
Abstract
Investigation of the pharmacokinetics (PK) of a compound is of significant importance during the early stages of drug development, and therefore several in vitro systems are routinely employed for this purpose. However, the need for more physiologically realistic in vitro models has recently fueled the emerging field of tissue-engineered 3D cultures, also referred to as organs-on-chips, or microphysiological systems (MPSs). We have developed a novel fluidic platform that interconnects multiple MPSs, allowing PK studies in multi-organ in vitro systems along with the collection of high-content quantitative data. This platform was employed here to integrate a gut and a liver MPS together in continuous communication, and investigate simultaneously different PK processes taking place after oral drug administration in humans (e.g., intestinal permeability, hepatic metabolism). Measurement of tissue-specific phenotypic metrics indicated that gut and liver MPSs can be fluidically coupled with circulating common medium without compromising their functionality. The PK of diclofenac and hydrocortisone was investigated under different experimental perturbations, and results illustrate the robustness of this integrated system for quantitative PK studies. Mechanistic model-based analysis of the obtained data allowed the derivation of the intrinsic parameters (e.g., permeability, metabolic clearance) associated with the PK processes taking place in each MPS. Although these processes were not substantially affected by the gut-liver interaction, our results indicate that inter-MPS communication can have a modulating effect (hepatic metabolism upregulation). We envision that our integrative approach, which combines multi-cellular tissue models, multi-MPS platforms, and quantitative mechanistic modeling, will have broad applicability in pre-clinical drug development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Investigation of the pharmacokinetic (PK) properties of a drug is particularly important during pre-clinical drug development, as it facilitates decision-making regarding the dosage regimen in early-phase clinical studies. Animal species are often used to study the PK of a compound under development; however, such studies are costly, can be considered unethical, and most importantly they often fail to accurately capture the human phenotype. For these reasons, several in vitro systems have been also developed and are routinely employed to investigate the absorption, distribution, metabolism, and excretion (ADME) of a compound (1). Although these systems have been extremely valuable in drug development, they are not without limitations and the need for more physiologically realistic and better predictive in vitro models is widely recognized.
The emerging field of microphysiological systems (MPSs), also known as organs-on-chips (OOC), holds promise for a transformative change in pre-clinical drug development (2–4). The term MPSs encompasses a range of compositionally complex (i.e., more than one cell type) and 3D cell cultures that are dynamically perfused, thus capturing more features of human organ or tissue function compared to the traditional static 2D cell cultures (3–8). In addition, the use of micro-machined biomimetic reactor platforms allows in vitro re-creation of the mechanical, fluidic, spatial, and chemical stimuli and cues that a tissue is exposed to in vivo (9). To better recapitulate human physiology at a systemic level and establish better pharmacologic pre-clinical models that translate more accurately to human outcomes, multi-MPS platforms have been developed to interconnect several MPSs representing facets of different organs together and thus allow organ-organ interaction and cross-signaling (10–18). It is important to highlight that neither single- nor multi-MPS technologies are aiming to reproduce an entire organ or the entire human body, respectively, but rather to mimic specific organ functions, microarchitecture, and organ-organ crosstalk relevant to the biological question of interest.
This emerging field has the potential to offer novel cutting-edge in vitro systems to explore the PK properties of a drug pre-clinically. For such investigations, the integration of gut and liver MPSs is crucial as these two organs play a central role on the bio-distribution and bioavailability of an orally administrated compound (through processes such as intestinal permeability and hepatic metabolism). Over the last years, significant progress has been made towards the development of such integrated gut-liver fluidic platforms (7,12,14,19–23). However, all these technologies published to date have at least one of the following limitations with respect to their application for PK investigations: (1) they employ materials (e.g., PDMS) that non-specifically adsorb lipophilic compounds; (2) they use relatively low culture volumes and cell numbers that can negatively affect the output biological signal and the collection of high-content measurements; (3) they do not allow continuous access to the MPS compartments for direct and frequent sampling of circulating drugs/metabolites and thus data-rich quantitative PK profiles across all platform compartments cannot always be obtained; and (4) they are usually not coupled with a mathematical modeling methodology to disentangle the biology-related parameters (e.g., intestinal permeability, intrinsic hepatic clearance) from system-specific processes and parameters (e.g., flow rates, surface areas), a step particularly crucial for the subsequent in vitro to in vivo translation.
In this work, we used a novel mesofluidic multi-MPS platform coupled with computational modeling to address these limitations. Specifically, this platform was used to interconnect a gut MPS with a well-characterized liver MPS that has repeatedly demonstrated its value for PK-related investigations (hepatic clearance determination (6,24), assessment of population variability in drug metabolism (24), effects of inflammation on drug metabolism (25), and drug-drug interactions (26)). The overall objective of this work is to introduce and illustrate the applicability of this novel multi-MPS technology for quantitative PK investigations, and also provide the necessary methodological foundation for its robust use in the future.
To do that, we first practically illustrate the operation of this integrated gut-liver system to explore the PK properties of drugs in vitro. Specifically, the kinetics of two compounds, diclofenac (DCF) and hydrocortisone (HC), were studied by collecting serial concentration-time measurements across the different compartments of the employed multi-MPS platform. Secondly, we investigated the effects of the interaction/crosstalk between the gut and liver MPSs on the on-platform PK-related processes (e.g., permeability through the epithelial barrier of the gut MPS, intrinsic metabolic clearance of the liver MPS) and other MPS functionality metrics (e.g., transepithelial electrical resistance (TEER) levels, albumin production). To do so, kinetic experiments were performed under several experimental perturbations (only the liver MPS on-platform, only the gut MPS on-platform, both the gut and liver MPSs on-platform). Finally, we employed mechanistic modeling demonstrating the value of this computational approach for the analysis of the experimental data obtained from such a complex in vitro system. All the above are critical to enable the future use of these systems to predict the PK properties of a compound before proceeding to clinical studies in humans.
METHODS
Multi-MPS Platform Description
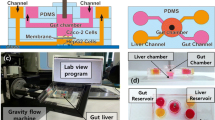
The multi-MPS platform employed in this work (see Fig. 1, Table I) is similar in size to a regular multi-well plate and has been developed with the aim to allow quantitative pharmacology studies in interconnected MPSs of up to four different organs. The top plate (fluidic plate) of this micro-machined platform is constructed from polysulfone plastic, taking advantage of its superior properties in terms of minimal drug adsorption. The platform incorporates five distinct compartments to accommodate a transwell-style gut MPS, a 3D-perfused liver MPS, two additional transwell-style MPSs of organs of interest, and a mixing chamber (mixer) that aims to represent the systemic circulation compartment (equivalent to blood in the human body). The different platform compartments are interconnected with microfluidic channels and peristaltic micropumps that are fabricated on the bottom of the fluidic plate. The respective flow rates can be dynamically controlled through a graphical user interface, with pumps being remotely actuated through pneumatic tubing attached to the bottom plate (pneumatic manifold plate). Programmable control of pump rate and direction allows easy reconfiguration of the hardware to accommodate various experimental designs and applications.

