
Abstract
Introduction
Mesenchymal stromal cells (MSC) are an integral cellular component of the tumor microenvironment. Nevertheless, very little is known about MSC originating from human malignant tissue and modulation of these cells by tumor-derived factors. The aim of this study was to isolate and characterize MSC from head and neck squamous cell carcinoma (HNSCC) and to investigate their interaction with tumor cells.
Methods
MSC were isolated from tumor tissues of HNSCC patients during routine oncological surgery. Immunophenotyping, immunofluorescence and in vitro differentiation were performed to determine whether the isolated cells met the consensus criteria for MSC. The cytokine profile of tumor-derived MSC was determined by enzyme-linked immunosorbent assay (ELISA). Activation of MSC by tumor-conditioned media was assessed by measuring cytokine release and expression of CD54. The impact of MSC on tumor growth in vivo was analyzed in a HNSCC xenograft model.
Results
Cells isolated from HNSCC tissue met the consensus criteria for MSC. Tumor-derived MSC constitutively produced high amounts of interleukin (IL)-6, IL-8 and stromal cell-derived factor (SDF)-1α. HNSCC-derived factors activated MSC and enhanced secretion of IL-8 and expression of CD54. Furthermore, MSC provided stromal support for human HNSCC cell lines in vivo and enhanced their growth in a murine xenograft model.
Conclusions
This is the first study to isolate and characterize MSC from malignant tissues of patients with HNSCC. We observed cross-talk of stromal cells and tumor cells resulting in enhanced growth of HNSCC in vivo.
Similar content being viewed by others
Introduction
Mesenchymal stromal cells (MSC) are multipotent, fibroblast-like progenitor cells that can differentiate into multiple lineages, including osteogenic, adipogenic and chondrogenic cell types. MSC express specific cell surface markers such as CD29, CD73, CD90 and CD105 while lacking hematopoietic markers such as CD14, CD34 and CD45 [1]. Initially described as multipotent stromal cells originating from the bone marrow, MSC were later isolated from various fetal and adult tissues, including the salivary gland, placenta, umbilical cord blood, muscle, adipose tissue, connective tissue and peripheral blood [2–7]. Recently, MSC were also identified in pathological tissues and were found to be actively recruited towards tumors and inflammatory microenvironments [8–11].
The functions of MSC within the tumor microenvironment are very complex and still require extensive characterization [12]. For instance, MSC can differentiate into endothelial or vascular smooth muscle cells and might be critically involved in angiogenic processes [13]. Furthermore, MSC might have strong immunoregulatory effects on both innate and adaptive immune cells as they were shown to modulate the proliferation of T cells, B cells or natural killer cells by releasing various cytokines and metabolites of the arachidonic acid pathway [14–16]. The strong modulatory potential of MSC is further highlighted by their ability to release either proinflammatory or anti-inflammatory factors in response to different types of stimulation [17]. Altogether, these findings indicate that MSC engage in a multidirectional communication with the other cells of the microenvironment and are likely to play an important role in the progression of solid tumors.
Indeed, a number of recent studies demonstrated that MSC enhanced tumor growth and metastasis in several types of cancer [9, 18–23]. MSC exerted these tumor-promoting effects by enhancing angiogenesis, inducing immunosuppression or inhibiting the apoptosis of tumor cells [22, 24, 25]. In sharp contrast, other studies found that MSC had potent antitumor effects by inhibiting angiogenesis, promoting antitumor immune responses and inducing apoptosis of the cancer cells [26–33]. A possible explanation for this discrepancy may be the variability of MSC depending on the tissue and the microenvironment from which they were isolated [34]. Additionally, some variability might exist between human MSC and MSC derived from murine tissues – which have been used in many of the previous studies. Because of these caveats, the exact effects of human tumor-resident MSC on the progression of human tumors remain unclear and need to be further investigated.
In this study, we isolated for the first time MSC from tumor tissues of patients with head and neck squamous cell carcinoma (HNSCC). The isolated cells met the consensus criteria for MSC [35] and were successfully maintained and propagated in vitro. These tumor-derived mesenchymal stromal cells (TuMSC) exhibited a proinflammatory phenotype and responded to stimulation by tumor cells with a strong interleukin (IL)-8 release and with CD54 upregulation, respectively. Most importantly, TuMSC significantly enhanced the growth of human HNSCC lines when xenografted into recipient animals in vivo. Our data thus suggest a bidirectional interaction of MSC and tumor cells that, ultimately, results in the progression of HNSCC.
