
Abstract
Background
Melanoma cancer causes serious health problem worldwide because of its rapid invasion to other organs and lack of satisfactory chemotherapy. The pGI50 anticancer activity values of 70 compounds from the NCI (National Cancer Institute) on MALME-3M cell line was modeled to describe the quantitative structure-activity relationships (QSARs) of the compounds, and some selected compounds were docked.
Results
The generated QSAR model was found to be statistically significant based on the obtained values of the validation keys such as R2 (0.885), \( {R}_{\mathrm{adjusted}}^2 \) (0.868), Q2cv (0.842), and \( {R}_{pred}^2 \) (0.738) required to evaluate the strength and robustness of QSAR model. Compound 39 was selected as a template due to its good pGI50 (9.205) and was modified to design new potent compounds. The predicted pGI50 activity of the designed compounds by the built model was N1 (9.836), N2 (12.876), N3 (10.901), and N4 (11.263) respectively. These proposed compounds were docked with V600E-BRAF receptor and the result shows that, N1, N2, N3, and N4 with free binding energy (FBE) of − 11.7 kcal mol−1, − 12.8 kcal mol−1, − 12.7 kcal mol−1, and − 12.9 kcal mol−1 respectively were better than the parent structure of the template (compound 39, FBE = − 7.0 kcal mol−1) and the standard V600E-BRAF inhibitor (Vemurafenib, FBE = − 11.3 kcal mol−1). Additionally, these compounds passed the drug-likeness criteria successfully to be orally bioavailable.
Conclusion
The proposed compounds were considered optimal as their performances are comparable to vemurafenib and possessed enhanced physicochemical properties. Thus recommends further research such as synthesis, in vivo, and ex-vivo evaluation.
Similar content being viewed by others