Design and operation of the platform employed for gut and liver MPS integration. a Exploded view of the multi-MPS platform. The fluidic plate (in yellow), constructed from polysulfone plastic, contains distinct compartments to accommodate different MPSs. These compartments are interconnected with the aid of microfluidic channels and pumps that are fabricated on the bottom of this plate and allow circulation of culture medium. The pneumatic plate (clear acrylic) distributes compressed air and vacuum to small ports below each pump/valve chamber. A pumping membrane, made of translucent polyurethane, is clamped between the fluidic and the pneumatic plate in order to provide a sterile barrier and serve as the actuation layer of the pumps and valves. All the platform layers are fastened together with stainless steel screws and spring washers to form a single unit that can be treated as a traditional well plate. A cross-section of the multi-MPS platform (shown on the right) illustrates the topology of the gut MPS, liver MPS, and mixing chamber (mixer) compartments and the fact that these compartments can hold circulating medium. The two platform compartments that can house additional organ MPS (marked with “x”) were dynamically excluded through configuration of the respective flow rates in order to conduct PK studies in a dedicated gut-liver interaction system. b Schematic diagram illustrating the platform’s medium circulation flow pattern used in the current work. The systemic flow rate (Q syst) represents the total output from the mixing chamber and was partitioned as 75 and 25% towards the gut and the liver MPS compartments, respectively. c Schematic diagram of the perfused gut MPS on-platform. d Schematic diagram of the perfused liver MPS on-platform
In the current work, the two platform compartments that can house MPSs of additional organs were dynamically excluded (see Fig. 1) to allow the performance of PK studies in a dedicated gut-liver interaction system. The MPS interconnection scheme on-platform and the specification of the associated flow partitioning aimed to mirror as close as possible the organ physiology and flow partition in humans (27). Therefore, the gut and the liver MPS compartments were connected in series and the total output from the mixing chamber (systemic flow rate) was partitioned as 75 and 25% towards the gut and liver MPS compartments, respectively (see Fig. 1b). Consequently, the gut MPS receives its entire input flow directly from the mixing chamber; and the liver MPS receives 1/4 of its input flow directly from the mixing chamber (mimicking the hepatic artery blood flow) and the remaining 3/4 from the outflow of the gut MPS compartment (mimicking the portal vein blood flow). The systemic flow rate (Q syst) was set to 15 mL/day, based upon a computational model-guided approach (28) that indicated that this flow rate would be adequate to achieve distribution equilibrium conditions across the different MPS compartments and the mixing chamber in the time frame of the performed PK studies. Along with inter-MPS flows, each compartment employed self-circulation (1 μL/s for the mixing chamber and liver MPS compartments and 0.25 μL/s for the basal gut MPS compartment) to allow well-stirred conditions and efficient provision of oxygen and nutrients to the micro-tissues in each MPS compartment (Fig. 1c, d).
A particular advantage of this multi-MPS platform for PK-related studies is the flexibility it offers with regard to drug dosing (route of administration). Specifically, a compound can be administered to the apical side of the gut MPS (mimicking oral administration) to be absorbed and consequently distributed firstly to the liver MPS compartment (as in the human body) and then to the mixing chamber. Alternatively, it can be administered directly into the mixing chamber (mimicking an IV administration). To illustrate this dosing flexibility and the way that the platform operates under these different modes of drug administration, time-lapse videos of dye distribution were generated for each mode and can be found in the Online Supplement. For these dye distribution experiments, the platform’s flow configuration was set as described in the earlier paragraph, phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA) was added to each platform compartment at nominal volumes (see the “Liver and Gut MPS Description” section), and at t = 0, 100 μL of 4 mM Fast Green FCF (FDA-approved food dye) in water was added to either the gut MPS apical compartment or the mixing chamber. Each video frame represents 24 s, such that 1 min of video spans 12 h. The reader is also referred to the Supplementary Material for extensive details regarding the cleaning, assembly, and preparation of the multi-MPS platforms.
Liver and Gut MPS Description
The liver MPS employed in this work is a functionally equivalent version of the liver micro-bioreactor previously described by Domansky et al. (29), with the oxygenation conduit folded around the reactor well to reduce the physical footprint. This bioreactor has been previously utilized in a number of drug-related studies (6,24–26). Briefly, the reactor houses a scaffold in the form of a thin (0.25 mm) polystyrene disc permeated with an array of 301 channels (0.3-mm diameter each) that is seated in the flow path on a microporous membrane. Hepatocytes are seeded into the scaffold, where they attach and form 3D structures. In this work, the liver MPS consisted of a 3D co-culture of human primary cryopreserved hepatocytes and Kupffer cells at a 10:1 ratio (600,000 hepatocytes:60,000 Kupffer cells), with the aim to recapitulate metabolic and immune function. The cells of the liver MPS were seeded on-platform 3 days prior to the start of the PK experiments to allow for micro-tissue formation and recovery from any seeding-related stress responses. The liver MPS preparation protocol is extensively described in the Supplementary Material and in prior publications (25,26).
The gut MPS consisted of Caco2-BBe epithelial cells and mucin-producing goblet cells (HT29-MTX) at a 9:1 ratio, plus an immune component consisting of primary monocyte-derived dendritic cells at a 10:1 ratio (1,000,000 epithelial cells at monolayer maturation:100,000 dendritic cells). The gut MPS was seeded in a standard 12-well transwell and matured for 3 weeks off-platform (static) prior to its integration in the multi-MPS platform at the start of the PK experiments (see Supplementary Material for all the details regarding the gut MPS preparation). Both gut and liver MPSs include an immune component with the scope to enable various biological and pharmacological future applications. The investigation of the role and contribution of the immune component to the function of these MPSs is outside the scope of the current paper as it has been extensively described previously (25,26,30,31).
For all the on-platform PK experiments, a serum-free common medium was circulating between the mixing chamber, the liver MPS compartment, and the basolateral side of the gut MPS compartment (Fig. 1). This common medium contained 500 mL Williams E medium, 20 mL Gibco Cocktail B, 80 nM HC, and 1% penicillin/streptomycin (P/S). The apical side of the gut MPS compartment was provided with serum-free apical medium that contained phenol red-free DMEM with 1× ITS, 1× NEAA, 1× GlutaMax, and 1% P/S. The mixing chamber and the liver MPS compartments hold nominal medium volumes of 1 and 1.4 mL, respectively. The apical and basolateral sides of the gut MPS compartments hold media volumes of 0.5 and 1.5 mL, respectively, while the surface area of the gut MPS epithelial monolayer was 1.12 cm2 (surface area of the gut MPS transwell). For both MPSs, tissue culture functionality was assessed by measurement of associated phenotypic metrics, specifically, albumin production for the liver MPS, TEER, and mucin production for the gut MPS (see Supplementary Material for extensive details on the methods for the quantification of these metrics).
Design and Execution of the PK Studies in the Multi-MPS Platform
To investigate the effect of organ-organ interaction/crosstalk on the PK processes taking place in the multi-MPS platform, the current study involved three experimental arms, each involving four replicate platforms (n = 4). In the first arm, referred to as “gut only” in this manuscript, only the gut MPS was present on-platform and no tissue was present in the liver compartment. In the second arm, referred to as “liver only,” only the liver MPS was present on-platform and no tissue was present in the gut compartment. Finally, in the last arm, referred to as “gut/liver,” both the gut and the liver MPSs were present on-platform.