Materials and methods
Ethic statement
Human HNSCC tissue was obtained from surgical specimens after patients’ written informed consent using guidelines approved by the Ethics Committee of the University Hospital of Essen.
Isolation of mesenchymal stromal cells
Head and neck cancer tissue samples were collected aseptically in sodium chloride (0.9%; Fresenius Kabi, Bad Homburg, Germany). Subsequently, tumor specimens were washed several times with Ringer’s solution (Braun, Melsungen, Germany) to remove the majority of erythrocytes. Tissues were cut into 1 to 2 mm pieces, washed extensively and digested in Ringer’s solution containing 5 mg/ml collagenase type II (CellSystems, Troisdorf, Germany) for 40 minutes at 37°C with gentle shaking. Tissues were centrifuged at 300 × g for 7 minutes, and the supernatant was discarded. The partially digested tissues were further treated with Ringer’s solution and 33 mg/ml dispase (Roche Applied Science, Mannheim, Germany) for 60 minutes at 37°C. The cell suspension was then centrifuged, and the pellet was resuspended in standard culture medium (high-glucose Dulbecco’s modified Eagle’s medium; Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (Biochrom, Berlin, Germany), 1% penicillin/streptomycin (Invitrogen) and 1% sodium pyruvate (Invitrogen) and was transferred to tissue culture flasks. Nonadherent cells were removed by washing with phosphate-buffered saline 48 hours later and fresh medium was added to the remaining cells. During the culture period, cells were maintained at 37°C in a humidified atmosphere of 5% carbon dioxide. Cells were continuously passaged after reaching subconfluency by StemPro® Accutase® Cell Dissociation Reagent (Invitrogen) treatment for 5 minutes at 37°C. Bone-marrow-derived mesenchymal stromal cells (BMMSC) were obtained by a standard procedure as described previously [36].
Flow cytometry
Direct immunofluorescence was performed for flow cytometric cell-surface marker immunophenotyping using the following specific monoclonal antibodies: CD14 PE (clone M5E2), CD19 PE (clone HD37), CD73 PE (clone AD2), CD90 (Thy-1) (clone 5E10) (all BD Bioscience, Heidelberg, Germany), CD34 fluorescein isothiocyanate (clone 581; Invitrogen/Molecular Probes), CD45 PE (clone 5B1; Miltenyi, Bergisch Gladbach, Germany) and CD105 fluorescein isothiocyanate (clone 166707; R&D Systems, Wiesbaden, Germany). To determine nonspecific signals, isotype controls were used at the same concentration as that used for the specific antibody. Analysis was performed using a FACS Canto II Flow Cytometer (BD Bioscience) and the resulting data were processed using Diva 6 software (BD Bioscience).
Immunofluorescence microscopy
Cells were immunostained for Vimentin (1:200; mouse monoclonal clone VIM13.2; Sigma-Aldrich, Taufkirchen, Germany), S100A4 (1:100, rabbit polyclonal antibody; Abcam, Cambridge, UK), secondary antibody goat anti-mouse IgG fluorescein isothiocyanate (1:100; Dianova, Hamburg, Germany) and goat anti-rabbit IgG Cy3 bis-NHS ester (Cy3) (1:1,000; Dianova). Cells were examined with an Axioskop 2 microscope with a Ph2 Plan-Neofluar 20×/0.5 objective lens (Carl Zeiss MicroImaging, Göttingen, Germany). Images were generated using an Axiocam MRc microscope camera and Axiovision AxioVS40 Software (Carl Zeiss MicroImaging).
Trilineage in vitro differentiation
Differentiation towards osteogenic, adipogenic and chondrogenic lineage was induced as described previously [37]. In brief, cells were seeded at a density of 3 × 103 cells/cm2 on round glass slides in 12-well culture dishes (Greiner Bio-One, Frickenhausen, Germany). For osteogenic differentiation, cells were cultured for 21 days in Mesenchymal Stem Cell Osteogenic Differentiation Medium (PromoCell, Heidelberg, Germany). Medium change was performed every 3 to 4 days. Cells were finally stained with alizarin red S solution for 2 minutes to confirm the formation of calcium phosphate salts.
For adipogenic differentiation, we used Mesenchymal Stem Cell Adipogenic Differentiation Medium (PromoCell) for 14 days. To examine the generation of oil droplets in the cytoplasm after differentiation, cells were fixed with 10% formalin (Sigma-Aldrich) and stained with Sudan-III (Sigma-Aldrich) for 20 minutes at room temperature. Hematoxylin (Thermo Scientific, Bonn, Germany) was used to visualize nuclei.
