited for the detection of in vitro nephrotoxicity. Recently, the correlation of increased serum levels of selected miRNAs in patients with acute kidney injury has been published (12).
In this work, we sought to investigate changes in miRNA released to the cell culture medium as potential markers for nephrotoxicity. For this purpose, we cultured the conditionally immortalized PTEC line overexpressing the organic anion transporter 1 (ciPTEC-OAT1) cells in 2D monolayers and in a 3D microfluidics system (OrganoPlate). The selected cell line has been engineered to overexpress the transporter OAT1. In vitro models generally lack this transporter due to dedifferentiation of PTEC in culture (13). It has been reported that OAT-deficient cell models are not ideal for drug toxicity evaluation as many substances, including the nucleotide analogue reverse transcriptase inhibitor tenofovir, are organic anions and as such substrates for OAT1 or OAT3 (14).
We exposed ciPTEC-OAT1 cells to four known nephrotoxicants and evaluated the effects by determining cell viability and by measuring putative biomarkers in the medium: miRNA levels and NAG levels. All these endpoints can be multiplexed and are non-destructive, so that the cells can be maintained in culture for longer exposures or lysed for the measurement of other parameters (e.g., gene expression or immunohistochemistry).
The four selected compounds were the aminoglycoside antibiotic tobramycin, the cytostatic cisplatin, the antiretroviral agent tenofovir, and the immunosuppressant cyclosporine A. All these compounds have been associated with kidney injury in the clinic. Cisplatin can lead to acute kidney injury, characterized by PTEC injury, oxidative stress, inflammation, and vascular injury in the kidney (15). Cyclosporine A has been often associated with acute and chronic nephropathies. Acute nephropathy may be caused by a reduction in renal blood flow, although the pathophysiological mechanism leading to chronic kidney damage is not completely understood. Among other effects, upregulation of transforming growth factor beta 1 (TGFβ-1), inappropriate apoptosis, endoplasmic reticulum stress, and autophagy have all been implicated in the pathogenesis of chronic cyclosporine A-induced nephropathy (16). Aminoglycosides like tobramycin are known to induce nephrotoxicity that can lead to renal failure, initiated by their receptor-mediated uptake in PTEC, where they accumulate in endosomal and lysosomal vacuoles, and Golgi complex (17). Tenofovir-induced injury has been associated with accumulation in the renal tubule cells due to rapid drug uptake via the human organic anion transporters and limited efflux into tubular lumen (18). Once in the cells, tenofovir has been shown to lead to mitochondrial damage, causing mitochondrial swelling, and impairment of the respiratory chain (19).
MicroRNAs (miRNAs) are a family of short, non-coding RNAs involved in the post-transcriptional gene regulation. These single-stranded RNAs bind complementary to transcripts and repress their translation or initiate degradation (20). miRNAs play a critical role in cellular and physiological processes and some were found to be expressed in a cell- or tissue-specific manner (21), for instance mir-122 (22) is enriched in the liver and mir-192 in the kidney cortex (23). Their dysregulation has been implicated with a wide range of diseases (24,25) as well as with organ toxicity. In particular, miRNA122 is increased after hepatocellular damage in vivo and in vitro (26,27). With regard to the kidney, some studies indicate that miRNAs may be suitable urinary biomarkers for the detection of drug-induced nephrotoxicity (28,29).
Despite this, the application of miRNAs for the detection of kidney toxicity is still in progress. In this study, we selected a panel of four miRNAs: mir-21, mir-29a, mir-34a, and mir-192 as potential biomarkers for proximal tubular damage. Moderate upregulation of mir-21 decreases cell death but massive overexpression may lead to increased cell death, resulting in severe inflammation and fibrosis (30). mir-29a and mir-192 are highly expressed in the kidney, including PTEC, and their expression is regulated by TGF-β1 (29,31); mir-34a, on the other hand, has been related to apoptosis during disease and nephrotoxicity (32,33).
Our data show that the panel of four selected miRNAs is released from ciPTEC upon exposure to nephrotoxicants, making them promising biomarkers for toxicity to kidney proximal tubular cells in vitro. In addition, the studies performed in two different laboratories clearly demonstrate that ciPTEC-OAT1 grown on the OrganoPlate represent a robust platform for the assessment of nephrotoxicity in vitro. Additional research will be needed to determine their sensitivity and specificity.
