
Abstract
For assessment of biosimilarity, the US Food and Drug Administration (FDA) recommends a stepwise approach for obtaining the totality-of-the-evidence for demonstrating biosimilarity between a proposed biosimilar product and an innovative (reference) biological product. The stepwise approach starts with analytical studies for functional and structural characterization at various stages of manufacturing process of the proposed biosimilar product. Analytical similarity assessment involves identification of critical quality attributes (CQAs) that are relevant to clinical outcomes. FDA proposes first classifying the identified CQAs into three tiers according to their criticality or risking ranking relevant to clinical outcomes and then performing equivalence test (for CQAs in Tier 1), quality range approach (for CQAs in Tier 2), and raw data or graphical presentation (for CQAs in Tier 3) for obtaining totality-of-the-evidence for demonstrating biosimilarity between the proposed biosimilar product with the reference product. In practice, some debatable issues are evitably raised due to this complicated process of analytical similarity assessment. In this article, these debatable are described and discussed.
Similar content being viewed by others

INTRODUCTION
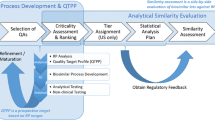
In recent years, the assessment of biosimilarity for biosimilar products has received much attention by scientists, researchers, and reviewers from the pharmaceutical industry (biosimilar sponsors), academia, and regulatory agencies such as the US Food and Drug Administration (FDA) and China Food and Drug Administration (CFDA). As indicated in its recent guidance, the FDA recommends a stepwise approach for obtaining the totality-of-the-evidence for demonstrating biosimilarity between a proposed biosimilar product and an innovative (reference) biological product (1,2). The stepwise approach starts with analytical studies for functional and structural characterization of critical quality attributes (CQAs) that are relevant to clinical outcomes at various stages of manufacturing process followed by animal studies for assessment of toxicity, clinical pharmacology pharmacokinetics (PK) or pharmacodynamics (PD) studies, and clinical studies for assessment of immunogenicity, safety/tolerability, and efficacy. CQAs in manufacturing process of drug products are referred to as chemical, physical, biological, and microbiological attributes that can be defined, measured, and continually monitored to ensure final product outputs remain within acceptable quality limits (see, e.g., (3)). For the analytical studies, FDA suggests that CQAs should be identified and classified into three tiers according to their criticality or risk ranking based on mechanism of action (MOA) or PK using appropriate statistical models or methods. CQAs with CQAs that are most relevant to clinical outcomes will be classified to Tier 1, while CQAs that are less (mild-to-moderate) or least relevant to clinical outcomes will be classified to Tier 2 and Tier 3, respectively (4,5).
FDA proposes equivalence test for CQAs in Tier 1, quality range approach for CQAs in Tier 2, and raw data or graphical presentation for CQAs in Tier 3 to assist sponsors in analytical similarity assessment for obtaining totality-of-the-evidence for demonstrating similarity between the proposed biosimilar product and the reference product. As indicated by the FDA, equivalence test for Tier 1 CQAs is more statistically rigorous than that of quality range approach for Tier 2 CQAs, which is in turn more rigorous than that of raw data or graphical presentation for Tier 3 CQAs (5). In practice, however, for a given CQA, there is no guarantee that passing Tier 1 test will pass Tier 2 test and vice versa. This has raised a number of debatable (controversial) issues in analytical similarity assessment (see also (6,7)). These issues include, but are not limited to, (1) fundamental similarity assumption, (2) primary assumptions for tiered approach, (3) statistical properties of FDA’s recommended Tier 1 equivalence test, (4) criticism of fixed approach for margin/range selection, (5) inconsistencies between tired approaches, (6) sample size requirement, (7) heterogeneity within lots and across lots within and between test product and reference product, (8) interpretation of FDA’s current thinking on scientific input, (9) relationship between similarity limit and variability, and (10) a proposed unified tiered approach.
Analytical similarity assessment is a complicated problem that involves characterization of bioactivity and protein content whose relationship with clinical outcomes may not be fully explored and understood. This uncertainty has made analytical similarity assessment even more complicated. The purpose of this article is to give a brief summary of these controversial issues in analytical similarity assessment rather than provide solutions. Recent development of these issues with discussion will be provided whenever possible.
FUNDAMENTAL SIMILARITY ASSUMPTION
For small molecule drug products, as indicated by Chow and Liu (2008), bioequivalence studies are necessarily conducted for regulatory review and approval of small molecule generic drug products, see also FDA (8). This is because it constitutes legal basis (from the Hatch-Waxman Act) under the Fundamental Bioequivalence Assumption, which states that
If two drug products are shown to be bioequivalent, it is assumed that they will reach the same therapeutic effect or they are therapeutically equivalent.