Chondrogenic differentiation was induced after 48 hours of culture in standard medium supplemented with dexamethasone, 1 × 10-3 M l-proline (Sigma-Aldrich), 10 ng/ml transforming growth factor-β3 (Sigma-Aldrich) and 1% BD ITS Culture supplement (BD Bioscience). Medium change was performed every 3 to 4 days. To demonstrate the presence of glycosaminoglycans, Alcian blue staining was used. Dried 5 μm cryosections of the micromass pellets were fixed with formalin and washed with phosphate-buffered saline. Staining with Alcian blue 8GX (Roth, Karlsruhe, Germany) was performed at room temperature for 60 minutes.
Cytokine profiling of mesenchymal stromal cells
After isolation and cultivation as described above, medium was exchanged and supernatant was collected from three different patient samples of MSC over a period of 24 hours. Cytokine secretion was analyzed with a bead-based multiplex assay (Bio-Plex; Bio Rad, Hercules, CA, USA) according to the manufacturer’s instructions.
Stimulation of MSC with tumor-conditioned medium
TuMSC from different patient samples (see Table 1) were incubated in the presence of supernatants obtained from the HNSCC cell lines FaDu (ATCC HTB-43) and UM-SSC-22B (kindly provided by TK Hoffmann, University of Ulm, Germany). After 36 to 48 hours, tumor-conditioned medium was discarded and replaced by standard culture medium (high-glucose Dulbecco’s modified Eagle’s medium; Invitrogen) supplemented with 10% fetal bovine serum (Biochrom), 1% penicillin/streptomycin (Invitrogen) and 1% sodium pyruvate (Invitrogen). The control group was incubated for the same time period in standard culture medium. After 24 hours, medium was collected or cells were harvested. IL-8 secreted by activated MSC was measured by enzyme-linked immunosorbent assay (R&D Systems). Expression of CD54 (clone HA58; eBioscience, Frankfurt, Germany) on MSC was analyzed by flow cytometry.
Murine xenograft model
Equal numbers of MSC isolated from three different patient samples were pooled and resuspended in fresh medium. TuMSC/BMMSC and HNSCC FaDu cells were mixed in a 1:1 ratio, resuspended in phosphate-buffered saline at a concentration of 106 cells/100 μl and injected subcutaneously into the right flank of immunodeficient nude mice (TuMSC/BMMSC, n = 8/n = 4). A control group (n = 8) was injected with 0.5 × 106 FaDu cells only. The tumor volume was calculated each day using the following formula:
where V is volume, w is width, and l is length. After 35 days (45 days for control group), mice were sacrificed and the tumors were analyzed. Early termination at day 35 for the MSC/HNSCC group was required by local animal ethics regulations. Animal experiments were approved by the responsible animal ethics committee of the state of North Rhine-Westphalia and carried out according to German guidelines for experimental animal welfare.
Histology
Murine xenograft tumors were stained for Ki-67 and terminal deoxynucleotidyl transferase-dUTP nick end-labeling (TUNEL) as markers for proliferation rates and cell death. Anti-Ki-67 antibody MIB-1 mouse anti-human (1:100; Dako, Hamburg, Germany) was used as primary antibody, followed by peroxidase-conjugated rabbit anti-mouse (1:50; Dianova) and goat anti-rabbit (1:50; Dianova) secondary antibodies. Hematoxylin and eosin counterstaining was performed for nucleus identification. Frequency of Ki-67-positive cells as percent of total cells was calculated using three representative images per sample.
Staining for TUNEL was performed with the Apo-Direct Kit© (BD Bioscience) according to the manufacturer’s instructions. The TUNEL-positive area was calculated with Image J (Fiji, open source, http://imagej.net/Fiji) software.
Results
Isolation and characterization of MSC from HNSCC tissues
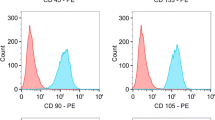
We first explored whether MSC can be obtained from malignant tissue of HNSCC patients employing standard isolation protocols for MSC. The isolated cells showed plastic adherence under standard culture conditions with a typical fibroblast-like morphology. To test whether the isolated cells expressed surface markers characteristic for MSC, these cells were subjected to flow cytometric analysis. We found that the isolated cells expressed MSC surface markers such as CD73, CD90 and CD105 and lacked the expression of hematopoietic markers such as CD14, CD19, CD34, CD45 and HLA class II (Figure 1a). Immunofluorescence analysis revealed the expression of further mesenchymal markers characteristic for MSC, such as vimentin and S100A4 (Figure 1b). An important criterion for MSC characterization is defined by the multilineage differentiation capacity. Under differentiation-inducing conditions, the isolated cells were tested for their chondrogenic, osteogenic and adipogenic multipotency. As shown in Figure 2, these cells could be induced to differentiate into all three lineages. Taken together, these studies established for the first time the expansion of MSC isolated from malignant tissue of patients with HNSCC.