MATERIAL AND METHODS
Cell Lines and Cell Culture Conditions
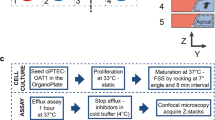
Human ciPTEC-OAT1 generated at Radboudumc (Nijmegen, Netherlands) (34,35) were cultured using standard protocols in 2D (monolayer) and 3D (OrganoPlate). Cells (passage number 55–60), were expanded and maintained in T75 flasks, in DMEM-HAM’s F12 supplemented with 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml selenium, 10 ng/ml epidermal growth factor, 36 ng/ml hydrocortisone, and 10% FBS, at 33 °C and 5% v/v CO2.
For 2D experiments, ciPTEC-OAT1 were seeded in uncoated 96-well plates (Falcon) at a density of 10,000 cells/well and cultured at 33 °C for 3 days to enable cell proliferation. Cells were then cultured for a further 7 days at 37 °C and 5% v/v CO2 to stimulate differentiation and formation of an epithelial monolayer.
For 3D experiments, ciPTEC-OAT1 cells were cultured under flow in a 3D, 3-lane OrganoPlate (Mimetas BV, 4003 400B) with a channel width of 400 μm and a height of 220 μm was used. First, 1.6 μL extracellular matrix (ECM), composed of a mixture of collagen I from rat tail (4 mg/ml, AMSbio), sodium bicarbonate (3.7 mg/ml, Sigma-Aldrich), and HEPES (100 mM, Life Technologies), was applied to the middle lane and let to gel during approximately 30–45 min. Subsequently, 40,000 ciPTEC-OAT1 cells/chip were seeded in the cell channel, next to the ECM channel. The OrganoPlate was placed on an interval rocker platform (± 7° angle, 8-min interval) enabling a bidirectional flow through the perfusion channels. Cells were cultured for 3 days at 33 °C and 5% v/v CO2 to allow proliferation and for approximately 7–8 days at 37 °C to allow differentiation before application of the test substances.
Exposures to Treatments
During the compound treatment, ciPTEC-OAT1 were cultured in DMEM-HAM’s F12 supplemented with, 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml selenium, 10 ng/ml epidermal growth factor, 36 ng/ml hydrocortisone, and 10% FBS. Cells were treated with four different nephrotoxic compounds: cisplatin (Sigma, P4394, stock: 5 mM in 0.9% NaCl), tenofovir (Santa-Cruz, SC-204335 in medium), cyclosporine A (in DMSO), and tobramycin (Sigma, T1783, in medium). Cytotoxicity of the four compounds (EC50 values based on cell viability assays) was determined after 48 h exposure on 2D cultures. For subsequent experiments, two to three compound concentrations were chosen. Compounds were kept on the cells for either 24 or 48 h. Cell treatment for 48 h was expected to elicit cell toxicity, whereas 24 h was selected as an early time point for the release of miRNAs from 3D cultures. After treatment, the medium was collected for determination of miRNAs and NAG content; cell viability was measured using the WST-8 viability assay.
NAG Measurement
The levels of β-N-acetylglucosaminidase (NAG) in supernatant were determined using the NAG-Assay Kit (BioVision, K733-100). For both, 2D and 3D experiments, 30 μL medium was collected and used for the assay following the provider’s instructions. Absorbance at 400 nm was measured in a Flexstation 3 (Molecular devices).
Cell Viability
At the end of the exposure period, the medium was removed for analysis and cells were incubated in fresh medium containing the Cell Counting Kit-8 (WST-8, Sigma #96992) to assess cell viability. In 2D experiments, 100 μL reagent was added per well and cells incubated during approximately 2 h at 37 °C. In the OrganoPlate, 50 μl of WST solution (1:11 in Medium) was added to each medium inlet and outlet of bottom and top channels. OrganoPlates were incubated at 37 °C for approximately 30–90 min. After a 2-min motionless incubation, absorbance in the top in- and outlets was measured with the Multiskan™ FC Microplate Photometer (Thermo scientific) or a FlexStation 3 (Molecular devices) at 450 nm.
For the determination of EC50 values, a subset of samples was used to determine intracellular ATP content using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega) and following the provider’s instructions. Briefly, 50 μl of prepared solution was added to 50 μl of HBSS in each well. After 2 min of shaking and 10 min of stabilization time, the supernatant was pipetted into an opaque 96-well plate and the luminescence was measured in a FlexStation 3 (Molecular devices).