Background
Melanoma is one of the most aggressive forms of skin tumor and a serious health issue worldwide because of its increasing incidence and the lack of satisfactory chemotherapy for the advanced stages of the disease [1, 2]. It has a high ability of metastasis and rapid invasion of other organs, e.g., lymph node, lung, liver, brain, etc. [3]. The oncoprotein BRAF, as discovered in 1988 is responsible for nearly sixty-six percent (66%) of melanomas [4]. The BRAF kinase is the main target of the therapies for as it is the most regularly mutated protein kinase in human cancers [5]. The most frequent mutation of BRAF, among more than 30 mutations of BRAF, is V600E [6, 7]. The V600E-BRAF mutation ended in 500-fold greater constitutive kinase activity when compared to other BRAF wild kind, and many inhibitors of V600E-BRAF have been designed [8, 9].
Despite thorough research and partial successes achieved by the use of several drugs approved for the for treatment of melanoma cancer by the U.S. Food and Drug Administration (FDA) such as ZM336372, benzylideneoxindoles, sorafenib, isoquinolones, triarylimidazoles, XL281, and Vemurafenib (PLX4032) [10], currently, the effective chemotherapy against invasive melanoma is still lacking. Therefore, it is necessary to search for new therapeutic approaches with better effectiveness and fewer side effects. Ligand-based is one of the most widely practiced approaches in drug discovery and drug design by medicinal chemists. The ligand-based method includes the well-known quantitative structure-activity relationships (QSARs) models [11, 12], which are based on changes in structural features of molecules such as steric, electrostatic, and hydrophobic properties. QSAR approaches have been utilized to identify vital structural features responsible for the anticancer activity of compounds [13, 14]. QSAR is an important factor in the drug design; therefore, it is quite evident why many users of QSAR are found mostly in the research units of industries [15,16,17].
It is, therefore, necessary to construct a QSAR model for the prediction of the activity of the designed leads before their synthesis. Because a successful QSAR model not only helps in understanding relationships between the structural features and biological activity of any class of compounds but also provides researchers a deep analysis of the lead compounds to be used in further studies [13]. Furthermore, understanding the mechanism of the ligand/receptor interactions is very significant in drug development, and the molecular docking simulation method is a proper tool for gaining such understanding. Molecular docking simulation is a computational technique used to predict the binding ability of the active site residues to specific groups on the receptor and to reveal the strength of interaction [5]. Molecular docking is a very useful and popular tool used in the drug discovery arena to evaluate the binding of small molecules (inhibitors) to the receptor (macromolecule) [18, 19]. This study was aimed to design new potent compounds on the MALME-3M cell line through QSAR modeling followed by molecular docking simulation based on the compounds collected from the National Cancer Institute (NCI).
Methods
Data collection and structure preparation
Seventy (70) sets of compounds and their pGI50 activities on MALME-3M melanoma cell line were retrieved from the National Cancer Institute (NCI) database. The anticancer activity, chemical name, and NSC number of the studied compounds are presented in Table 1. The 2D structures were firstly converted into the 3D structure using Spartan 14. Then, the structures were cleaned by checking and minimizing using a molecular mechanic force field (MM2) in order to remove all strain from the molecular structure. In addition, this will ensure a well-defined conformer relationship among compounds of the study. Secondly, the calculation was further set to equilibrium geometry at the ground state using the density functional theory at the B3LYP level of theory and 6-311G (d) basis set for the geometrical optimization of the cleansed structures. The optimized 3D structure was formatted to the SD file and then taken to the PaDEL descriptor tool kit to generate required descriptors for further studies [20].
QSAR model development and validation
The data set was splits into two subsets, the training set and test set using Kennard-Stone Algorithm [21, 22]. The training set is used in building the QSAR model which contains 70% of the data and the remaining 30% is for the test set that was used to evaluate the predictive ability of the model [23]. All the studied compounds were screened through the derived QSAR model for pGI50 activity prediction.
The genetic function algorithm (GFA) was used in the selection of proper descriptors as this improves the model accuracy [24]. Multiple linear regression (MLR) was used on the training set to determine the relationship between the dependent variable Y (pGI50) and independent variable X (molecular descriptors). In regression analysis, the contingent mean of the dependent variable (pGI50) Y relies on (descriptors) X. The best QSAR model was chosen based on the validation parameters such as the correlation coefficient (R2), adjusted R2 (R2adj), cross-validation coefficient (Q2CV), and correlation coefficient for an external prediction set (R2pred) all are represented in Eqs. (1, 2, 3, 4):
where p is the number of independent variables in the model and N is the sample size. Yexp, Ypred, and Ymtraining are the experimental activity, the predicted activity, and the mean experimental activity of the compounds in the modeling set, respectively [23].
Applicability domain and in-silico screening
The applicability domain (AD) of the QSAR model is the theoretical space in the chemical region comprising of both the descriptors of the model and modeled response. This domain permits prediction of uncertainty in the identification of a particular compound based on the data set of compounds used in the development of the model. The AD is also used to define the X-outliers in case of the training set and identify the molecules residing outside the defined AD in case of the test set utilizing the basic theory of standardization approach [11]. Several techniques have been used to define AD of QSAR models [25]. The commonly used one was demonstrated by Gramatica [26] which employed the leverages for each compound of the data set. The leveraged approach enables the evaluation of the position of a new compound in the QSAR model [26]. Therefore, leverage method is utilized and is shown as hi in Eq. (5):
where x refers to the descriptor vector of the considered compound and X represents the descriptor matrix derived from the training set descriptor values. The warning leverage (h*) was determined as in Eq. (6):
where N is the number of training compounds and p is the number of descriptors in the model.
The defined AD was then viewed via a Williams plot, the plot of the standardized residuals against the leverage values (h). A compound with hi > h* seriously influences the model performance and may be eliminated from the AD applicability, but it does not appear to be an outlier since its standardized residual could be small. Furthermore, a value range of ± 3 standardized residuals is often used as a cutoff value for accepting predictions of a compound, because points which lie within ± 3 standardized residuals from the mean cover ninety-nine percent (99%) of the normally distributed data [27]. Thus, the leverage and the standardized residuals were used jointly for the characterization and determination of the applicability domain.
Ligand-protein preparation and docking studies
The selected ligands (compounds) were optimized and formatted to PDB files for docking utilizing Spartan 14. The x-ray structure of the V600E-BRAF kinase (receptor) in complex with PLX4032 (PDB CODE: 3OG7) [5, 28, 29] was retrieved from (www.rcsb.org). The PDB file of V600E-BRAF was prepared using Discovery studio by deleting the excess water molecules contained in the x-ray structure and optimizing the hydrogen molecules and the bound ligand (vemurafenib) was also removed from the target before for the docking process. This complex structure comprises of two homo-dimeric chains (A and B). Our goal was to target the mutated chain (chain A) of V600E-BRAF. Thus, chain B was removed from the structure of 3OG7 and the bound ligand also removed from chain A. All the selected compounds (ligands) were docked into the active kinase domain of V600E-BRAF using Autodock Vina of Pyrex docking program software.
Prediction of drug-likeness properties
The application of computational tools for identifying the novel drug candidate assist to lessen the number of experimental researches and for increasing the success rate. For this purpose, we applied Lipinski’s rule of five for drug-likeness as an initial screening step for oral bioavailability and synthetic accessibility using SwissADME (www.swissadme.ch/) online tool.
Results
To give a systematic prediction of the studied molecules as antimelanoma agents on MALME-3M cell line, the QSAR model was built using Material Studio 8.0. The modeling and prediction sets were selected using the Kennard stone algorithm, in which 49 compounds was used as a modeling data set and 21 compounds as the prediction data set. The GFA lead to the selection of six (6) descriptors and MLR was applied to generate a new QSAR model for the prediction of pGI50 activities of compounds on the MALME-3M cell line. The developed model and the corresponding statistics are shown in Eq. (7):
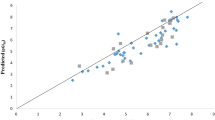
The generated QSAR model was employed to predict the pGI50 activity of the test (prediction) set molecules, and the outcomes are displayed (Table 1). The predicted activity values (pGI50) for the molecules in the training data set and test data set for the MALME-3M melanoma cell line was plotted versus the observed (pGI50) values (Fig. 1). Also, the ME values of all the descriptors that appear in the model are listed in Additional file 1: Table S1. To further validate the proposed QSAR model in this study, the pGI50 of 31 Flavone-based arylamides (FBA) retrieved from the literature [30] was predicted using the developed QSAR model (Eq. 7). The predicted pGI50 results of the FBA derivatives are presented in Additional file 1: Table S2.