Under the Fundamental Bioequivalence Assumption, bioavailability (defined as the rate and extent of drug absorbed into the blood stream and become available) serves as surrogate endpoint for clinical outcomes (safety and efficacy). Thus, under the Fundamental Bioequivalence Assumption, an approved generic drug product can serve as a substitute to the innovative (brand-name) drug product. Although this Fundamental Bioequivalence Assumption constitutes legal basis, it has been challenged by many researchers. In practice, there are following four possible scenarios:
-
(1)
Drug absorption profiles are similar and they are therapeutic equivalent.
-
(2)
Drug absorption profiles are not similar but they are therapeutic equivalent.
-
(3)
Drug absorption profiles are similar but they are not therapeutic equivalent.
-
(4)
Drug absorption profiles are not similar and they are not therapeutic equivalent.
The Fundamental Bioequivalence Assumption is considered scenario (1). Scenario (1) works if the drug absorption (in terms of the rate and extent of absorption) is predictive of clinical outcome. In this case, PK responses such as AUC (area under the blood or plasma concentration-time curve for measurement of the extent of drug absorption) and Cmax (maximum concentration for measurement of the rate of drug absorption) serve as surrogate endpoints for clinical endpoints for assessment of efficacy and safety of the test product under investigation. Scenario (2) is the case where generic companies use to argue for generic approval of their drug products especially when their products fail to meet regulatory requirement for bioequivalence. In this case, it is doubtful that there is a relationship between PK responses and clinical endpoints. The innovator companies usually argue with the regulatory agency to against generic approval with scenario (3). However, more studies are necessarily conducted in order to verify scenario (3). There are no arguments with respect to scenario (4).
It should be noted that a generic drug contains identical active ingredient(s) as the brand-name drug. Thus, it is reasonable to assume that generic (test) drug products and the brand-name (reference) drug have identical means, i.e., μ T = μ R . In addition, bioequivalence testing focuses on mean difference (i.e., μ T − μ R ) or ratio of means (i.e., μ T /μ R ) and ignores heterogeneity in variability of the test and reference product (i.e., σ T ≠ σ R ). As a result, a generic drug product may fail the bioequivalence testing when σ R is relatively large (say > 30%) even when μ T = μ R .
Following similar idea, SSAB (9) proposed the following Fundamental Similarity Assumption:
When a follow-on biologic product is claimed to be biosimilar to an innovator product in some well-defined study endpoints, it is assumed that they will reach similar therapeutic effect or they are therapeutically equivalent.
Although the above-proposed Fundamental Similarity Assumption does not constitute legal basis, the FDA seems to adopt the assumption for analytical similarity assumption without verifying the validity of the assumption. In other words, FDA assumes that analytical similarity in terms of CQAs identified at various stages such as functional and structural characterization of the manufacturing process is predictive of clinical outcomes.
Unlike small molecule drug products, biosimilar products are large molecule drug products which are made of living cells or living organisms. As a result, it is expected that μ T ≠ μ R , i.e., μ T = μ R + Δ, where Δ is the true mean difference. Chow et al. (10) indicated that there are fundamental differences between small molecule drug products and biosimilar products. For example, biosimilar products are often very sensitive to environmental factors during the manufacturing process. A small change and variation may translate to a huge change in clinical outcomes. Consequently, biosimilar products are expected to have much larger variability as compared to that of generic drug products. In this case, statistical methods for similarity assessment following the concept of bioequivalence testing (i.e., focusing on mean difference or ratio of means but ignore variability) for assessing biosimilarity of biosimilar products may not be appropriate. Table I provides a comparison between bioequivalence test for generic drug products and biosimilarity test for biosimilar products.
PRIMARY ASSUMPTIONS FOR TIERED APPROACH
Tier 1 Equivalence Test
For CQAs in Tier 1, FDA recommends that an equivalency test be performed for to assess analytical similarity. As indicated by the FDA, for a given CQA, we may test for equivalence by the following interval (null) hypothesis:
where δ > 0 is the equivalence limit (or similarity margin), and μ T and μ R are the mean responses of the test (the proposed biosimilar) product and the reference product lots, respectively. Analytical equivalence (similarity) is concluded if the null hypothesis of non-equivalence (dis-similarity) is rejected. Note that Yu (12) defined inequivalence as when the confidence interval falls entirely outside the equivalence limits. Similarly to the confidence interval approach for bioequivalence testing under the raw data model, analytical similarity would be accepted for a quality attribute if the (1–2α) 100% two-sided confidence interval of the mean difference is within (−δ, δ). FDA further recommended that the equivalence acceptance criterion (EAC), δ = EAC = 1.5 * σ R , where σ R is the variability of the reference product be used based on extensive simulation studies and internal scientific input. Chow (7) provided statistical justification for the selection of c = 1.5 in EAC following the idea of scaled average bioequivalence (SABE) criterion for highly variable drug products proposed by the FDA.