For the arms that included the gut MPS on-platform (gut only, gut/liver), DCF powder was dissolved in the gut apical medium, filter-sterilized, and administered to the apical compartment of the gut MPS (mimicking an oral administration). For the liver only arm, DCF powder was dissolved in the common medium, filter-sterilized, and administered to the mixing chamber compartment (mimicking an IV administration). The same DCF concentration (40 μM) was administered in the dosed compartments across all experimental arms. Model-based simulations, utilizing prior knowledge from in-house single-MPS DCF experiments, indicated that such a DCF dose on-platform will likely result in mixing chamber maximum concentrations (C max) that are in the range of the clinically observed DCF C max in plasma following standard therapeutic dosing (32).
Corticosteroids, either HC or dexamethasone, are almost always included in liver cell culture media because their presence regulates CYP450 activity and other differentiated functions. As in the current work HC was a component of the circulating common medium (see the previous section), HC kinetics were examined on-platform simultaneously to the DCF kinetics. Every platform compartment in which a common medium was added at the beginning (t = 0) of the kinetic experiments (basolateral compartment of the gut MPS, liver MPS compartment, and mixing chamber compartment) was treated as receiving a HC dose.
For all experimental arms, media samples (75 μL) were taken from the different platform compartments at predetermined sampling times (see the “Results” section). At the end of the PK experiments (72-h post-dose), the liver micro-tissue was removed from the platform, snap-frozen, and lysed in 500 μL of 100% methanol for intracellular drug quantification (see Supplementary Material for further details). All the obtained media and lysate samples were analyzed for the quantification of DCF, 4-hydroxy-diclofenac (4-OH-DCF), and HC using a 3-in-1 LC-MS/MS method (see Supplementary Material for details).
Off-Platform PK Studies in Single MPS Technologies
In parallel to the PK studies performed in this multi-MPS platform (on-platform), kinetic studies were also performed in single MPS technologies off-platform to assess whether the hardware/operational-related differences have substantial impact on the underlying PK-related processes. Firstly, DCF and HC kinetics were investigated off-platform in a static gut MPS. This static gut MPS is exactly equivalent to on-platform gut MPS as described earlier (e.g., the same cell types/numbers/ratio, seeding, maturation, medium components, volumes) but is not incorporated into a fluidic device and thus lacks basolateral medium recirculation. Secondly, HC kinetics were investigated off-platform in a liver MPS device, the LiverChip™ (6,25,26), while the kinetics of DCF in the LiverChip™ have been previously reported (24). The liver MPS housed in the LiverChip™ device was identical to the liver MPS in our multi-MPS platform (e.g., the same cell types/numbers/ratio, seeding, medium components), except that in the multi-MPS platform, the oxygenation conduit is folded around the reactor. Similarly to the on-platform experiments, media samples were taken during the off-platform kinetic experiments at predetermined sampling times (see Supplementary Figures S3-S5) and subsequently analyzed for drug quantification with an LC-MS/MS method (see Supplementary Material). Cell culture health and function were assessed by measurement of associated metrics, more specifically, albumin production in the LiverChip™, TEER, and mucin production in the static gut MPS.
Model-Based Analysis of the Experimental PK Data
The experimentally observed DCF and HC kinetic data on-platform were analyzed with the aid of mechanistic PK models (see the “Results” section), the structure of which was guided by the MPS interconnection scheme and the physiological processes taking place on-platform. The developed mechanistic models were fitted to the experimental data (across all platform compartments) to estimate the model parameters that represent the PK processes of interest (e.g., intrinsic metabolic clearance of the gut and liver MPS, permeability across the epithelial barrier of the gut MPS). A similar approach was used for the analysis of the experimental data obtained off-platform in single MPS technologies (static gut, LiverChip™).
Model-based analysis was performed in NONMEM 7.3 (ICON Development Solutions, Ellicott City, MD) and the Markov-Chain Monte Carlo (MCMC) Bayesian estimation algorithm (BAYES) was used for parameter estimation. The mass balance differential equations were evaluated with an LSODA differential equation solver that can handle a mixed system of both stiff and non-stiff differential equations (ADVAN13). Additional investigations of the modeling output and graphical analyses were performed in Matlab R2015b (The MathWorks Inc., Natick, MA).
The parameters upon estimation were log-transformed (the natural log of the parameters was estimated) to allow sampling of the log-parameters from the multivariate normal distribution that has been assigned for the priors. In order to provide completely uninformative priors for the estimated parameters, the variance of the normal prior was set to the value of 10,000. Inter-platform variability of the estimated parameters was treated to be negligible and the residual (unexplained) variability in the observed data was modeled with an additive error model on the domain of the log-transformed data. Three separate MCMC chains were run simultaneously with dispersed initial estimates and 550,000 iterations were requested for each chain (the first 50,000 were treated as “burn-in phase” and were discarded). MCMC convergence was investigated by visual inspection of the MCMC chain histories (trace plots) to assess mixing and by the calculation of the Gelman-Rubin diagnostic (33) (potential scale reduction factor metric) using the “CODA” package in R (34). If this calculated metric had a value close to 1 (<1.1), the within- and between-chains variability can be considered identical and it was assumed that convergence has been achieved (35). The samples from the “stationary phase” (500,000) of the three different chains were pooled to summarize the posterior distribution of the estimated parameters and the posterior marginal probability densities were obtained by fitting a kernel density function to the posterior histogram.
RESULTS
Assessment of Gut and Liver MPS Functionality
Specific phenotypic markers were measured at the end of the kinetic experiments (3 days post drug administration) to assess cell culture functionality for the gut and the liver MPSs on-platform (Fig. 2). It was evident that TEER (indicating the integrity of the gut MPS epithelial barrier) and mucin production (indicating functionality of the HT29-MTX cells in the gut MPS) were comparable in the gut only and gut/liver arms (Fig. 2a, b). Similarly, albumin production (indicating hepatocyte functionality) was comparable between the liver only and gut/liver arms (Fig. 2c). These results indicate that the gut-liver crosstalk in the multi-MPS platform does not affect the functionality metrics monitored in this work for either the gut or the liver MPS. Additional data regarding these phenotypic markers either on-platform before drug administration, or off-platform in single MPS technologies (static gut and LiverChip™) are provided in Supplementary Table S1.