The predicted pGI50 against the experimental values for the training and test sets
The Williams plot for the built QSAR is shown in Fig. 2. The warning leverage (h*) was found to be 0.430 for the developed QSAR model. Furthermore, an in silico screening method was used for the design of new potent compounds with pGI50 activity on the MALME-3M cell line. The structure of compound 39 as a template used for modifications is shown in Figs. 3 and 4. The predicted pGI50 and leverage limit of the designed compounds are presented in Table 2.

The Williams plot, the plot of the standardized residuals versus the leverage value

Structure of the template (compound 39) used for design

3D diagram of the interaction between compound 39 with V600E-BRAF
Furthermore, molecular docking simulation studies were conducted between the V600E-BRAF protein kinase, compound 39 (AC1L2OAS), designed compounds, and the standard drug (Vemurafenib) approved for the treatment of melanoma cancer (V600E-BRAF inhibitor). Firstly, the template compound 39 (AC1L2OAS) was docked, then the designed compounds and vemurafenib into the active kinase domain of V600E-BRAF using Autodock Vina in Pyrex version 4.0 software docking program. The best docking pose of vemurafenib were superimposed upon co-crystal structure of ligand as shown in Additional file 1: Figure S1. The detail docking results are reported in Table 3 and the best docking poses of the docked ligands are shown in Figs. 3, 4, 5, 6, 7, 8 and 9. In addition, to ensure that the designed molecules are viable drugs, the drug-likeness properties was predicted using SwissADME (online tool) as presented Table 4.