For the establishment of EAC, FDA made the following assumptions. First, FDA assumes that the true difference in means is proportional to σ R , i.e., μ T − μ R is proportional to σ R . Second, FDA adopts the similarity limit as EAC = 1.5 * σ R and recommended that σ R be estimated by the sample standard deviation of test values from reference lots (one test value from each lot). Third, in the interest of achieving a desired power of the similarity test, FDA further recommends that an appropriate sample size be selected by evaluating the power under the alternative hypothesis at \( {\mu}_T-{\mu}_R=\frac{1}{8}{\sigma}_R \). The assumption that μ T − μ R is proportional to σ R , the selection of c = 1.5, and the allowed mean shift of \( {\mu}_T-{\mu}_R=\frac{1}{8}{\sigma}_R \) have generated tremendous discussion among FDA, biosimilar sponsors, and academia, and they are debatable.
To provide a better understanding of the debatable issue and FDA’s proposal, we would like to point out the following which may be helpful to resolve the debatable issues: (1) unlike the traditional bioequivalence test, FDA’s intention is to take variability into consideration by considering the effect size adjusted for variability, i.e.,
which is half-way between 1 and 1.25 (unity to the upper equivalence limit of 125%), (2) the EAC for effect size adjusted for variability becomes fixed, i.e., EAC = c = 1.5, and (3) if the true difference falls on the half-day between 1 and 1.25, the worst possible observed difference could fall on the 1.25 (this may happen if the worst possible reference lot is selected for comparison). In this case, the original EAC bioequivalence testing for generic drug products with μ T = μ R ) is necessarily shifted by 0.25. Thus, upper limit is shifted from 1.25 to c = 1.25 + 0.25 = 1.5.
Tier 2 Quality Range Approach
For CQAs in Tier 2, FDA suggests that analytical similarity be performed based on the concept of quality ranges, i.e., ± x * σ R , where σ R is the standard deviation of the reference product and x a constant which should be appropriately justified. Thus, the quality range of the reference product for a specific quality attribute is defined as \( \left({\widehat{\mu}}_R-x{\widehat{\sigma}}_R,{\widehat{\mu}}_R+x{\widehat{\sigma}}_R\right) \). Analytical similarity would be accepted for the quality attribute if a sufficiently large percentage of test lot values falls within the quality range. Under normality assumption, if x = 1.645, we would expect 90% of the test results from reference lots to lie within the quality range. If x is chosen to be 1.96, we would expect that about 95% test results of reference lots will fall within the quality range. Thus, the selection of x could have an impact on the width of the quality range and consequently the percentage of test lot values that will fall within the quality range.
At the 2015 Duke-Industry Statistics Symposium held in Duke Campus on October 22–23, one of FDA speakers indicated that x should be selected between 2 and 3 to guarantee that majority of test values of the test lots will fall within the quality range established based on test values of the reference lots. Under the normality assumption, in practice, we would expect that there are about 95% of data would fall below and above 2 (i.e., x = 2) standard deviations (SD) of the mean and about 99.7% of data would fall within ± 3 SDs (i.e., x = 3)) of the mean. Under the normality assumption, the FDA recommended quality range approach is considered a reasonable approach only under the assumption that μ T ≈ μ R and σ T ≈ σ R . In this case, it can be expected that majority of test values obtained from the test lots will fall within ± x SD of the range established based on the test values of the reference lots.
In practice, however, the assumptions that μ T ≈ μ R and σ T ≈ σ R are usually not true due to the nature of biosimilar products. Thus, one of the major criticisms of the quality range approach is that it ignores the fact that there are differences in population mean and population standard deviation between the proposed biosimilar product and the reference product, i.e., μ T ≠ μ R and σ T ≠ σ R . In practice, it is recognized that biosimilarity between a proposed biosimilar product and a reference product could be established even under the assumption that μ T ≠ μ R and σ T ≠ σ R . Thus, under the assumption that μ T ≈ μ R and σ T ≈ σ R , quality range approach for analytical similarity assessment for CQAs from Tier 2 is considered more stringent as compared to equivalence testing for CQAs from Tier 1 (most relevant to clinical outcomes) regardless they are mild-to-moderate relevant to clinical outcomes. This is because that equivalence testing allows a possible mean shift of σ R /8, while the quality range approach does not. In practice, there are several possible scenarios that include the cases where (1) μ T ≈ μ R or there is a significant mean shift (either a shift to the right or a shift to the left) and (2) σ T ≈ σ R , σ T > σ R , or σ T < σ R .