Phenotypic markers measured at the end of the kinetic experiments (3 days post-dose) to assess cell culture functionality for the gut (TEER (a), secreted mucin (b)) and the liver MPS (albumin production (c)) on-platform. Red lines represent the mean across the measurements obtained in four replicate platforms in each experimental arm; purple boxes extend the mean by ±1 SD (standard deviation). “Gut/Liver” refers to the experimental arm where both the gut and liver MPSs are present in the platform; “Gut only” and “Liver only” refer to the experimental arms where only the gut or only the liver MPS is present in the platform, respectively
DCF Kinetics in the Multi-MPS Platform
The experimental data and modeling results regarding DCF PK on-platform are presented in Figs. 3, 4, and 5 for the gut only, liver only, and gut/liver study arms, respectively. In the gut only arm, DCF was absorbed by the gut epithelium from the apical to the basolateral side of the gut MPS and then distributed to the “tissue-free” liver compartment and the mixing chamber of the platform (Fig. 3a, b). DCF metabolism in this arm was minimal and the concentration-time profiles appeared to reach a plateau approximately 24 h after the dose (Fig. 3b). In the liver only arm, DCF after being dosed to the mixing chamber quickly distributed to the tissue-free gut compartment and the liver MPS compartment (Fig. 4a, b). In contrast to the gut only experimental arm, DCF metabolism in the liver only arm was substantial and diminishing drug concentrations were observed across the 3-day experiment (Fig. 4b). Two discrete phases were observed in the mixing chamber concentration-time profile: a rapid decline in the first few hours when both distribution to the other platform compartments and elimination were taking place and a less steep decline in the remaining time when elimination was the main driving force of the concentration-time profile (Fig. 4b). In the gut/liver arm, DCF after being dosed to the apical side of the gut MPS was absorbed through the epithelial barrier to the basolateral side and then distributed to the liver MPS compartment (main site of elimination) and the mixing chamber of the platform (Fig. 5a, b). Consequently, the obtained concentration-time profiles in the mixing chamber have a clear ascending (absorption) and descending (elimination) phase, similar to a typical plasma concentration-time profile observed in humans after an oral drug administration.

Model structure, observed DCF data, and model fit referring to the experimental arm where only the gut MPS is present in the platform (“Gut only”). a Structure of the mechanistic PK model to describe the “Gut only” arm DCF data. The red square marker indicates the site of DCF administration (apical side of the gut MPS). P DCF permeability coefficient across the epithelial barrier of the gut MPS, CLint g intrinsic metabolic DCF clearance of the gut MPS, Q syst systemic flow rate during platform operation. b Mechanistic PK model fit (red line) on top of the experimentally observed DCF data (closed gray circles) across the different platform compartments. c Trace plots of three different MCMC chains started from different initial estimates (plotted in different colors); values for each estimated parameter (in the log-transformed domain) are plotted against the iteration number; on the top right corner of each subplot, the potential scale reduction factor (R) from the Gelman-Rubin diagnostic is reported. d Posterior marginal probability density for each estimated model parameter

Model structure, observed DCF data, and model fit referring to the experimental arm where only the liver MPS is present in the platform (“Liver only”). a Structure of the mechanistic PK model to describe the “Liver only” arm DCF data. The red square marker indicates the site of DCF administration (mixing chamber). CLint l intrinsic metabolic DCF clearance of the liver MPS, Q syst systemic flow rate during platform operation. b Mechanistic PK model fit (red line) on top of the experimentally observed DCF data (closed gray circles) across the different platform compartments. c Trace plots of three different MCMC chains started from different initial estimates (plotted in different colors); values for the estimated parameter (in the log-transformed domain) are plotted against the iteration number; on the top right corner, the potential scale reduction factor (R) from the Gelman-Rubin diagnostic is reported. d Posterior marginal probability density for the estimated model parameter

Model structure, observed DCF data, and model fit referring to the experimental arm where both the gut and the liver MPSs are present in the platform (“Gut/Liver”). a Structure of the mechanistic PK model to describe the “Gut/Liver” arm DCF data. The red square marker indicates the site of DCF administration (apical side of the gut MPS). P DCF permeability coefficient across the epithelial barrier of the gut MPS, CLint g intrinsic metabolic DCF clearance of the gut MPS, CLint l intrinsic metabolic DCF clearance of the liver MPS, Q syst systemic flow rate during platform operation. b Mechanistic PK model fit (red line) on top of the experimentally observed DCF data (closed gray circles) across the different platform compartments. c Trace plots of three different MCMC chains started from different initial estimates (plotted in different colors); values for each estimated parameter (in the log-transformed domain) are plotted against the iteration number; on the top right corner of each subplot, the potential scale reduction factor (R) from the Gelman-Rubin diagnostic is reported. d Posterior marginal probability density for each estimated model parameter
The employed mechanistic PK models for the three different experimental arms (Figs. 3a, 4a, and 5a) accurately described the observed experimental DCF data across all platform compartments (Figs. 3b, 4b, and 5b). The reader is referred to the Supplementary Material for further details on the model assumptions and mass balance differential equations. Convergence of the parameter estimation procedure was indicated for all experimental arms by MCMC trace plots and the calculation of the Gelman-Rubin diagnostic (Figs. 3c, 4c, and 5c). All the model-related parameters (both fixed and estimated) for the studied experimental arms are summarized in Table I and the complete posterior distributions with regard to the estimated parameters are shown in Figs. 3d, 4d, and 5d.
Model-based analysis of the experimental data across the three different arms allowed mechanistic investigation of the effect of gut-liver interaction on the PK-specific parameters on-platform. Specifically, the parameter posterior distributions derived from the arm where both the gut and liver MPSs were present on the platform (gut/liver arm) were compared with those from the arms where only the gut or only the liver MPS was on-platform. There was no evidence (Fig. 6) that the gut-liver interaction/crosstalk alters the DCF permeability across the gut MPS epithelial barrier (P) or the gut MPS intrinsic metabolic clearance (CLintg). In contrast, our results indicate with enough confidence (non-overlapping 90% credible intervals) that the gut-liver crosstalk increases the liver MPS intrinsic metabolic clearance (Clintl), although the magnitude of the increase is marginal (Fig. 6).

Effect of gut-liver interaction/crosstalk on DCF PK model parameters. For each parameter, the posterior distributions referring to the arm where both the gut and liver MPS were present in the platform (“Gut/Liver”) are highlighted with red, while those referring to the arms where only the gut MPS (“Gut only”) or only the liver MPS (“Liver only”) was present are highlighted with gray. The line above each posterior distribution indicates the 90% credible interval of the parameter in each arm (red color for “Gut/Liver” arm and black for “Gut Only” or “Liver only” arm). P DCF permeability coefficient across the epithelial barrier of the gut MPS, CLint g intrinsic metabolic DCF clearance of the gut MPS, CLint l intrinsic metabolic DCF clearance of the liver MPS
The production of a major DCF CYP2C9 metabolite, 4-OH-diclofenac (4-OH-DCF), was also monitored on-platform and the associated mixing chamber concentration-time profiles are presented in Fig. 7a. 4-OH-DCF was produced in all experimental arms, however as expected, its levels were very low (<0.05 μM) when only the gut MPS was present on-platform. On the contrary, significant 4-OH-DCF production was observed in the arms where the liver MPS was on-platform (Fig. 7a). The area under the mixing chamber concentration-time curve (AUC0-72h) was calculated for both DCF and 4-OH-DCF and the metabolite/parent AUC0-72h ratio was calculated to quantify the degree of parent-to-metabolite conversion in each experimental arm (Fig. 7a). While this ratio was on average only 0.01 for the gut only arm, it was 0.28 for the liver only arm and 0.31 when both the gut and liver MPSs were present in the platform (gut/liver arm).