Detection of miRNAs in Medium
Total RNA was extracted from 80 μL supernatant using the miRNeasy® Serum/Plasma Kit (Qiagen, 217,184) following the manufacturer’s protocol. miRNAs were reverse-transcribed using the TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems®, 4366596) and the miRNA-specific stem-loop primers for the miRNAs, mir-21, mir-34a, mir-29a, and mir-192 (Applied Biosystems™, TaqMan microRNA Assays, listed in Table I). The reaction mix was prepared according to the manufacturer’s instructions for a final reaction volume of 10 μL with 3 μL RNA extract. The PCR conditions were set for 30 min at 16 °C followed for 30 min at 42 °C and 5 min at 85 °C.
The real-time PCR was carried out using a fluorometric thermal cycler (Qiagen, Rotor-Gene Q 3000 or Celtic Diagnostics, Corbett research PCR Rotorgene 6000). The reaction mix contained TaqMan® Fast Advanced Master Mix 1X (Applied Biosystems™, 4444557), TaqMan microRNA Assay primer 1X (Applied Biosystems™, see Table I), and 1.3 μL cDNA in a final reaction volume of 20 μl. The PCRs were run at 95 °C for 20 s followed by 40 cycles of 1 s at 95 °C and 20 s at 60 °C. All extracted RNAs were analyzed in technical duplicates; Ct values of these two measurements were averaged and considered a single value.
Data Analysis
Viability data (WST-8 assay and ATP content) and NAG release were calculated following the provider’s instructions and expressed as percentage of the vehicle control group (set to 100%). EC50 values were calculated using a 4-parameter fit using GraphPad Prism (version 5.03). All PCR data were analyzed with the Rotor-Gene 6000 software version 1.7. Changes of miRNA levels in supernatants were determined by comparing the Ct values in the treated samples in comparison to the control samples and expressed a −ΔCt.
For statistical analysis of the effects, we applied ANOVA, followed by Dunnetts’ multiple comparison test for pair-wise comparison, using GraphPad Prism (version 5.03). Significance levels are *p ≤ 0.05, **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
RESULTS
Dose-Range Finders in ciPTEC-OAT1
As described in “Material and Methods”, ciPTEC-OAT1 were exposed to several concentrations of the four test substances to determine the EC50s and select appropriate concentrations for the follow-up experiments. As depicted in Fig. 1, all four compounds led to loss of cell viability assessed by WST-8 assay and ATP content. The obtained EC50 are listed in Table II. The concentrations for the subsequent experiments were chosen to be between 5 and 30 μM for cisplatin and cyclosporine A, between 15.6 and 1000 μM for tenofovir, and up to 15,000 μM for tobramycin.

Cell viability and determination of EC50 by WST-8 assay and ATP content after 48 h exposure of ciPTEC-OAT1 to a cisplatin, b tenofovir, c tobramycin, and d cyclosporine A. Data represent average ± SD of four (WST-8) and two (ATP) independent experiments with three replicates and are expressed as percentage of control values
Toxicity Responses and miRNA Release in 2D and 3D
Based on the concentrations selected from the EC50 values, we compared the responses of ciPTEC-OAT1 cells to cisplatin (high toxicity, EC50 ~ 20 μM) and tenofovir (medium toxicity, EC50 ~ 250 μM) in 2D and in 3D (OrganoPlate). As depicted in Fig. 2, side-by-side comparison of the effects on cell viability shows that both compounds lead to dose-dependent decreased viability at similar concentrations in both systems. The extent of the loss of viability was greater in 2D than in 3D cultures.

Cell viability determined by the WST-8 assay after 48 h exposure of cells to cisplatin and tenofovir in 2D (96-well plates) and 3D (OrganoPlates). In 2D, data represent average ± SD of six replicates. In 3D, data represent average ± SD of two independent experiments with four to five replicates each. All data are expressed as percentage of control values
In order to evaluate secreted miRNAs as potential biomarkers, the medium of the cells exposed to cisplatin or tenofovir for 48 h was collected and processed as described in “Material and Methods”. As depicted in Fig. 3, both compounds elicited increases in all four measured miRNAs (mir-34a, mir-192, mir-21 and -29a) in the cell culture medium. Cisplatin led to significant increased miRNA concentrations at 5 and 15 μM in 2D and 3D, respectively. Tenofovir, showed significantly higher miRNA release at 250 μM in both 2D and 3D. Concordant with the overt cytotoxicity, the levels of miRNAs released after treatment of cells with cisplatin at 15 μM in 2D was significantly higher than the control but lower than the values measured at 5 μM.