Thus, one of the most controversial issues for quality range approach for CQAs in Tier 2 is that the approach does not reflect the real practice that μ T ≠ μ R and σ T ≠ σ R . As a result, the test results are somewhat misleading and not reliable.
Tier 3 Raw Data and Graphical Comparison
For CQAs in Tier 3 with the lowest risk ranking, FDA recommends an approach that uses raw data/graphical comparisons. The examination of similarity for CQAs in Tier 3 by no means is less stringent, which is acceptable because they have least impact on clinical outcomes in the sense that a notable dis-similarity will not affect clinical outcomes.
Evaluation based on raw data and graphical presentation, it is not only somewhat subjective, but also biased. Tier 1 equivalence test and Tier 2 quality range similarity test are supposed to be more rigorous than Tier 3 raw data and graphical comparison. That is, passing Tier 1 equivalence test and Tier 2 quality range similarity test will pass Tier 3 raw data graphical comparison test. In practice, however, there is no guarantee that a given CQA which passes Tier 1 equivalence test or Tier 2 quality range similarity test will pass Tier 3 raw data graphical comparison test and vice versa. Since CQAs in Tier 3 are considered least relevant to clinical outcomes, it is necessary that all Tier 3 CQAs pass the test. If not, it is of interest to know about what percentage of CQAs needs to pass in order to pass Tier 3 test. Figures 1, 2, and 3 exhibit graphical comparison for the cases where (1) μ T = μ R and σ T ≠ σ R , (2) μ T ≠ μ R and σ T = σ R , and (3) μ T ≠ μ R and σ T ≠ σ R , respectively.

Graphical comparison for the case where μ T = μ R and σ T ≠ σ R

Graphical comparison for the case where μ T ≠ μ R and σ T = σ R

Graphical comparison for the case where μ T ≠ μ R and σ T ≠ σ R
STATISTICAL PROPERTIES OF FDA’S RECOMMENDED TIER 1 EQUIVALENCE TEST
For equivalence test for Tier 1 CQAs, FDA recommends testing one sample from each reference lot for obtaining an estimate of σ R . Wang and Chow (7) evaluated statistical properties of the FDA’s recommended method. Without loss of generality, suppose multiple test samples from each reference lot are available. Let x Rij be the test value of the jth test sample from the ith reference lot, i = 1, …, k, j = 1, …, n i , and it follows a normal distribution with mean μ i and variance σ 2 i , where μ i and σ 2 i are also random variables. The expectations of μ i and σ 2 i are μ and σ 2, and the variances are σ 2 μ and σ 2 σ . Then, the variance of x Rij is given by
Define \( {\overline{x}}_{Ri\cdot }=\frac{1}{n_i}{\displaystyle \sum_{j=1}^{n_i}}{x}_{Rij},{\overline{\overline{x}}}_{R\cdot \cdot }=\frac{1}{k}{\displaystyle \sum_{i=1}^k}{\overline{x}}_{Ri\cdot } \). Then
where
If we assume that n 1 = … = n k = n, then we have
Thus, it can be seen that the FDA’s approach is an unbiased estimate of σ R , however, when n > 1, the FDA’s recommended approach with multiple test samples per lot is a biased estimate of σ R . In the interest of having an unbiased estimate with multiple test samples per lot, Wang and Chow (13) proposed an alternative approach to correct the biasedness. As indicated by Wang and Chow (13), multiple test samples per lot provide valuable information regarding the heterogeneity across lots, which is useful especially when extreme lots (i.e., lots with extremely low or high variability) are selected for equivalence test.
CRITICISM OF FIXED APPROACH FOR MARGIN/RANGE SELECTION
In tiered approach, FDA seems to prefer a fixed margin approach for EAC by treating \( s={\widehat{\sigma}}_R \) as the true σ R (i.e., 1.5 * σ R ) for Tier 1 CQAs. The fixed margin approach is also referred to as a fixed standard deviation (SD) approach for similarity quality range by treating the estimate of SD (which is obtained based on test values of reference lots) as the true σ R (i.e., x * SD) for Tier 2 CQAs. The fixed margin or SD is in fact a statistic, which is a random variable rather than a fixed constant. In other words, it may vary depending upon the selected reference lots for tiered testing. The fixed SD approach is a conditional approach rather than an unconditional approach. In practice, if reference lots with less variability are selected for tiered testing, the proposed biosimilar product is most likely to fail the test. As a result, the scientific validity of fixed SD approach is questionable.