Additional PK-related investigations. a The two top and the bottom left subplot illustrate DCF (gray) and 4-OH-DCF (red) concentration-time profiles measured in the mixing chamber compartment of the platform across the different experimental arms. The subplot on the bottom right illustrates the metabolite to parent (M/P) area under the mixing chamber concentration-time curve (AUC0-72h) ratio across the different experimental arms. Red lines represent the mean value across four replicate platforms in each experimental arm; purple boxes extend the mean by ±1 SD (standard deviation). “Gut only” and “Liver only” refer to the arms where only the gut MPS or only the liver MPS was present on-platform and “Gut/Liver” refers to the arm where both the gut and liver MPS were present on-platform. b Ratio of compound (DCF or 4-OH-DCF) concentration in the liver MPS cells to compound concentration in the surrounding liver compartment medium (liver Kp) across the experimental arms where the liver MPS was on-platform (“Gut/Liver” and “Liver only” arms). Red lines represent the mean value across four replicate platforms in each experimental arm; purple boxes extend the mean by ±1 SD (standard deviation)
Finally, to quantify the liver MPS intracellular accumulation of DCF and 4-OH-DCF, cell lysis was performed at the end of the PK experiment (72 h after dosing) and the ratio (Kp) of compound concentration in the cells to extracellular compound concentration in the liver compartment medium was calculated (Fig. 7b). It was evident that for both DCF and 4-OH-DCF, intracellular accumulation was substantial and the cellular concentrations were many-fold higher than the extracellular concentrations measured in the liver compartment of the platform (Fig. 7b). In addition, our results suggest that gut-liver interaction does not substantially affect drug partition to the liver micro-tissue (Fig. 7b). Specifically, DCF and 4-OH-DCF liver Kp values for the gut/liver arm were not substantially different from the respective values when only the liver MPS was on-platform (liver only arm).
HC Kinetics in the Multi-MPS Platform
HC was contained in the circulating common media, and thus its kinetics on-platform were simultaneously investigated across all the studied experimental arms. Mechanistic PK models were developed with respect to both the gut only and the liver only arms (Supplementary Figures S1, S2). These models provided an accurate description of the respective experimental HC data and allowed the estimation of mechanistic PK parameters of interest (P, CLintg, CLintl) for HC in the absence of any gut-liver interaction (Supplementary Figures S1, S2 and Table S2). Subsequently, these parameters were used as inputs into a mechanistic PK model that assumes that both the gut and liver MPSs are on-platform (Fig. 8a). Model simulations were then performed to assess the success in predicting the experimental HC data in the gut/liver arm using parameters derived from the gut only and liver only arms. It was evident that model predictions (without any parameter fitting) accurately capture the observed HC experimental data in the gut/liver arm (Fig. 8b), providing further validation to the hypothesis that the effects of gut-liver interaction/crosstalk, if any, are not substantially affecting the PK processes (permeability, drug metabolism) taking place in the gut or the liver MPS.

Model structure, observed HC data, and model predictions referring to the experimental arm where both the gut and the liver MPSs were present in the platform (“Gut/Liver”). a Structure of the mechanistic PK model to describe the “Gut/Liver” arm HC data. The red square markers indicate the site of HC administration (HC is contained in the common medium, thus all platform compartments where common medium was added at the beginning (t = 0) of the kinetic experiments, were treated as receiving a HC dose). P HC permeability coefficient across the epithelial barrier of the Gut MPS, CLint g intrinsic metabolic HC clearance of the gut MPS, CLint l intrinsic metabolic HC clearance of the liver MPS, Q syst systemic flow rate during platform operation. b Mechanistic PK model predictions (red line) on top of the experimentally observed HC data (closed gray circles) across the different platform compartments
Off-Platform DCF and HC Kinetics in Single MPS Technologies
DCF and HC kinetics were also investigated in a static gut MPS off-platform. The observed data and their model-based analysis are presented in Supplementary Figures S3 and S4 (for DCF and HC, respectively). In addition, HC kinetics were investigated in the LiverChip™ (isolated liver MPS off-platform). The derived data and their model-based analysis are presented in Supplementary Figure S5. The kinetics of DCF in the LiverChip™ have been extensively studied and reported in a previous work (24). The values of the mechanistic PK parameters of interest (P, CLintg, CLintl) obtained from the model-based analysis of all these off-platform experimental data are summarized in Supplementary Table S3. These values were generally comparable to those derived on-platform in the gut only or liver only arms as the ratio of the off-platform to the respective on-platform values was between 0.77 and 1.19 for all PK parameters of interest and for both DCF and HC with the sole exception of DCF CLintg (value obtained in the static gut MPS was 1.92 times the value obtained in the gut only arm on-platform). Note that the on-platform values are not expected to exactly match those derived off-platform due to nominal system (hardware) and operational differences (e.g., inter- and intra-MPS flow rates). However, a rough agreement provides further confidence in the developed multi-MPS platform as it illustrates that the hardware employed does not have any negative impact on the underlying biological processes taking place.
DISCUSSION
The emerging field of MPS coupled with recent advances in the development of bio-engineered fluidic platforms not only allows more physiologically realistic in vitro models of human organ/tissue function but also permits their interconnection towards a “physiome-on-a-chip” paradigm (also referred to as “human-on-a-chip”) (36,37). Multi-organ MPS technologies, encompassing organ-organ interaction and communication, can more closely capture the complexity of in vivo human physiology, and thus hold promise for significant applications related to disease biology, personalized pharmacotherapy, and drug development. In the current work, we focused on the application of a novel multi-MPS platform to interconnect a gut and a liver MPS, and investigate different important PK processes taking place after oral drug administration (e.g., intestinal absorption, hepatic metabolism).
The multi-MPS platform employed in this work has unique properties favoring its use for quantitative PK and, in extension, pharmacodynamic (PD) investigations, as it was specifically designed and developed with this purpose in mind. In contrast to other existing individual MPS and multi-MPS devices that are constructed from polydimethylsiloxane (PDMS), the fluidic plate in this platform is constructed from polysulfone (PSF). PDMS is a material known to adsorb and partition hydrophobic and high logD compounds (38), which can significantly hinder quantitative pharmacology and drug development-related applications (3,39). PSF reduces these concerns because of its superior properties in terms of minimal surface adsorption and bulk absorption of hydrophobic/lipophilic molecules (40). We recently illustrated using the LiverChip™ technology (made with the same material as the multi-MPS platform employed in this work) the absence of non-specific binding to PSF for six drugs, including DCF, spanning a range of different physicochemical properties (24).
Additionally, in contrast to the majority of the available multi-MPS technologies, which are configured as closed systems and operate with very small fluid volumes (μL range) using microfluidic chips, our multi-MPS platform has an open system configuration and operates at the mesofluidic scale (mL range). This offers significant advantages in the context of quantitative PK/PD investigations. Firstly, the open system configuration allows direct, easy, and frequent sampling of circulating drugs, metabolites, and other biomarkers across the different platform compartments and consequently permits the generation of concentration-time profiles that can be computationally analyzed (e.g., see Figs. 3, 4, and 5). Secondly, the meso-scale medium volumes and tissue sizes allow quantitative high-content measurements with frequent sampling.