Immunophenotyping of head and neck squamous cell carcinoma-derived mesenchymal stromal cells. Mesenchymal stromal cells (MSC) were isolated from malignant tissue of patients with head and neck squamous cell carcinoma and were expanded. (a) Immunophenotyping was performed by flow cytometry. Data depicted as histograms. (b) Tumor-derived MSC were seeded on cover slips and immunofluorescence staining for vimentin and S100A4 was performed. Data and images from one representative experiment are shown.

Trilineage differentiation of head and neck squamous cell carcinoma-derived mesenchymal stromal cells. Tumor-derived mesenchymal stromal cells were harvested and subjected to trilineage differentiation using standard differentiation-inducing culture conditions. Top row: adipogenic differentiation after 14 days, staining performed with Sudan III. Middle row: chondrogenic differentiation after 14 days, staining with Alcian blue. Bottom row: osteogenic differentiation after 21 days, staining with Alizarin red. Images from one representative donor are shown.
Cytokine profiling of TuMSC and modulation by tumor-derived factors
MSC exert their immunoregulatory activity partly via release of cytokines. Cytokine profiling of TuMSC revealed high constitutive expression of inflammatory cytokines (IL-6, IL-8, tumor necrosis factor alpha), homeostatic chemokines (stromal cell-derived factor-1α) and T-helper type 1 cytokines (interferon gamma) (Figure 3b). A number of other T-cell-modulating cytokines either were not expressed (IL-5, IL-7, IL-10, IL-12, IL-13, IL-17; data not shown) or were expressed at low levels (IL-1β, IL-2, IL-4, granulocyte–macrophage colony-stimulating factor, macrophage inflammatory protein-1β; Figure 3a).

Cytokine profile of tumor-derived mesenchymal stromal cells. Tumor-derived mesenchymal stromal cells obtained from three individual patients were cultured for 24 hours and the culture supernatant was harvested. Concentration of cytokines was analyzed with a bead-based multiplex assay. Secretion was classified as no secretion (IL-5, IL-7, IL-10, IL-12, IL-13, IL-17; data not shown), (a) low secretion (<100 pg/ml) and (b) high secretion (>100 pg/ml). Data depicted as mean ± standard deviation. G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; MIP-1 β, macrophage inflammatory protein-1β; SDF-1α, stromal cell-derived factor-1α; TNFα, tumor necrosis factor alpha; VCAM1, vascular cell adhesion molecule 1.
In the tumor microenvironment, TuMSC are likely to be engaged in cross-talk with tumor cells. To investigate a possible modulation of MSC by tumor cells, we exposed TuMSC to conditioned media obtained from the HNSCC cell lines FaDu and UM-SSC-22B and determined the subsequent release of cytokines. Specifically, we assessed the levels of IL-8 because this proinflammatory cytokine is known to be released by MSC from various sources and is involved in the modulation of the tumor microenvironment [38]. As illustrated in Figure 4a, HNSCC cell line supernatant strongly enhanced the release of IL-8 by MSC. The cell–cell adhesion molecule CD54 (ICAM-1) is a robust marker of activation in a large number of cell types. We therefore examined regulation of CD54 expression in TuMSC exposed to tumor-conditioned medium. Our results show that tumor-derived factors strongly upregulated surface expression of CD54 in all tested donors (Figure 4b) both for FaDu and UM-SSC-22B supernatant. As shown in Table 1, this activation occurred in MSC obtained from tumor samples of various histopathological background. In sum, these studies indicate that TuMSC from HNSCC patients release large amounts of immunomodulatory cytokines and are strongly activated if exposed to tumor-derived factors from HNSCC cell lines.

Activation of tumor-derived mesenchymal stromal cells by head and neck squamous cell carcinoma-conditioned medium. Individual tumor-derived mesenchymal stromal cell (TuMSC) lines were incubated in the presence of culture supernatants obtained from head and neck squamous cell carcinoma cell lines. After 36 to 48 hours, the FaDu/UM22B conditioned supernatant was aspirated and replaced by fresh standard culture medium. After another 24 hours, the TuMSC culture supernatant (a) or TuMSC (b) was collected. Concentration of interleukin (IL)-8 was measured by enzyme-linked immunosorbent assay (a) and expression of CD54 on activated TuMSC was analyzed by flow cytometry (b, c). The experiment was performed with TuMSC from seven independent donors designated #1 to #7. APC, Allophycocyanin; MFI, median fluorescence intensity; SN, supernatant.