miRNA levels (mir-21, mir-29a, mir-34a, and mir-192) released into the medium after 48 h exposure of ciPTEC-OAT1 cells to cisplatin and tenofovir in 2D (96-well plates: graphs a and b) 3D (OrganoPlates: graphs c and d). Data are expressed as log2 fold-change (−ΔCT) using the vehicle-treated samples as baseline. Data represent average ± SD of three or four replicates (for 2D and 3D, respectively)
These results show that the release of specific miRNAs into the medium was indicative of toxicity. An additional finding of these experiments was that the increase in mir-192 was generally the highest observed while mir-21 seemed less sensitive. These findings are very similar in 2D and 3D cultures (Fig. 3).
Toxicity Responses and miRNA Release in 3D at an Earlier Time Point
The results obtained in 2D and in 3D after exposure of ciPETC-OAT1 to cisplatin and tenofovir after 48 h exposure were strongly indicative of the potential of this miRNA panel as potential biomarkers of nephrotoxicity. Next, we assessed the effect of cisplatin, tenofovir, cyclosporine A, and tobramycin in an independent set of experiments that included 24 h exposure as an earlier time point and 48 h exposure to replicate the previous results.
Exposure of ciPTEC-OAT1 in OrganoPlate to all four compounds caused a time- and concentration-dependent decrease in cell viability, as depicted in Fig. 4a, with the lowest viability levels caused by exposures to cisplatin and cyclosporine A. Concordantly, only cisplatin and cyclosporine A caused significant time- and concentration-dependent increases in NAG activity in medium (Fig. 4b). Tobramycin led to a significant, yet moderate decrease in viability (69% at 24 h). Also, tenofovir led to a concentration-dependent decrease in viability at 24 h. The turnover of WST was increased by the low concentration of tenofovir. However, the values were significantly lower 48 h after treatment with 1000 μM (compared with 15.6 μM). With regard to NAG, tobramycin and tenofovir only caused marginal effects (not significant) and only at the highest concentration and after 48 h of exposure (Fig. 4b).

Cell viability WST-8 (a) and NAG release into the medium (b) after 48 h exposure of ciPTEC-OAT1 cells to cisplatin and tenofovir in 3D (OrganoPlates) from two independent experiments with four to ten replicates. Data represent average ± SD and are expressed as percentage of control values
The medium of the treated cells was collected after 24 h of exposure and miRNA levels determined as described before. All four tested compounds led to concentration-dependent increases in the miRNA panel (Fig. 5a–c). Specifically, tobramycin and cyclosporine A caused a dose-dependent increase of all tested miRNAs, while cisplatin and tenofovir led to an increase in three of the four miRNAs; mir-21 did not show significant changes with these compounds. These results are concordant with the results obtained in the previous experiments in 2D and in 3D (Fig. 3).

miRNA levels of mir-192 (a), mir-34a (b), mir-21 (c), and mir-29a (d) released into the medium after 24 h exposure of ciPTEC-OAT1 cells to cisplatin, tenofovir, tobramycin, and cyclosporine A in 3D (OrganoPlates). Data are expressed as log2 fold-change (−ΔCT) using the vehicle treated samples as baseline and represent average ± SD of three to five replicates
DISCUSSION
In this work, we evaluated the suitability of a novel in vitro system to detect the potential of compounds of causing renal proximal tubule damage in man. In our approach, we exposed renal tubular cells to four nephrotoxic compounds in two different cell culture systems, 2D monolayers and 3D microfluidic OrganoPlates. We assessed cytotoxicity, NAG release, and miRNA release into the medium. Preliminary experiments showed that all compounds exert cytotoxicity on the ciPTEC-OAT1, with EC50 ranging from relatively low for cisplatin and cyclosporine A to medium for tenofovir or very high with tobramycin. For cyclosporine A, cisplatin, and tobramycin, the EC50 values are in agreement with results obtained in another laboratory using a different cell line (10). Based on the cell viability results, we selected concentrations to perform 2D vs 3D comparisons and biomarker assessment.