Finally, another unique advantage of the employed multi-MPS platform for quantitative PK/PD studies is that it can be flexibly configured through the dynamic control of the flows inter-connecting the different platform compartments. This allowed in this work the exclusion of two of the platform compartments and execution of PK studies in a dedicated gut-liver interaction system that has an MPS interconnection scheme that mimics the analogous physiology and flow partition in humans. This set-up can be easily altered to incorporate additional organ(s) MPS referring to the efficacy or the toxicity site of a drug under investigation. Dynamic control of the different flow rates can enable precise modulation of concentration-time profiles in such downstream organ MPSs of pharmacological/PD interest.
A gut-liver interaction system is an important building block for any multi-MPS technology focusing on PK/PD investigations. With that in mind, we demonstrated in this work that a gut and a liver MPS can be fluidically coupled with circulating common medium without compromising MPS functionality. More specifically, we quantified tissue-specific phenotypic metrics associated with these two MPSs and illustrated acceptable functionality and no difference between experimental arms where both the gut and liver MPSs were on-platform and arms where only the gut or only the liver MPS was on-platform (Fig. 2). In addition to that, the absolute values of these on-platform metrics were not substantially different compared to the respective off-platform measurements in conventional single-MPS technologies (static gut, LiverChip™) and were also numerically comparable to previously reported values (24,30,41–43). All the above clearly support that the gut-liver interaction and crosstalk on the multi-MPS platform is not negatively affecting the individual MPS tissue health and function.
In order to practically illustrate the value of the developed multi-MPS platform for in vitro PK studies, we investigated the kinetics of DCF and HC in an integrated gut-liver system. Our model-guided experimental design allowed us to successfully select a DCF dose that yielded (across the different experimental arms) concentrations in the mixing chamber of the platform that are in the range of the clinically observed DCF plasma concentrations following standard therapeutic dosing (32). This allowed the investigation of DCF PK-related processes in pharmacologically relevant concentrations. A model-based approach was used in this work not only to guide the experimental design but also to analyze the obtained experimental data. Our mechanistic PK models were developed taking into account the operational characteristics of the platform (e.g., flow rates, volumes) and the MPS interconnection scheme. By fitting these models to the experimental data, we estimated the intrinsic PK-related parameters of interest (intrinsic metabolic clearance of the gut and liver MPS, permeability across the epithelial barrier of the gut MPS) disentangled to the greatest possible degree from non-biological platform-specific processes (e.g., circulation and distribution across the different platform compartments). The model-obtained PK parameters from this integrated gut-liver system (e.g., DCF: P = 14.7 × 10−6 cm/s, Clintl = 17 μL/min/106 cells) were in agreement with previous reports in Caco-2 cell cultures (DCF: P = 17.8 × 10−6 cm/s (44)) and human hepatocyte cultures (DCF: Clintl = 17.8 μL/min/106 cells (24). This agreement highlights the value of mechanistic modeling for the analysis and robust interpretation of the output obtained from such a complex in vitro system.
In addition, model-based analysis of the data obtained across different on-platform experimental arms (gut only, liver only, gut/liver) indicated that (for both DCF and HC) the intrinsic PK-related processes of interest (e.g., permeability, metabolism) are not substantially affected from the interaction/communication between the gut and the liver MPSs. This finding builds further confidence with regard to the application of multi-MPS technologies for quantitative PK studies, as prior to this work, there was no direct evidence to robustly support that the interconnection of MPSs of different organs does not impede intrinsic PK-related processes such as hepatic metabolism. In contrast, our results suggested that the liver MPS intrinsic metabolic clearance for DCF was upregulated to a small degree by the on-platform crosstalk between the gut and liver MPSs (Fig. 6). This observation was followed up and further validated in independent work in the context of long-term cell culture (30). Specifically, it was found that the production of 4-OH-DCF (a CYP2C9 metabolite of DCF) was moderately but statistically significantly higher in liver MPSs that have been previously interacting with a gut MPS for a 2-week period, compared to liver MPSs that were maintained in isolation for the same period. These findings illustrate that the fluidic coupling of the gut and liver MPSs may allow exchange of biochemical signals that modulate hepatic metabolism. It should be noted, however, that the available data to date indicate that even if the effect of such a gut-liver crosstalk exists, its magnitude is not enough to dramatically affect the in vitro-obtained intrinsic hepatic clearance and subsequently the in vivo clearance predictions. Further work is needed across a large and diverse set of compounds to thoroughly understand the effects of gut-liver communication on different metabolic pathways and investigate the degree to which gut-liver communication can improve in vivo PK predictions.
Two additional PK-related aspects were also explored in this work. Firstly, by measuring the formation and kinetics of 4-OH-DCF on-platform, it was illustrated that the in vivo DCF primary metabolite in humans was detectable in our in vitro system, and that this metabolite (formed mainly in the liver MPS) circulated within the common medium across the different compartments of the multi-MPS platform. Given that off-target drug toxicity in humans frequently occurs due to metabolites, the ability of multi-MPS systems to circulate the hepatocyte-formed metabolites to other MPS through a common medium gives the ability to study such toxicity effects early during the development of a new compound. Secondly, by measuring DCF concentrations in the liver MPS tissue lysate, it was illustrated that the intracellular MPS drug concentrations can be substantially higher than the concentrations measured in the surrounding medium, which is in line with previous literature suggesting high intracellular binding of DCF (45). Thus, it becomes evident that investigation of the medium-to-tissue drug partition is essential if quantitative understanding of drug efficacy/toxicity in an intracellular target is desired using these multi-MPS platforms.
A significant challenge during the development of MPS technologies and multi-MPS platforms is their appropriate scaling to create an in vitro system that recapitulates the desired aspects of in vivo human physiology. Scaling is the specification of the in vitro system characteristics and operating parameters relative to the in vivo system that one is trying to model. Several approaches, including direct linear, allometric, and functional scaling, have been proposed (36,46–49) and utilized in various efforts to reverse-engineer MPS technologies from human physiology. This aspect was carefully considered in this work. Firstly, the cell type compositions in our liver MPS (10:1 hepatocytes to Kupffer cells) and gut MPS (9:1 absorptive enterocytes (Caco2-BBe) to mucus-producing Goblet cells (HT29-MTX)) were selected to mimic the same cell ratios in vivo (8,50). Secondly, our system was designed so that the ratio of the surface area of the gut MPS epithelial barrier (1.12 cm2) to the number of hepatocytes in the liver MPS (600,000) is similar to the ratio of the small intestinal epithelium surface area (30 m2 (51)) to the number of hepatocytes (2.16 × 1011 (27,52)) in humans (ratio is 1.87 × 10−10 in our in vitro system and 1.39 × 10−10 in vivo), while utilizing a commercially available transwell system for the gut MPS. Finally, our integrated gut-liver system was designed so that the MPS interconnection scheme and flow partition closely mirror human physiology (27) (see the “Methods” section and Fig. 1).