3D diagram of the interaction between N1 with V600E-BRAF

3D diagram of the interaction between N2 with V600E-BRAF

3D diagram of the interaction between N3 with V600E-BRAF

3D diagram of the interaction between N4 with V600E-BRAF

3D diagram of the interaction between vemurafenib with V600E-BRAF
Discussion
Quantitative evaluation of the structure-activity relationship (QSAR) was conducted on 70 molecules with unique organic moiety acting as anti-melanoma agents to know a quantitative relationship between their molecular structures and anti-melanoma activity. The nature of the QSAR model generated was described by its fitting and predictive ability. The correlation between experimental and predicted activity based on the developed QSAR model was highly significant for our data set as indicated by statistical analysis. The closeness of coefficient of determination (R2) to its absolute value of 1.0 is an indication that the model explained a very high percentage of the response variable (descriptor) variation, high enough for a robust QSAR model. Its 0.885 value illustrates that 88.5% of the variation is residing in the residual meaning that the model is very good.
The high adjusted R2 (R2adj) value as seen in the model and its closeness in value to the value of R2 implies that the model has excellent explanatory power to the descriptors in it. It also demonstrates the real influence of applied descriptors on the pGI50. Also, the high and closeness of Q2cv to R2 revealed that the model was not over-fitted. The high R2pred as seen in the model is an indication that the model is capable of providing valid prediction for new compounds. Generally, a good QSAR model has the following characteristics: R2 and R2adj values close to one. Q2cv > 0.5, R2 - Q2cv ≤ 0.3, R2pred ≥ 0.6, and Npred ≥ 5 [11, 23, 31]. The built QSAR model satisfied these criteria and was therefore statistically acceptable. Therefore, we can conclude with confidence that the model will correctly predict the anti-melanoma activity of a given compound.
Molecular descriptors are the physicochemical and structural information in the form of numerical values, each descriptor represents specific information that can be implore to improve the overall biological effect of a compound. By interpreting descriptors that appear in the model, it is possible to understand the factors which are related to the anti-melanoma activity. Also, the significance and contribution of each descriptor of the developed model was assessed by calculating the value of mean effect, (ME) [32] of each descriptor by applying Eq. (8). Therefore, brief descriptions of the selected descriptors and their ME values are presented in Additional file 1: Table S1 and an acceptable interpretation is provided.
MEj is the ME (mean effect) of descriptor j, βj signifies the coefficient of descriptor j, dij denotes the value of the chosen descriptor for each molecule, and m represents the number of descriptors that appear in the model. The ME value shows the relative importance of each descriptor in compare to the other descriptors. Descriptors that influence anti-melanoma activity and show high values of ME increase anti-melanoma activity (pIG50). The pIG50 changes with the MF values of the descriptor, as shown in Additional file 1: Table S1. Based on ME values, the associated descriptors are arranged in a sequence about their contribution toward overall pGI50 of the compounds, in the following increasing order of pGI50 of compounds.
SpMax1_Bhs > SpMin3_Bhm > SM1_Dzi > VE1_Dzp > TIC1 > SpMax6_Bhe
SpMax1_Bhs is defined as the largest absolute eigenvalue of Burden modified matrix—n 1 / weighted by relative I-state. The positive ME value of the descriptor (1.20) shows that an increase in the value of this descriptor will increase the pGI50 of the compounds. SpMin3_Bhm descriptor has been proposed as the chemical structure descriptors derived from a new representation of the molecular structure. SpMin3_Bhm is the smallest absolute eigenvalue of Burden modified matrix—n 3 / weighted by relative mass. The SpMin3_Bhm has a positive mean effect (1.13). This sign suggests that the anti-melanoma activity is directly related to this descriptor. SM1_Dzi is defined as the Spectral moment of order 1 from the Barysz matrix / weighted by first ionization potential. The ME value of SM1_Dzi is 0.22 as shown in Additional file 1: Table S1. This positive value suggests that the increase of value for this descriptor will increase the anti-melanoma activity of a compound and vice versa.
VE1_Dzp is defined as the coefficient sum of the last eigenvector from the Barysz matrix/weighted by polarizabilities. The low positive value of the mean effect (Additional file 1: Table S1) for VE1_Dzp suggests a positive contribution to the activity, though insignificant since the value is low. TIC1 is defined as the total information content index (neighborhood symmetry of 1-order). The mean effect value for this descriptor has a negative sign (Additional file 1: Table S1). This sign suggests that the anti-melanoma activity will increase with a decrease in its value. SpMax6_Bhe a Burden modified descriptor is defined as the largest absolute eigenvalue of Burden modified matrix—n 6/weighted by relative Sanderson electronegativities. It is related to electronegative atoms of the drug. The ME of this descriptor was − 1.27. Its negative sign indicates that a decrease in the number of electronegative atoms in the molecular structure of the compound increases the pGI50 values of the compounds. The descriptors used for the constructed QSAR model in this work encoded topological, electronic, and geometrical aspects of molecules. Appearances of these descriptors in the model reveal the role of electronic and steric interactions in inducing anti-melanoma pGI50 activity on the MALME-3M cell line.
To further validate the predictive ability of the proposed model, a total of 31 Flavone-based arylamides (new anticancer compounds) was chosen from the literature [30]. Their structures were subsequently optimized and molecular descriptors calculated using the same methods as used for the NCI dataset used in building the model. Thereafter, the proposed QSAR model for MALME melanoma cell line (Eq. 7) was applied to predict the activity values of these compounds and the results (Additional file 1: Table S2) showed that the model could accurately predict the pGI50 of the compounds.