The results on side-by-side comparison of 2D and 3D obtained with cisplatin and tenofovir demonstrated that ciPTEC-OAT1 cells respond with dose-dependent cell death to both compounds in monolayer cultures and in the OrganoPlate under flow conditions after 48 h of exposure. Growing cells in more physiological conditions has been shown to improve performance, making the system more comparable with the situation in vivo. In addition, culturing proximal tubular cells in OrganoPlates allows for additional functional measurements such as transport function and barrier integrity (Vormann M, et al. (2018) Nephrotoxicity and kidney transport assessment on 3D perfused proximal tubules, Vriend J, et al. (2018) High-throughput screening of drug-transporter interactions in a 3D microfluidic renal proximal tubule on a chip). The selected cell line overexpressing OAT1 is particularly suited for these experiments, as tenofovir is known to be internalized via the OAT transporters (35,36).
The release of the four selected miRNAs into the medium was significantly increased upon treatment with cisplatin and tenofovir for 48 h in 2D and 3D cultures. This suggested that miRNA release of mir-21, mir-29a, mir-34a and mir-192 may be good indicators of compound-induced cell death of proximal tubular cells. Both compounds led to a concentration-dependent decrease in viability and to a dose-dependent increase in miRNAs. It is noteworthy, that the miRNA release of the cells exposed to 15 μM of cisplatin for 48 h was less marked that the cells treated with 5 μM. A plausible explanation for this finding is that at this time point and concentration, overt cytotoxicity acts as a confounding factor. Thus, subsequent experiments were performed in which miRNA was collected earlier, 24 h post-exposure. In this set of experiments, we also assessed the effects of cyclosporine A and tobramycin.
The experiments performed with on ciPTEC-OAT1 treated with cisplatin, tenofovir, tobramycin, or cyclosporine A in the OrganoPlate corroborated the toxicity of the compounds to ciPTEC-OAT1, as shown by the decreased viability observed after 24 or 48 h exposure. The commonly used nephrotoxicity biomarker NAG appeared to be less sensitive than viability assays, as only cisplatin and cyclosporine A led to a significant increase in activity at the highest tested concentrations. The levels of the selected miRNAs in the medium, on the other hand, were consistently and dose-dependently increased by all four test compounds. Hence, the changes in miRNAs in the medium outperform conventional viability assays.
Altered miRNA levels have been associated with the pathogenesis of various renal diseases, such as acute kidney injury and renal fibrosis (30,37). Also, circulating miRNAs and exosomal miRNAs are known to be released as a response to stress or during injury (38) and have been proposed to have great potential as biomarkers for prognosis and diagnosis of several diseases (39,40). Whether the changes in released miRNAs detected in our study are due to incipient cell damage (leakage) or are causally related to the ensuing cytotoxicity cannot be concluded from our experiments. However, it is known that in the kidney, circulating miRNAs and exosomal miRNAs are released as a response to stress or during injury, and may be suitable biomarkers in vivo (38,40). This would make miRNAs potential translational (in vitro-in vivo) biomarkers of nephrotoxicity. Also, our data show that increased release of miRNAs may be detectable before significant toxicity and/or leakage of NAG becomes apparent. This indicates better sensitivity, but targeted investigations on the mechanisms of release of the miRNAs would be necessary to clarify this point. Cultures of ciPTEC-OAT1 can be maintained for at least 2 weeks on the OrganoPlate (unpublished data), which would allow time-course experiments to be performed in the future.