One of the major criticisms of the fixed approach for margin/range selection in tiered analysis is that the fixed approach does not take into consideration the variability of the estimate of the standard deviation. Thus, it is considered bad luck to the biosimilar sponsors if reference lots with less variability are selected for Tier 1 equivalence test. Another criticism is that the reference product cannot pass Tier 1 equivalence test itself if we divide all of the reference lots into two groups: one group with less variability and the other group with large variability. In this case, the group with large variability may not pass Tier 1 equivalence test with EAC established based on test values from the reference lots with less variability. It would be a concern that reference product cannot pass Tier 1 equivalence test when comparing to itself.
INCONSISTENT TEST RESULTS BETWEEN TIERED APPROACHES
As indicated in Tsong (5), Tier 1 equivalence test is considered more rigorous than Tier 2 quality range approach, which is in turn more rigorous than Tier 3 raw data and graphical comparison. The primary assumptions for these tired approaches, however, are different. Thus, under different assumptions, there is no guarantee that passing Tier 1 equivalence test will pass Tier 2 quality range approach and vice versa although these tests are conducted based on same data set collected from the test and reference lots under study. In practice, it is of interest to evaluate inconsistencies regarding the passages between Tier 1 equivalence test and Tier 2 quality range approach.
For a given CQA, the inconsistencies between Tier 1 equivalence test and Tier 2 quality range approach can be assessed by means of clinical trial simulation as follows. Let p ij be the probability of passing the ith tier test given that the CQA has passed the jth tier test. Thus, we have the following 2×2 contingency table for comparison between Tier 1 equivalence test and Tier 2 quality range approach.
Let μ T = μ R + Δ and σ T = Cσ R . It is then suggested that the inconsistencies between Tier 1 equivalence test and Tier 2 quality range approach be evaluated at various combinations of (1) \( \varDelta =0\left(\mathrm{nomeanshift}\right),\frac{1}{8}{\sigma}_R\left(\mathrm{FDArecommendedmeanshiftallowed}\right),\mathrm{and}\frac{1}{4}{\sigma}_R \) (the worst possible scenario), and (2) C = 0.8 (deflation), 1.0, and 1.2 (inflation) to provide a complete picture of the relative performance of Tier 1 equivalence test and Tier 2 quality range approach.
SAMPLE SIZE REQUIREMENT
One of the most commonly asked questions for analytical similarity assessment is probably that how many reference lots are required for establishing an acceptable EAC for achieving a desired power. For a given EAC, formulas for sample size calculation under different study designs are available in Chow et al. (14). In general, sample size (the number of reference lots, k) required is a function of (i) overall type I error rate (α), (ii) type II error rate (β) or power (1 − β), (iii) clinically or scientifically meaningful difference (i.e., μ T − μ R ), and (iv) the variability associated with the reference product (i.e., σ R ) assuming that σ T = σ R . Thus, we have
In practice, we select an appropriate k for achieving a desired power of 1 − β for detecting a clinically meaningful difference of μ T − μ R at a pre-specified level of significance α assuming that the true variability is σ. If α, μ T − μ R , and σ are fixed, the above equation becomes k = f(β).. We can then select an appropriate k for achieving the desired power. FDA’s recommendation attempts to control all parameters at the desired levels (e.g., α = 0.05 and 1 − β = 0.8) by knowing that μ T − μ R and σ are varying. In practice, it is often difficult, if not impossible, to control (or find a balance point among) α (type I error rate), 1 − β (power), μ T − μ R = Δ (clinically meaningful difference), and σ (variability in observing the response) at the same time. For example, controlling α at a pre-specified level of significance may be at the risk of decreasing power with a selected sample size.