However, other aspects of meso-scale systems such as ours are substantially different than in vivo, in particular with respect to medium-to-tissue ratio, which is larger than if it was linearly scaled from the in vivo blood-to-tissue volume ratio. Our selection of the larger ratio was driven by the needs of fluidics control, sampling requirements, cell resources, and a desire to use where possible commercially available components (e.g., transwell culture chambers). Nonetheless, it is important to recognize that strict adherence to mimic in vivo physiological relationships (linear or allometric scaling) is not always necessarily desirable and rather that modifications might be needed to reproduce function for specific MPS applications (49). In addition, it is inevitable that multi-MPS technologies will have components of the human physiology that are missing or neglected and thus the associated crude in vitro output (e.g., measured circulating drug concentrations on-platform) is not expected to be directly representative of the in vivo output (e.g., plasma concentrations in humans) or directly scalable to it (e.g., simply by a factor of miniaturization). Due to these inherent differences of MPSs compared to humans as well as needs driven by research applications, our framework for utilizing MPS is to include an additional step of in vitro-to-in vivo translation, as was described in our previous work (24). Specifically, we use model-based analysis of the in vitro output to derive the biological parameters of interest, disentangled as much as possible from the in vitro system characteristics. Then, to translate the results to an in vivo context, we use computational systems pharmacology/biology approaches to integrate these parameters with the characteristics of the human body (e.g., number of hepatocytes in the human liver) and other in vivo processes that are missing from the in vitro system (e.g., drug binding to human plasma proteins) (24).
Recently, we illustrated in an independent work that the developed multi-MPS platform supports long-term functional maintenance of interacting gut and liver MPSs for over 2 weeks (30). Combined with the present work, it becomes evident that the presented gut-liver system has the potential for several PK-related applications that are particularly challenging with the current routinely employed in vitro systems, including bioavailability prediction, investigation of low-clearance compounds, drug-drug interactions at both the intestinal and hepatic level, effects of repeated drug exposure and metabolite accumulation, and effects of inflammatory chronic diseases on drug PK. The developed system has, however, limitations that need to be overcome in order for this technology to reach its maximum potential. Firstly, the gut MPS in this study consisted of Caco-2/HT29-MTX cells, which do not fully recapitulate the metabolic functions of the intestinal tissue in vivo. Ongoing efforts are under way in our lab and others to develop primary intestinal monolayer cultures using organoids derived from patient biopsies. Once fully characterized, the primary gut MPS can be easily incorporated into the developed platform for improved functionality. Secondly, the liver MPS technology used in this work, similar to other conventional hepatic in vitro systems (e.g., hepatocyte suspensions, microsomes) (53), has the tendency to under-predict in vivo hepatic clearance (24). This trend of under-prediction and its origins is a subject of ongoing research in the drug metabolism field (54,55) and we are also working towards directions that can improve this aspect of the liver MPS performance (e.g., control of micro-tissue oxygenation, medium composition, programmable medium exchange and waste removal).
CONCLUSION
This work builds further confidence with regard to the application of multi-MPS technologies for quantitative PK and in extension PD investigations. We envision that our integrative approach, which combines multi-cellular tissue models, multi-MPS bioreactor platforms, and quantitative mechanistic modeling, will have broad applicability in pre-clinical drug development, to better bridge the gap between animal and human studies.
References
Zhang D, Luo G, Ding X, Lu C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm Sin B. 2012;2(6):549–61.
Sutherland ML, Fabre KM, Tagle DA. The national institutes of health microphysiological systems program focuses on a critical challenge in the drug discovery pipeline. Stem Cell Res Ther. 2013;4(1):I1.
Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32(8):760–72.
Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nat Rev Drug Discov. 2015;14(4):248–60.
Nam KH, Smith AS, Lone S, Kwon S, Kim DH. Biomimetic 3D tissue models for advanced high-throughput drug screening. J Lab Autom. 2015;20(3):201–15.
Vivares A, Salle-Lefort S, Arabeyre-Fabre C, Ngo R, Penarier G, Bremond M, et al. Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica. 2015;45(1):29–44.
Ouattara DA, Choi S-H, Sakai Y, Péry ARR, Brochot C. Kinetic modelling of in vitro cell-based assays to characterize non-specific bindings and ADME processes in a static and a perfused fluidic system. Toxicol Lett. 2011;205(3):310–9.
Ebrahimkhani MR, Neiman JAS, Raredon MSB, Hughes DJ, Griffith LG. Bioreactor technologies to support liver function in vitro. Adv Drug Deliv Rev. 2014;69–70:132–57.
Zheng F, Fu F, Cheng Y, Wang C, Zhao Y, Gu Z. Organ-on-a-chip systems: microengineering to biomimic living systems. Small. 2016;12(17):2253–82.
Sung JH, Shuler ML. A micro cell culture analog ([small micro]cca) with 3-d hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip. 2009;9(10):1385–94.
Oleaga C, Bernabini C, Smith AS, Srinivasan B, Jackson M, McLamb W, et al. Multi-organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci Rep. 2016;6:20030.
Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip. 2015;15(12):2688–99.
Materne EM, Maschmeyer I, Lorenz AK, Horland R, Schimek KM, Busek M, et al. The multi-organ chip—a microfluidic platform for long-term multi-tissue coculture. J Vis Exp. 2015;98:e52526.
Esch MB, Mahler GJ, Stokol T, Shuler ML. Body-on-a-chip simulation with gastrointestinal tract and liver tissues suggests that ingested nanoparticles have the potential to cause liver injury. Lab Chip. 2014;14(16):3081–92.
Sin A, Chin KC, Jamil MF, Kostov Y, Rao G, Shuler ML. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol Prog. 2004;20(1):338–45.
Viravaidya K, Sin A, Shuler ML. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnol Prog. 2004;20(1):316–23.
Sung JH, Kam C, Shuler ML. A microfluidic device for a pharmacokinetic-pharmacodynamic (pk-pd) model on a chip. Lab Chip. 2010;10(4):446–55.
Esch MB, Ueno H, Applegate DR, Shuler ML. Modular, pumpless body-on-a-chip platform for the co-culture of GI tract epithelium and 3D primary liver tissue. Lab Chip. 2016;16(14):2719–29.
Maschmeyer I, Hasenberg T, Jaenicke A, Lindner M, Lorenz AK, Zech J, et al. Chip-based human liver-intestine and liver-skin co-cultures—a first step toward systemic repeated dose substance testing in vitro. Eur J Pharm Biopharm. 2015;95(Pt A):77–87.
Prot JM, Maciel L, Bricks T, Merlier F, Cotton J, Paullier P, et al. First pass intestinal and liver metabolism of paracetamol in a microfluidic platform coupled with a mathematical modeling as a means of evaluating ADME processes in humans. Biotechnol Bioeng. 2014;111(10):2027–40.
Imura Y, Yoshimura E, Sato K. Micro total bioassay system for oral drugs: evaluation of gastrointestinal degradation, intestinal absorption, hepatic metabolism, and bioactivity. Anal Sci. 2012;28(3):197–9.