Tumor-derived MSC promote HNSCC growth in a murine xenograft model
In the final part of the study we analyzed the impact of TuMSC on the progression of HNSCC in vivo. In this experiment we included BMMSC, for which tumor-promoting properties have been described in some models [39, 40], as a reference group. TuMSC/BMMSC were coinjected with HNSCC cells (FaDu) into immunodeficient nude mice. As a control, a group of animals was injected with HNSCC cells only. Tumor growth was assessed in a time course. The results show a significant difference in tumor growth between the three groups of mice (Figure 5). Specifically, the majority of mice injected with HNSCC cells and MSC developed palpable tumors already between days 7 and 20, and reached a high tumor volume around day 30. Owing to local animal ethics regulations, animals with FaDu/MSC tumors had to be sacrificed at day 35 because of tumor size and progression. In contrast, HNSCC cells lacking support by MSC displayed significantly delayed growth, with tumor growth starting around day 35 post injection. Histologic sections showed differences in proliferation rates and apoptosis between tumors coinjected with MSC and FaDu-only tumors. As shown in Figure 5b, coinjected tumors had a larger proliferative activity in comparison with FaDu-only tumors. This difference was also observed for TUNEL staining. These data demonstrate that primary MSC isolated from human HNSCC tissues and bone marrow provide substantial stromal support to promote HNSCC growth in vivo.

Effect of mesenchymal stromal cells on head and neck squamous cell carcinoma progression in a xenograft murine model. Equal numbers of mesenchymal stromal cells isolated from three different patient samples were pooled and admixed to HNSCC FaDu cells in a 1:1 ratio. Mixtures of tumor-derived mesenchymal stromal cells (TuMSC)–FaDu cells (n = 8) and bone marrow-derived mesenchymal stromal cells (BMMSC)–FaDu cells (n = 4) were injected into the right flank of immunodeficient nude mice. The control group was injected with only FaDu cells (n = 8). (a) The tumor volume was monitored by caliper measurements. Gray vertical line, sacrifice of mice according to ethic requirements. Cryosection slides of excised tumors (n = 3 for each group) were analyzed for Ki-67 staining (b) and apoptosis TUNEL staining (c). Representative pictures were chosen and analyzed under a 400-fold magnification. p.i., post injection (of tumor); TUNEL, terminal deoxynucleotidyl transferase-dUTP nick end-labeling.
Discussion
The histopathology of HNSCC is characterized by large stromal compartments, which surround the tumor islands. Only recently has it been fully recognized that the growth and progression of carcinomas are not only determined by the intrinsic properties of the tumor cells but critically depend on their interaction with the stromal cells. More recently, MSC have been recognized as important cellular components of the tumor stroma [18, 19]. As such, MSC can promote tumor development and progression by various means [12]. For instance, MSC directly modulate tumor cell biology and enhance proliferation, invasion and metastasis. MSC can also enhance angiogenesis by differentiating into or activating endothelial cells [13]. Finally, MSC have very potent immunoregulatory properties [41] and may downregulate antitumor immunity by a large variety of immunosuppressive mechanisms [14–16].
One way in which MSC interact with the surrounding cells in the tumor microenvironment is via release of cytokines and chemokines. Consistent with this idea, MCS isolated from our HNSCC patients released large amounts of cytokines. It is also noteworthy that, on a per-cell basis, this release may well exceed production of such cytokines by tumor-infiltrating leukocytes. Cytokine profiling revealed that IL-6, IL-8 and stromal cell-derived factor-1 were the most abundant cytokines secreted by the TuMSC in our study. Interestingly, this group of cytokines has previously been reported to be involved in the recruitment of MSC to the tumor site [8, 11, 42]. Still a matter of debate, however, is whether MSC are actively recruited to the tumor site or derive from tissue-resident precursors. While it appears to be unlikely that the entire stroma of carcinomas is actively recruited from a distant site, elegant studies by several groups indicate at least a partial contribution of bone-marrow derived cells to certain components of the tumor stroma [9, 20].
Irrespective of their origin, once present in the malignant tissue MSC are exposed to tumor cells and are likely to be engaged in a bidirectional interaction with those malignant cells. In support of this hypothesis, we found a dramatic upregulation of IL-8 release and of CD54 expression when TuMSC were exposed to tumor-conditioned medium. These findings are in agreement with a number of other studies demonstrating that MSC can be functionally modulated by tumor-derived factors [43–45].