HETEROGENEITY WITHIN LOTS AND ACROSS LOTS WITHIN AND BETWEEN TEST PRODUCT AND REFERENCE PRODUCT
Suppose there are n R and n T lots for analytical similarity assessment. For a given reference (test) lot, assume that the test value follows a distribution with mean μ Ri (μ Ti ) and variance σ 2 Ri (σ 2 Ti ). FDA’s recommended approach assumes that μ Ri = μ Rj and σ 2 Ri = σ 2 Rj for i ≠ j, i, j = 1, …, n R and μ Ti = μ Tj and σ 2 Ti = σ 2 Tj for i ≠ j, i, j = 1, …, n T for equivalence test in Tier 1 and quality range approach in Tier 2. Now let σ 2 R and σ 2 T be the variabilities associated with the reference product and the test product, respectively. Thus, we have
where σ 2 WR , σ 2 BR and σ 2 WT , σ 2 BT are the within-lot variability and between-lot (lot-to-lot) variability for the reference product and the test product, respectively. In practice, it is very likely that σ 2 R ≠ σ 2 T and often σ 2 WR ≠ σ 2 WT and σ 2 BR ≠ σ 2 BT even when σ 2 R ≈ σ 2 T . This has posted a major challenge to the FDA’s proposed approaches for the assessment of analytical similarity for CQAs from both Tier 1 and Tier, especially when there is only one test sample from each lot from the reference product and the test product. FDA’s proposal ignores within lot variability, i.e., when σ 2 WR = 0 or σ 2 R = σ 2 BR . In other words, sample variance based on x i , i = 1, .., n R from the reference product may underestimate the true σ 2 R and consequently may not provide a fair and reliable assessment of analytical similarity for a given quality attribute.
In practice, it is well recognized that μ Ri ≠ μ Rj and σ 2 Ri ≠ σ 2 Rj for i ≠ j, where μ Ri and σ 2 Ri are the mean and variance of the ith lot of the reference product. A similar argument is applied to the proposed biosimilar (test) product. As a result, the selection of reference lots for the estimation of σ R is critical for the proposed approach. The selection of reference lots has an impact on the estimation of σ R and consequently on the EAC. Assuming that n R > n T , FDA suggested using the remaining n R − n T lots to establish EAC to avoid selection bias. It sounds a reasonable approach if n R ≫ n T .. In practice, however, there might be a few lots available. Alternatively, it is suggested that all of the n R lots be used to establish EAC.
The assumption that μ Ri = μ Rj and σ 2 Ri = σ 2 Rj for i ≠ j; i, j = 1, …, n R and μ Ti = μ Tj and σ 2 Ti = σ 2 Tj for i ≠ j; i, j = 1, …, n T is a strong assumption which does not reflect real practice. Since biosimilar products are made of living cell and/or living organisms, it is expected that
Heterogeneity within lots and across lots between the test product and the reference product has posted the following controversial issues in analytical similarity assessment. First, suppose two extreme reference lots, one lot with the smallest within-lot variability and the other lot has the largest within-lot variability, are randomly selected for analytical similarity assessment. In this case, chances are that the reference product (the two selected extreme lots) may not even pass the equivalence test itself. Thus, analytical similarity between a test product and the reference product is not comprehensive. The other controversial issue is that if reference lots selected for establishment of EAC are extreme lots with smallest variability, the established EAC could be too narrow to penalize good test products.
FDA’S CURRENT THINKING ON SCIENTIFIC INPUT
In his recent presentation, Tsong (15) indicated that FDA’s current thinking for establishment of EAC is to consider 1.5 * σ R + Δ where Δ is a regulatory allowance depending upon scientific input. From statistical perspective, we may interpret the scientific input as scientific justification for accounting for the worst possible reference lot (i.e., a reference lot with extremely large variability) in establishment of EAC. Thus, 1.5 * σ R + Δ can be rewritten as 1.5 * σ ′ R , where σ ′ R = σ R + ε. Thus, we have
FDA’s original proposal is to estimate σ ′ R using sample standard deviation (s) of the test values obtained from the reference lots assuming that there is only one single test value per lot. Although Wang and Chow (7) showed that s is an unbiased estimate of σ R , it underestimates σ ′ R because it does not take the variability associated with s into consideration. Thus, Chow (7) suggested using the 95% upper confidence bound to estimate σ ′ R , i.e.,
This leads to
One of the controversial issues for establishment of EAC is that whether the margin should be fixed. FDA seems to recommend using the estimate of σ R as the true σ R without taking into consideration of the variability associated with the observed sample variance. The variability associated with the observed sample variance depends upon the sample size (i.e., the number of reference lots) used for analytical similarity assessment. As a result, the result of equivalence test may not be reproducible.
RELATIONSHIP BETWEEN SIMILARITY LIMIT AND VARIABILITY
As it can be seen from Table I, similarity (equivalence) limit for assessment of similarity (equivalence) depends upon the variability associated with the drug product. For example, if the variability is less than 10%, (90, 111%) similarity limit is recommended, while (80, 125%) similarity limit is used for drug product with variability between 20 and 30%. For drug products exhibit high variability such as highly variable small molecule drug products or large molecule biological products including biosimilars, it is suggested a scaled similarity limit adjusted for variability be considered. As an alternative to the scaled similarity limit and in the interest of one-size-fits-all criterion, some researchers suggest (70, 143%) be considered. The selection of similarity limits based on the associated variability is somewhat arbitrary without scientific/statistical justification. In what follows, we attempt to describe the relationship between similarity limit and variability for achieving a desired probability of claiming similarity.