Altogether, the data presented here point out that miRNAs (mir-21, mir-29a, mir-34a, and mir-192) are suitable biomarkers for drug-induced toxicity of proximal tubular cells in vitro. These miRNAs play different roles in tissue damage, which further supports their usefulness as PTEC injury markers. In vivo, mir-29a in serum correlates with the severity of acute kidney injury (12) and has also been associated with the modulation of early events that facilitate tissue remodeling in tendon (41). mir-192 and mir-34a have been associated with tissue fibrosis and apoptosis (32,33,37). mir-34a is induced in kidneys of mice (in vivo) and in a mouse proximal tubular cell, BUMPT-306 (in vitro) after cisplatin-induced activation of p53. Experimentally blocking the induction of mir-34a led to an exacerbation of the tissue damage, indicating that this miRNA may play a cytoprotective role (33). mir-192 is considered kidney-specific and is highly expressed in the renal proximal convoluted tubule. It has been reported that knockdown of mir-192 in mice led to upregulation of Atp1b1 protein, suggesting a mechanism by which mir-192 suppresses Na+/K+-ATPase and contributes to renal handling of fluid balance (42). Moreover, toxicological investigations with acetaminophen showed an increase in the release of mir-192 that did not correlate with hepatotoxicity and suggested a direct association with acetaminophen-induced kidney damage (43). From all four miRNAs determined in our study, mir-21 seems to yield the most variable performance, as it was only significantly increased by some treatments. This may be due to the fact that in acute kidney injury, mir-21 is involved in the prevention of apoptosis by the epithelial cells and in the development of fibrosis by promoting myofibroblast differentiations (30). Thus, some of the changes in mir-21 observed in disease progression may involve myofibroblasts, a cell type not represented in the ciPTEC-OAT1 cultures. Hence, despite its implication as a potential clinically relevant marker for kidney damage, the results obtained with mir-21 may lead ultimately to its exclusion from the panel of in vitro biomarkers. Similarly, in our hands, changes in NAG released into the medium showed a relatively poor performance as an in vitro biomarker. Despite the fact this tubular enzyme has emerged as a predictor of tubular damage in serum and in urine (44), it was a more variable and less sensitive indicator of PTEC damage than WST-8 viability assay or miRNA release.
Our data further demonstrate that the panel of miRNAs is applicable to 2D (96-well) and 3D (OrganoPlate under flow conditions) PTEC cultures. Based on the results obtained with four compounds known to be toxic to the proximal tubule, we are not yet able to claim that these biomarkers will apply to a larger panel of nephrotoxicants. This point would require a larger number of test substances, including positive and negative compounds. However, the data strongly suggest that the release of the selected miRNAs into the medium is a good indicator of cellular damage to proximal tubular cells that can be implemented in vitro. Both these systems are compatible with medium- to high-throughput screening and image analysis and could be promptly implemented in the drug screening process. Cell culture in the OrganoPlate offers the additional advantages of allowing culture under more physiological flow conditions and the parallel assessment of functional aspects such as epithelial barrier function (45). miRNAs are attractive biomarkers since they can be measured without interrupting the exposure and are relatively easy to quantify using accessible molecular biology methods such as qPCR and NexGen sequencing. Moreover, the performance of similar experiments at two sites (FHNW and Mimetas) showed the technical site-to-site transferability of the cell culture system and of the selected markers, a key requirement for their successful implementation. As shown in this paper, the profiles of miRNAs secreted into the medium showed similar profiles after exposure of ciPTEC-OAT1 to cisplatin and tenofovir in 2D and in 3D. Consistent with this, the cytotoxicity caused by cisplatin and tenofovir was comparable in the different experimental settings.
CONCLUSION
The results presented in this manuscript strongly support the use of miRNAs (mir-21, mir-29a. mir-34a, and mir-192) in cell culture medium as early and sensitive biomarkers of damage to renal proximal tubule cells. This biomarker panel, in combination with the 3D cultures of ciPTEC-OAT1 in the OrganoPlate, represents a promising novel tool for in vitro detection of nephrotoxicity.
References
Pannu N, Nadim MK. An overview of drug-induced acute kidney injury. Crit Care Med. 2008;36(4 Suppl):S216–23.
Huang J, Wu H. Drug-induced nephrotoxicity: pathogenic mechanisms, biomarkers and prevention strategies. Curr Drug Metab. 2017;18
Tiong HY, Huang P, Xiong S, Li Y, Vathsala A, Zink D. Drug-induced nephrotoxicity: clinical impact and preclinical in vitro models. Mol Pharm. 2014;11(7):1933–48.
Dieterle F, Sistare F, Goodsaid F, Papaluca M, Ozer JS, Webb CP, et al. Renal biomarker qualification submission: a dialog between the FDA-EMEA and predictive safety testing consortium. Nat Biotechnol. 2010;28(5):455–62.
Dieterle F, Marrer E, Suzuki E, Grenet O, Cordier A, Vonderscher J. Monitoring kidney safety in drug development: emerging technologies and their implications. Curr Opin Drug Discov Devel. 2008;11(1):60–71.