During recent years, MSC have been isolated from a variety of tissues. Many studies analyzing the interactions of MSC with tumor cells used MSC derived from adipose tissue (a readily available tissue from which MSC can be easily isolated) or from bone marrow (where MSC were originally identified and characterized). While MSC derived from different sources share many common features and properties, some functional heterogeneity does exist depending on the tissue of origin [2, 3, 34]. This heterogeneity might also explain the contradictory findings regarding the effect of MSC on cancer progression [34]. Here, we investigated the effect of MSC derived from bone marrow and from malignant tissues of HNSCC patients on the biology of the tumor cells. We clearly showed that both types of MSC strongly promoted tumor growth when coinjected with HNSCC cells into recipient mice. Histologic observations showed histomorphological differences between the tumors coinjected with MSC and the FaDu-only tumors. Notably, coinjection of MSC results in a higher proliferation and apoptosis throughout the sections, indicating a stimulating and metabolism promoting role of MSC in the tumor microenvironment. Because very few studies have thus far analyzed human tumor-resident MSC, our findings might shed new light on how MSC modulate tumor progression in cancer patients.
Conclusion
In this study, we isolated, expanded and characterized for the first time MSC from human malignant HNSCC tissues. This enabled us to assess basic biological functions of these cells, to investigate their modulation by tumor cells and to explore their effects on the growth of human cancer xenografts in mice. Our findings thus contribute to a better understanding of the tumor–stroma interactions and, ultimately, may stimulate the search for novel therapeutic strategies for HNSCC and other types of cancer.
Abbreviations
- BMMSC:
-
bone marrow-derived mesenchymal stromal cells
- HNSCC:
-
head and neck squamous cell carcinoma
- IL:
-
interleukin
- MSC:
-
mesenchymal stromal cells
- TuMSC:
-
tumor-derived mesenchymal stromal cells
- TUNEL:
-
terminal deoxynucleotidyl transferase-dUTP nick end-labeling.
References
Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, Deans RJ, Krause DS, Keating A: Clarification of the nomenclature for MSC: the International Society for Cellular Therapy position statement. Cytotherapy. 2005, 7: 393-395. 10.1080/14653240500319234.
Hass R, Kasper C, Bohm S, Jacobs R: Different populations and sources of human mesenchymal stem cells (MSC): a comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal. 2011, 9: 12-10.1186/1478-811X-9-12.
Miao Z, Jin J, Chen L, Zhu J, Huang W, Zhao J, Qian H, Zhang X: Isolation of mesenchymal stem cells from human placenta: comparison with human bone marrow mesenchymal stem cells. Cell Biol Int. 2006, 30: 681-687. 10.1016/j.cellbi.2006.03.009.
Nathan S, Das De S, Thambyah A, Fen C, Goh J, Lee EH: Cell-based therapy in the repair of osteochondral defects: a novel use for adipose tissue. Tissue Eng. 2003, 9: 733-744. 10.1089/107632703768247412.
Prockop DJ, Gregory CA, Spees JL: One strategy for cell and gene therapy: harnessing the power of adult stem cells to repair tissues. Proc Natl Acad Sci U S A. 2003, 100: 11917-11923. 10.1073/pnas.1834138100.
Rotter N, Oder J, Schlenke P, Lindner U, Bohrnsen F, Kramer J, Rohwedel J, Huss R, Brandau S, Wollenberg B, Lang S: Isolation and characterization of adult stem cells from human salivary glands. Stem Cells Dev. 2008, 17: 509-518. 10.1089/scd.2007.0180.
Vaananen HK: Mesenchymal stem cells. Ann Med. 2005, 37: 469-479. 10.1080/07853890500371957.
Gao H, Priebe W, Glod J, Banerjee D: Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells. 2009, 27: 857-865. 10.1002/stem.23.
Goldstein RH, Reagan MR, Anderson K, Kaplan DL, Rosenblatt M: Human bone marrow-derived MSCs can home to orthotopic breast cancer tumors and promote bone metastasis. Cancer Res. 2010, 70: 10044-10050. 10.1158/0008-5472.CAN-10-1254.
Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, Battula VL, Weil M, Andreeff M, Marini FC: Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009, 27: 2614-2623. 10.1002/stem.187.
Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D: Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp Cell Res. 2010, 316: 3417-3424. 10.1016/j.yexcr.2010.07.002.
Cuiffo BG, Karnoub AE: Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell Adh Migr. 2012, 6: 220-230. 10.4161/cam.20875.