Let (δ L , δ U ) be the similarity limits for evaluation of similarity between a test product (T) and a reference product (R). Current regulation indicates that we can claim similarity if the 90% confidence interval of difference in mean (i.e., μ T − μ R ) falls entirely within the lower and upper similarity limits. Let (L, U) be the 90% confidence interval for μ T − μ R . Define
where σ 2 R and σ 2 T are the variabilities associated with the reference product and the test product, respectively, and p is a desired probability of claiming similarity. Thus, for a given set of p, σ 2 R , and σ 2 T , appropriate (δ L , δ U ) can be determined.
A PROPOSED UNIFIED TIERED APPROACH
For biosimilar products, their population means and population variances are expected to be different. The relationship between a proposed biosimilar (test) product and an innovative (reference) product can be described as μ T = μ R + Δ and σ T = Cσ R , where Δ is a measure of a possible shift in population mean and C is an inflation factor. When there is a significant shift in mean (e.g., Δ > > 0) or notable heterogeneity between the proposed biosimilar product and the reference product (e.g., either C > > 1 or C < < 1), in this case, the validity of equivalence test for Tier 1 CQAs and the quality range approach for Tier 2 CQAs are questionable because the equivalence test and/or quality range approach are unable to handle major shift in population mean and significant change in population variance.
To overcome the problems of major mean shift and significant change in variability, alternatively, we may consider the equivalence test and quality range approach be applied to standardized test values (or effect size adjusted for standard deviation) rather than apply to untransformed raw data. For simplicity and without loss of generality, consider quality range approach for Tier 2 CQAs (Table II).
Define eff R = μ R /σ R and eff T = μ T /σ T and let y i , i = 1, …, n R be the test values of the reference lots, where n R is the number of reference lots considered for the test. Also, let z i , i = 1, …, n T be the test values of the reference lots, where n T is the number of test lots considered for the test. FDA’s quality range approach is applied on {y i , i = 1, …, n R } to establish the quality range with appropriate selection of x. Instead, we suggested the quality range approach be applied to {y i /ŝ R , i = 1, …, n R }, where ŝ R is the sample standard deviation of the test values of the reference lots. The quality range can then be established with the following adjustment on the selected x for achieving eff R ≈ eff T .. For simplicity and illustration purpose, consider the case that σ R ≈ σ T (i.e., C ≈ 1). In this case, under the assumption that eff R ≈ eff T , we have
This leads to
where \( 1+\frac{\varDelta }{\mu_R} \) is referred to as the adjustment factor for selection of x. Note that the left hand side of the above equation is for test lots and the right hand side is referred to the reference lots. Various selection of x and adjusted x when there is a shift in mean are given in Table III.
As it can be seen from the above table, under the assumption that eff R ≈ eff T , if there is a 20% shift in mean, i.e., \( \frac{\varDelta }{\mu_R}=0.2, \) the selection of x = 2.5 is equivalent to the selection of x = 3 without a shift in population mean.
Similar idea can be applied to the case where there is a shift in scale parameter (i.e., C ≠ 1). It should also be noted that the above proposal is similar to the justification (based on % of coefficient of variation) as described in your questions. It should be noted that for biosimilar products, the assumption that μ T ≈ μ R and σ T ≈ σ R is usually not true. In practice, it is reasonable to assume that eff R ≈ eff T . The above proposal accounts for possible shift in population mean and heterogeneity in variability.
CONCLUDING REMARKS
The concept of stepwise approach recommended by the FDA is well taken. The purpose is to obtain the totality-of-the-evidence in order for demonstration of biosimilarity between a proposed biosimilar product and an innovative biological product. The totality-of-the-evidence consists of evidence from analytical studies for characterization of the molecule, animal studies for toxicity, pharmacokinetics and pharmacodynamics for pharmacological activities, clinical studies for safety/tolerability, immunogenicity, and efficacy. FDA, however, does not indicate whether these evidences should be obtained sequentially or simultaneously. It is a concern that sequential approach may kill good products early purely by chance alone.
The stepwise approach starts with structural and functional characterization of critical quality attributes that may be relevant to clinical outcomes. FDA’s recommended tiered approach is to serve the purpose. The recommended tired approach depends upon the classification of identified CQAs based on their criticality (or risk ranking) relevant to clinical outcomes. The assessment of criticality, however, is somewhat subjective and often lack of scientific/statistical justification.