Zhang PL, Mashni JW, Sabbisetti VS, Schworer CM, Wilson GD, Wolforth SC, et al. Urine kidney injury molecule-1: a potential non-invasive biomarker for patients with renal cell carcinoma. Int Urol Nephrol. 2014;46(2):379–88.
Obermuller N, Geiger H, Weipert C, Urbschat A. Current developments in early diagnosis of acute kidney injury. Int Urol Nephrol. 2014;46(1):1–7.
Wang F, Yang B, Ling GH, Yao C, Jiang YS. Methylguanidine cytotoxicity on HK-2 cells and protective effect of antioxidants against MG-induced apoptosis in renal proximal tubular cells in vitro. Ren Fail. 2010;32(8):978–85.
Wilmer MJ, Ng CP, Lanz HL, Vulto P, Suter-Dick L, Masereeuw R. Kidney-on-a-chip technology for drug-induced nephrotoxicity screening. Trends Biotechnol. 2016;34(2):156–70.
Adler M, Ramm S, Hafner M, Muhlich JL, Gottwald EM, Weber E, et al. A quantitative approach to screen for nephrotoxic compounds in vitro. J Am Soc Nephrol. 2016;27(4):1015–28.
Aschauer L, Limonciel A, Wilmes A, Stanzel S, Kopp-Schneider A, Hewitt P, et al. Application of RPTEC/TERT1 cells for investigation of repeat dose nephrotoxicity: a transcriptomic study. Toxicol in Vitro. 2015;30(1 Pt A):106–16.
Aguado-Fraile E, Ramos E, Conde E, Rodriguez M, Martin-Gomez L, Lietor A, et al. A pilot study identifying a set of microRNAs as precise diagnostic biomarkers of acute kidney injury. PLoS One. 2015;10(6):e0127175.
Brown CD, Sayer R, Windass AS, Haslam IS, De Broe ME, D'Haese PC, et al. Characterisation of human tubular cell monolayers as a model of proximal tubular xenobiotic handling. Toxicol Appl Pharmacol. 2008;233(3):428–38.
Burckhardt G, Burckhardt BC. In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy. Handb Exp Pharmacol. 2011;201:29–104.
Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014:967826.
Yoon HE, Yang CW. Established and newly proposed mechanisms of chronic cyclosporine nephropathy. Korean J Intern Med. 2009;24(2):81–92.
Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43(5):1003–12.
Jafari A, Khalili H, Dashti-Khavidaki S. Tenofovir-induced nephrotoxicity: incidence, mechanism, risk factors, prognosis and proposed agents for prevention. Eur J Clin Pharmacol. 2014;70(9):1029–40.
Ramamoorthy H, Abraham P, Isaac B. Mitochondrial dysfunction and electron transport chain complex defect in a rat model of tenofovir disoproxil fumarate nephrotoxicity. J Biochem Mol Toxicol. 2014;28(6):246–55.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Babak T, Zhang W, Morris Q, Blencowe BJ, Hughes TR. Probing microRNAs with microarrays: tissue specificity and functional inference. RNA. 2004;10(11):1813–9.
Hu J, Xu Y, Hao J, Wang S, Li C, Meng S. MiR-122 in hepatic function and liver diseases. Protein Cell. 2012;3(5):364–71.
Tian Z, Greene AS, Pietrusz JL, Matus IR, Liang M. MicroRNA-target pairs in the rat kidney identified by microRNA microarray, proteomic, and bioinformatic analysis. Genome Res. 2008;18(3):404–11.
Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4(3):143–59.
Ha TY. MicroRNAs in human diseases: from cancer to cardiovascular disease. Immune Netw. 2011;11(3):135–54.
Starkey Lewis PJ, Dear J, Platt V, Simpson KJ, Craig DG, Antoine DJ, et al. Circulating microRNAs as potential markers of human drug-induced liver injury. Hepatology. 2011;54(5):1767–76.
Kia R, Kelly L, Sison-Young RL, Zhang F, Pridgeon CS, Heslop JA, et al. MicroRNA-122: a novel hepatocyte-enriched in vitro marker of drug-induced cellular toxicity. Toxicol Sci. 2015;144(1):173–85.
Pavkovic M, Riefke B, Ellinger-Ziegelbauer H. Urinary microRNA profiling for identification of biomarkers after cisplatin-induced kidney injury. Toxicology. 2014;324:147–57.