Kimura H, Ikeda T, Nakayama H, Sakai Y, Fujii T. An on-chip small intestine-liver model for pharmacokinetic studies. J Lab Autom. 2015;20(3):265–73.
Jie M, Li H-F, Lin L, Zhang J, Lin J-M. Integrated microfluidic system for cell co-culture and simulation of drug metabolism. RSC Adv. 2016;6(59):54564–72.
Tsamandouras N, Kostrzewski T, Stokes CL, Griffith LG, Hughes DJ, Cirit M. Quantitative assessment of population variability in hepatic drug metabolism using a perfused three-dimensional human liver microphysiological system. J Pharmacol Exp Ther. 2017;360(1):95–105.
Sarkar U, Rivera-Burgos D, Large EM, Hughes DJ, Ravindra KC, Dyer RL, et al. Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab Dispos. 2015;43(7):1091–9.
Long TJ, Cosgrove PA, Dunn RT, Stolz DB, Hamadeh HK, Afshari C, et al. Modeling therapeutic antibody-small molecule drug-drug interactions using a 3D perfusable human liver co-culture platform. Drug Metab Dispos. 2016;44(12):1940–8.
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13(4):407–84.
Yu J, Cilfone NA, Large EM, Sarkar U, Wishnok JS, Tannenbaum SR, et al. Quantitative systems pharmacology approaches applied to microphysiological systems (MPS): data interpretation and multi-MPS integration. CPT Pharmacometrics Syst Pharmacol. 2015;4(10):585–94.
Domansky K, Inman W, Serdy J, Dash A, Lim MHM, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 2010;10(1):51–8.
Chen WLK, Edington C, Suter E, Yu J, Velazquez JJ, Velazquez JG, et al. Integrated gut/liver microphysiological systems elucidates inflammatory inter-tissue crosstalk. Biotechnol Bioeng. 2017; doi:10.1002/bit.26370.
Sarkar U, Ravindra KC, Large E, Young CL, Rivera-Burgos D, Yu J, et al. Integrated assessment of diclofenac biotransformation, pharmacokinetics, and omics-based toxicity in a three-dimensional human liver-immunocompetent coculture system. Drug Metab Dispos. 2017;45(7):855–66.
Davies NM, Anderson KE. Clinical pharmacokinetics of diclofenac. Therapeutic insights and pitfalls. Clin Pharmacokinet. 1997;33(3):184–213.
Gelman A, Rubin DB. Inference from iterative simulation using multiple sequences. Stat Sci. 1992;7(4):457–72.
Plummer M, Best N, Cowles K, Vines K. Coda: convergence diagnosis and output analysis for MCMC. R News. 2006;6(1):7–11.
Gelman A. Inference and monitoring convergence, Markov chain monte carlo in practice. Boca Raton: Chapman & Hall; 1996. p. 131–43.
Stokes CL, Cirit M, Lauffenburger DA. Physiome-on-a-chip: the challenge of “scaling” in design, operation, and translation of microphysiological systems. CPT Pharmacometrics Syst Pharmacol. 2015;4(10):559–62.
Fabre KM, Livingston C, Tagle DA. Organs-on-chips (microphysiological systems): tools to expedite efficacy and toxicity testing in human tissue. Exp Biol Med. 2014;239(9):1073–7.
Toepke MW, Beebe DJ. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip. 2006;6(12):1484–6.
Low LA, Tagle DA. Microphysiological systems (“organs-on-chips”) for drug efficacy and toxicity testing. Clin Transl Sci 2017.
Ng S-F, Rouse J, Sanderson D, Eccleston G. A comparative study of transmembrane diffusion and permeation of ibuprofen across synthetic membranes using franz diffusion cells. Pharmaceutics. 2010;2(2):209–23.
Mahler GJ, Esch MB, Tako E, Southard TL, Archer SD, Glahn RP, et al. Oral exposure to polystyrene nanoparticles affects iron absorption. Nat Nanotechnol. 2012;7(4):264–71.
Nollevaux G, Devillé C, El Moualij B, Zorzi W, Deloyer P, Schneider Y-J, et al. Development of a serum-free co-culture of human intestinal epithelium cell-lines (Caco-2/HT29-5M21). BMC Cell Biol. 2006;7:20.
VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2015;64(6):911–20.
Paixão P, Gouveia LF, Morais JAG. Prediction of the in vitro permeability determined in Caco-2 cells by using artificial neural networks. Eur J Pharm Sci. 2010;41(1):107–17.
Mateus A, Matsson P, Artursson P. Rapid measurement of intracellular unbound drug concentrations. Mol Pharm. 2013;10(6):2467–78.
Wikswo JP, Curtis EL, Eagleton ZE, Evans BC, Kole A, Hofmeister LH, et al. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip. 2013;13(18):3496–511.
Ucciferri N, Sbrana T, Ahluwalia A. Allometric scaling and cell ratios in multi-organ in vitro models of human metabolism. Front Bioeng Biotechnol. 2014;2(74).
Abaci HE, Shuler ML. Human-on-a-chip design strategies and principles for physiologically based pharmacokinetics/pharmacodynamics modeling. Integr Biol. 2015;7(4):383–91.
Maass C, Stokes CL, Griffith LG, Cirit M. Multi-functional scaling methodology for translational pharmacokinetic and pharmacodynamic applications using integrated microphysiological systems (MPS). Integr Biol. 2017;9(4):290–302.
Noah TK, Donahue B, Shroyer NF. Intestinal development and differentiation. Exp Cell Res. 2011;317(19):2702–10.
Helander HF, Fandriks L. Surface area of the digestive tract—revisited. Scand J Gastroenterol. 2014;49(6):681–9.
Hakooz N, Ito K, Rawden H, Gill H, Lemmers L, Boobis AR, et al. Determination of a human hepatic microsomal scaling factor for predicting in vivo drug clearance. Pharm Res. 2006;23(3):533–9.
Hallifax D, Foster JA, Houston JB. Prediction of human metabolic clearance from in vitro systems: retrospective analysis and prospective view. Pharm Res. 2010;27(10):2150–61.
Galetin A. Rationalizing underprediction of drug clearance from enzyme and transporter kinetic data: from in vitro tools to mechanistic modeling. In: Nagar S, Argikar AU, Tweedie JD, editors. Enzyme kinetics in drug metabolism: fundamentals and applications. Totowa: Humana Press; 2014. p. 255–88.
Bowman CM, Benet LZ. Hepatic clearance predictions from in vitro-in vivo extrapolation and BDDCS. Drug Metab Dispos. 2016;44(11):1731–5.
Acknowledgements
This work was supported by the DARPA Microphysiological Systems Program (Grant W911NF-12-2-0039) and the National Institutes of Health Microphysiological Systems Program (Grant 4-UH3-TR000496-03). The authors thank the members of the MIT PhysioMimetics team for the comments and discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tsamandouras, N., Chen, W.L.K., Edington, C.D. et al. Integrated Gut and Liver Microphysiological Systems for Quantitative In Vitro Pharmacokinetic Studies. AAPS J 19, 1499–1512 (2017). https://doi.org/10.1208/s12248-017-0122-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-017-0122-4