The FDA-recommended tiered approach has raised a number of scientific and/or controversial issues. These controversial issues are related to difference in population means and heterogeneity within and across lots within and between the test product and the reference product. In practice, the primary assumption that μ T ≈ μ R and σ T ≈ σ R is usually not true. In this case, it is reasonable to assume that \( ef{f}_R=\frac{\mu_R}{\sigma_R}\approx \frac{\mu_T}{\sigma_T}=ef{f}_T \) so that assessment of similarity between the proposed biosimilar product and the reference product is possible.
As indicated by Tsong (15), Tier 1 equivalence test supposes to be more rigorous than Tier 2 quality range approach. Thus, we would expect passing Tier 1 test will pass Tier 2 test. In practice, however, there is no guarantee that a given CQA which passes Tier 1 test will pass Tier 2 test and vice versa. This may be due to difference in primary assumptions made for Tier 1 equivalence test and Tier 2 quality range approach. Since there may be a large number of CQAs in both Tier 1 and Tier 2, “Does FDA require all CQAs at either Tier pass the corresponding test in order to claim totality-of-the-evidence?” is probably the most commonly asked question. If not, are there any rules to follow?
Liao and Darken (16) and Chow (17) indicated that a good study design than can include different reference lots manufactured at different times with different shelf lives should be used in order to accurately and reliably quantitate different sources of variability for estimation of σ R . Under a valid study design, appropriate statistical model depending upon the nature of the CQAs (e.g., paired or non-paired) should be employed. A proposed biosimilar product with relatively smaller variability as compared to its innovative biological product should be rewarded (16).
References
Chow SC, Endrenyi L, Lachenbruch PA, Mentre F. Scientific factors and current issues in biosimilar studies. J Biopharm Stat. 2014;24:1138–53.
FDA. Guidance on scientific considerations in demonstrating biosimilarity to a reference product. Silver Spring: The United States Food and Drug Administration; 2015.
FDA. Guidance for industry – process validation: general principles and practices, Current Good Manufacturing Practices (CGMP), Revision 1, the United States Food and Drug Administration, Rockville, Maryland, USA; 2011.
Christl L. Overview of regulatory pathway and FDA’s guidance for development and approval of biosimilar products in US. Presented at FDA ODAC meeting, January 7, 2015, Silver Spring, Maryland. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM436387.pdf.
Tsong Y, Dong X, Shen M. Development of statistical methods for analytical similarity assessment. J Biopharm Stat. 2015. doi:10.1080/10543406.2015.1092038.
Chow SC. On assessment of analytical similarity in biosimilar studies. Drug Des Open Access. 2014;3:e124. doi:10.4172/2169-0138.1000e124.
Chow SC. Challenging issues in assessing analytical similarity in biosimilar studies. Biosimilars. 2015;5:33–9.
FDA. Guidance on bioavailability and bioequivalence studies for orally administrated drug products – general considerations, center for drug evaluation and research, the United States Food and Drug Administration, Rockville, Maryland, USA; 2003.
SSAB. A communication package submitted to the FDA. Thousand Oaks: Scientific Statistical Advisory Board on Biosimilars (SSAB) sponsored by Amgen; 2010.
Chow SC, Endrenyi L, Lachenbruch PA, Yang LY, Chi E. Scientific factors for assessing biosimilarity and drug interchangeability of follow-on biologics. Biosimilars. 2011;1:13–26.
Haidar SH, Davit B, Chen ML, Conner D, Lee L, Li QH, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res. 2008;25:237–41.
Yu LX. Bioinequivalence: concept and definition. Presented at Advisory Committee for Pharmaceutical Science of the Food and Drug Administration. April 13–14, 2004, Rockville, Maryland.
Wang TR, Chow SC. On establishment of equivalence acceptance criterion in analytical similarity assessment. Presented at Poster Session of the 2015 Duke-Industry Statistics Symposium, Durham, North Carolina, October 22–23, 2015.
Chow SC, Song FY, Endrenyi L. A note on Chinese draft guidance on biosimilar products. Chin J Pharmaceut Anal. 2015;35(5):762–7.
Tsong Y. Analytical similarity assessment. Presented at Duke-Industry Statistics Symposium, Durham, North Carolina. October 22–23, 2015.
Liao JJZ, Darken PF. Comparability of critical quality attributes for establishing biosimilarity. Stat Med. 2013;32:462–9.
Chow SC. Biosimilars: design and analysis of follow-on biologics. New York: Taylor & Francis: Chapman and Hall/CRC Press; 2013.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chow, SC., Song, F. & Bai, H. Analytical Similarity Assessment in Biosimilar Studies. AAPS J 18, 670–677 (2016). https://doi.org/10.1208/s12248-016-9882-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-016-9882-5