Kanki M, Moriguchi A, Sasaki D, Mitori H, Yamada A, Unami A, et al. Identification of urinary miRNA biomarkers for detecting cisplatin-induced proximal tubular injury in rats. Toxicology. 2014;324:158–68.
Li YF, Jing Y, Hao J, Frankfort NC, Zhou X, Shen B, et al. MicroRNA-21 in the pathogenesis of acute kidney injury. Protein Cell. 2013;4(11):813–9.
Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012;23(2):252–65.
Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17(2):193–9.
Bhatt K, Zhou L, Mi QS, Huang S, She JX, Dong Z. MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol Med. 2010;16(9–10):409–16.
Wilmer MJ, Saleem MA, Masereeuw R, Ni L, van der Velden TJ, Russel FG, et al. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010;339(2):449–57.
Nieskens TT, Peters JG, Schreurs MJ, Smits N, Woestenenk R, Jansen K, et al. A human renal proximal tubule cell line with stable organic anion transporter 1 and 3 expression predictive for antiviral-induced toxicity. AAPS J. 2016;18(2):465–75.
Kohler JJ, Hosseini SH, Green E, Abuin A, Ludaway T, Russ R, et al. Tenofovir renal proximal tubular toxicity is regulated by OAT1 and MRP4 transporters. Lab Investig. 2011;91(6):852–8.
Chung AC, Lan HY. MicroRNAs in renal fibrosis. Front Physiol. 2015;6:50.
Villarroya-Beltri C, Baixauli F, Gutierrez-Vazquez C, Sanchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol. 2014;28:3–13.
Etheridge A, Lee I, Hood L, Galas D, Wang K. Extracellular microRNA: a new source of biomarkers. Mutat Res. 2011;717(1–2):85–90.
Nassirpour R, Mathur S, Gosink MM, Li Y, Shoieb AM, Wood J, et al. Identification of tubular injury microRNA biomarkers in urine: comparison of next-generation sequencing and qPCR-based profiling platforms. BMC Genomics. 2014;15:485.
Watts AE, Millar NL, Platt J, Kitson SM, Akbar M, Rech R, et al. MicroRNA29a treatment improves early tendon injury. Molecular therapy : the journal of the American Society of Gene Therapy. 2017;25(10):2415–26.
Mladinov D, Liu Y, Mattson DL, Liang M. MicroRNAs contribute to the maintenance of cell-type-specific physiological characteristics: miR-192 targets Na+/K+-ATPase beta1. Nucleic Acids Res. 2013;41(2):1273–83.
Baker LA, Lee KC, Palacios Jimenez C, Alibhai H, Chang YM, Leckie PJ, et al. Circulating microRNAs reveal time course of organ injury in a porcine model of acetaminophen-induced acute liver failure. PLoS One. 2015;10(5):e0128076.
Bosomworth MP, Aparicio SR, Hay AW. Urine N-acetyl-beta-D-glucosaminidase—a marker of tubular damage? Nephrol Dial Transplant. 1999;14(3):620–6.
Trietsch SJ, Naumovska E, Kurek D, Setyawati MC, Vormann MK, Wilschut KJ, et al. Membrane-free culture and real-time barrier integrity assessment of perfused intestinal epithelium tubes. Nat Commun. 2017;8(1):262.
Acknowledgements
The OrganoPlate® is a registered trademark of MIMETAS BV.
Funding
This project was financially supported by NC3Rs (National Center for the Replacement, Refinement and Reduction of Animals in Research), UK, under the Crack-it challenge 15 (NephroTube project no. 37497-25920).
Author information
Authors and Affiliations
Contributions
LSD: Conception and planning of the study, interpretation of the data, writing of the manuscript
MC, DR, MV, SH: Performance of the cell culture experiments and treatments in 2D and 3D
LM: Extraction and analysis of miRNA, data and statistical analysis, writing of the manuscript
MW, RM, HL, JV: Scientific discussions on planning and executing the project, support with interpretation of data, revision of the manuscript
Corresponding author
Additional information
Guest Editors: Rosalinde Masereeuw and Raymond Lai
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Suter-Dick, L., Mauch, L., Ramp, D. et al. Combining Extracellular miRNA Determination with Microfluidic 3D Cell Cultures for the Assessment of Nephrotoxicity: a Proof of Concept Study. AAPS J 20, 86 (2018). https://doi.org/10.1208/s12248-018-0245-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-018-0245-2