Silva GV, Litovsky S, Assad JA, Sousa AL, Martin BJ, Vela D, Coulter SC, Lin J, Ober J, Vaughn WK, Branco RV, Oliveira EM, He R, Geng YJ, Willerson JT, Perin EC: Mesenchymal stem cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a canine chronic ischemia model. Circulation. 2005, 111: 150-156. 10.1161/01.CIR.0000151812.86142.45.
Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, Risso M, Gualandi F, Mancardi GL, Pistoia V, Uccelli A: Human mesenchymal stem cells modulate B-cell functions. Blood. 2006, 107: 367-372. 10.1182/blood-2005-07-2657.
Sheng H, Wang Y, Jin Y, Zhang Q, Zhang Y, Wang L, Shen B, Yin S, Liu W, Cui L, Li N: A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. 2008, 18: 846-857. 10.1038/cr.2008.80.
Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M: Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells. 2006, 24: 74-85. 10.1634/stemcells.2004-0359.
Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM: A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One. 2010, 5: e10088-10.1371/journal.pone.0010088.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA: Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007, 449: 557-563. 10.1038/nature06188.
Muehlberg FL, Song YH, Krohn A, Pinilla SP, Droll LH, Leng X, Seidensticker M, Ricke J, Altman AM, Devarajan E, Liu W, Arlinghaus RB, Alt EU: Tissue-resident stem cells promote breast cancer growth and metastasis. Carcinogenesis. 2009, 30: 589-597. 10.1093/carcin/bgp036.
Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B, Wang TC: Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011, 19: 257-272. 10.1016/j.ccr.2011.01.020.
Tsukamoto S, Honoki K, Fujii H, Tohma Y, Kido A, Mori T, Tsujiuchi T, Tanaka Y: Mesenchymal stem cells promote tumor engraftment and metastatic colonization in rat osteosarcoma model. Int J Oncol. 2012, 40: 163-169.
Yu JM, Jun ES, Bae YC, Jung JS: Mesenchymal stem cells derived from human adipose tissues favor tumor cell growth in vivo. Stem Cells Dev. 2008, 17: 463-473. 10.1089/scd.2007.0181.
Zhu W, Xu W, Jiang R, Qian H, Chen M, Hu J, Cao W, Han C, Chen Y: Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp Mol Pathol. 2006, 80: 267-274. 10.1016/j.yexmp.2005.07.004.
Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, Noel D, Jorgensen C: Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003, 102: 3837-3844. 10.1182/blood-2003-04-1193.
Suzuki K, Sun R, Origuchi M, Kanehira M, Takahata T, Itoh J, Umezawa A, Kijima H, Fukuda S, Saijo Y: Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol Med. 2011, 17: 579-587.
Cousin B, Ravet E, Poglio S, De Toni F, Bertuzzi M, Lulka H, Touil I, Andre M, Grolleau JL, Peron JM, Chavoin JP, Bourin P, Pénicaud L, Casteilla L, Buscail L, Cordelier P: Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PLoS One. 2009, 4: e6278-10.1371/journal.pone.0006278.
Lim YS, Lee JC, Lee YS, Lee BJ, Wang SG: Growth inhibitory effect of palatine tonsil-derived mesenchymal stem cells on head and neck squamous cell carcinoma cells. Clin Exp Otorhinolaryngol. 2012, 5: 86-93. 10.3342/ceo.2012.5.2.86.
Lu YR, Yuan Y, Wang XJ, Wei LL, Chen YN, Cong C, Li SF, Long D, Tan WD, Mao YQ, Zhang J, Li YP, Cheng JQ: The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol Ther. 2008, 7: 245-251. 10.4161/cbt.7.2.5296.
Ohlsson LB, Varas L, Kjellman C, Edvardsen K, Lindvall M: Mesenchymal progenitor cell-mediated inhibition of tumor growth in vivo and in vitro in gelatin matrix. Exp Mol Pathol. 2003, 75: 248-255. 10.1016/j.yexmp.2003.06.001.
Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J: Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood. 2009, 113: 4197-4205. 10.1182/blood-2008-09-176198.
Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC, Ye L, Zhang X: Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008, 18: 500-507. 10.1038/cr.2008.40.
Qiao L, Xu ZL, Zhao TJ, Ye LH, Zhang XD: Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008, 269: 67-77. 10.1016/j.canlet.2008.04.032.
Zhu Y, Sun Z, Han Q, Liao L, Wang J, Bian C, Li J, Yan X, Liu Y, Shao C, Zhao RC: Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. 2009, 23: 925-933. 10.1038/leu.2008.384.
Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F: Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth?. Stem Cells. 2011, 29: 11-19. 10.1002/stem.559.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E: Minimal criteria for defining multipotent mesenchymal stromal cells, the International Society for Cellular Therapy position statement. Cytotherapy. 2006, 8: 315-317. 10.1080/14653240600855905.
Hemeda H, Jakob M, Ludwig AK, Giebel B, Lang S, Brandau S: Interferon-gamma and tumor necrosis factor-alpha differentially affect cytokine expression and migration properties of mesenchymal stem cells. Stem Cells Dev. 2010, 19: 693-706. 10.1089/scd.2009.0365.
Jakob M, Hemeda H, Janeschik S, Bootz F, Rotter N, Lang S, Brandau S: Human nasal mucosa contains tissue-resident immunologically responsive mesenchymal stromal cells. Stem Cells Dev. 2010, 19: 635-644. 10.1089/scd.2009.0245.
Waugh DJ, Wilson C: The interleukin-8 pathway in cancer. Clin Cancer Res. 2008, 14: 6735-6741. 10.1158/1078-0432.CCR-07-4843.
Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Onoyama M, Ohnishi M, Ohara E, Higashi Y, Tanaka S, Yasui W, Chayama K: Stroma-directed imatinib therapy impairs the tumor-promoting effect of bone marrow-derived mesenchymal stem cells in an orthotopic transplantation model of colon cancer. Int J Cancer. 2013, 132: 813-823. 10.1002/ijc.27735.
Annabi B, Naud E, Lee YT, Eliopoulos N, Galipeau J: Vascular progenitors derived from murine bone marrow stromal cells are regulated by fibroblast growth factor and are avidly recruited by vascularizing tumors. J Cell Biochem. 2004, 91: 1146-1158. 10.1002/jcb.10763.
Manferdini C, Maumus M, Gabusi E, Piacentini A, Filardo G, Peyrafitte JA, Jorgensen C, Bourin P, Fleury-Cappellesso S, Facchini A, Noël D, Lisignoli G: Adipose-derived mesenchymal stem cells exert antiinflammatory effects on chondrocytes and synoviocytes from osteoarthritis patients through prostaglandin E2. Arthritis Rheum. 2013, 65: 1271-1281. 10.1002/art.37908.
Picinich SC, Glod JW, Banerjee D: Protein kinase C zeta regulates interleukin-8-mediated stromal-derived factor-1 expression and migration of human mesenchymal stromal cells. Exp Cell Res. 2010, 316: 593-602. 10.1016/j.yexcr.2009.11.011.
Al-toub M, Almusa A, Almajed M, Al-Nbaheen M, Kassem M, Aldahmash A, Alajez NM: Pleiotropic effects of cancer cells secreted factors on human stroma (mesenchymal) stem cells. Stem Cell Res Ther. 2013, 4: 114-10.1186/scrt325.
Menon LG, Picinich S, Koneru R, Gao H, Lin SY, Koneru M, Mayer-Kuckuk P, Glod J, Banerjee D: Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. 2007, 25: 520-528. 10.1634/stemcells.2006-0257.
Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D: Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68: 4331-4339. 10.1158/0008-5472.CAN-08-0943.
Acknowledgements
The authors would like to acknowledge Anne-Marie Heider and Petra Altenhoff for providing technical assistance. They are grateful to all patients and clinical colleagues who donated or collected clinical samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
BAK provided the data analysis, interpretation and manuscript writing, conducted the immunohistology and obtained clinical material. PAD performed the murine xenograft experiments and cytokine profiling, analyzed data and helped to write the manuscript. KB conducted flow cytometry for phenotyping MSC and analyzed data. HH and AMW performed immunostaining and trilineage differentiation experiments. VJ conducted substantial parts of the murine xenograft experiments and helped during cytokine analysis. PS and KK helped to draft the manuscript and analyzed the data. SL provided clinical management and advice and obtained clinical material. CAD conducted the enzyme-linked immunosorbent assay and flow cytometry, analyzed data, and revised the manuscript. SB provided the conception and design of the study and the manuscript, analyzed and interpreted data, and revised the manuscript. All authors were involved in drafting the manuscript and all authors read and approved the final manuscript.
Benjamin A Kansy, Philip A Dißmann contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kansy, B.A., Dißmann, P.A., Hemeda, H. et al. The bidirectional tumor - mesenchymal stromal cell interaction promotes the progression of head and neck cancer. Stem Cell Res Ther 5, 95 (2014). https://doi.org/10.1186/scrt484
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/scrt484