The good pGI50 values predicted for these compounds only shows which structures should be targeted for use as antimelanoma agents on the basis that they approach the optimal values for the chosen descriptors in the model developed in the present study. Also, the in silico screen based on the developed QSAR model clearly achieved its objective in identifying derivatives with improved predicted activity while simultaneously identifying structures that were out of the models domain of applicability and therefore the scope of the model’s reliability. This study thus demonstrates the usefulness of constructing QSAR model which can aid in identifying new viable targets for drug discovery.
Moreover, the Williams plot for the built QSAR is shown in Fig. 2 and the warning leverage (h*) was found to be 0.430 for the developed QSAR model. Based on the leverages (hi > 0.430), only three prediction set compounds (48, 52, and 66) were found to be outside of the defined AD (Fig. 2) of the QSAR model; so, they were identified as structurally influential chemical based on their large leverage values (hi > h*). Furthermore, an in silico screening method was used for the design of new potent compounds with pGI50 activity on the MALME-3M cell line according to the developed QSAR model and was validated by the developed QSAR model. For this purpose, compound 39 (AC1L2OAS, NSC-376,128) listed in Table 1 (pGI50 = 9.205) was chosen as a template due to its high pGI50 activity and low residual value (− 0.001) between the experimental and predicted activity (pGI50). The structure of compound 39 as a template used for modifications is shown in Fig. 3 and 4.
The compound was altered in a way that will make its synthesis experimentally possible. Then, the in-silico screening was applied by the insertion and substitution of different groups at R1 and R2 positions as presented in Fig. 3 and 4; the results of this are presented in Table 2. The model endures various AC1L2OAS substituents since all the designed analogous were within the applicability domain (hi < 0.430). The predicted pGI50 of all the designed analogous were more than the lead compound 39 used for the design and among which compound N2 showed the best activity (pGI50 = 12.876). Thus, it is clear that using a simple QSAR model, there is a possibility to simultaneously predict and identify compounds with better activity and to determine which of the structural modifications do not fall within the AD.
Further, molecular docking simulation studies were conducted between the V600E-BRAF protein kinase, compound 39 (AC1L2OAS), designed derivatives, and vemurafenib (V600E-BRAF inhibitor). All the prepared ligands were docked into the active kinase domain of V600E-BRAF using Autodock Vina in Pyrex version 4.0 software docking program and the desirable conformations of the studied compounds were identified. Validation of docking protocol is an essential step before performing molecular docking based virtual screening and to determine threshold parameters [33]. In order to validate the docking protocol and productivity, the co-crystalized ligand (vemurafenib) was also docked to the binding site of V600E-BRAF kinase with the binding affinity of − 11.3 kcal/mol and the RMSD value for both upper and lower bounds were measured (0.0) which confirmed the docking protocol and productivity. The best docking pose of vemurafenib were superimposed upon co-crystal structure of ligand (vemurafenib) (superimposed co-crystal structure is given in Additional file 1: Figure S1). Based on the RMSD value and binding affinity, the best docking pose of vemurafenib was selected and analyzed.
Based on the free binding energies of the docked ligands and the type of interactions involved (Table 3), it was found that these compounds were sufficiently bonded to the active site. To further analyze the interaction, the values of the free binding energies were used to select the best inhibitors that were found to have good free binding energy and to some extent show similar interactions as the standard drug with the receptor. The selected template (Comp. 39, AC1L2OAS) for the design was docked on the active segment of V600E-BRAF kinase with the free binding energy of − 7.0 kcal mol−1 (Table 3). This docking simulation research revealed that AC1L2OAS was found to bind in the active segment on the protein dimer due to the formation of four carbon-Hbond that occurred between carbonyl oxygen to LYS483, methoxy group to SER465, and other two with GLY534 and ILE463 as shown in Fig. 3 and 4. The benzene ring moiety group intercalated in space to form pi–pi interaction stacked with PHE583 residue similar to vemurafenib. Pi-cation electrostatic interaction was also observed with LYS483 residue. Two alkyl hydrophobic interactions occurred from between Comp. 39 to ILE463 and VAL471 residues. Seven pi-alkyl hydrophobic interaction also occurred in the complex, four were formed from Comp. 39 to ALA481, CYS532, LYS483, and LEU514, while the other three occurred from TYR538 and PHE583 (2) residues to Comp. 39. The obtained results of this molecular docking simulation suggest that the selected active Comp. 39 (AC1L2OAS) can inhibit the growth of the melanoma cell lines by inhibiting the V600E-BRAF kinase which is supported by its high experimental pGI50 (9.205) obtained from NCI as presented in Table 1.
On conducting molecular docking simulation for the newly designed compounds, the best four compounds in terms of their obtained free binding energy values and the type of interactions involved were identified and it was found that the binding energy of the template, Comp. 39 (AC1L2OAS) was increased from − 7.0 kcal mol−1 to − 11.7 kcal mol−1 for N1, − 12.8 kcal mol−1 for N2, − 12.7 kcal mol−1 for N3, and − 12.9 kcal mol−1 for N4 respectively as shown in Table 3. Therefore, the new compounds are the novel V600E-BRAF inhibitors, thus their docking results were compared to the docking result of vemurafenib the standard V600E-BRAF inhibitor.
In Fig. 5, N1 binding to V600E-BRAF was presented. N1 was bound to the active site of the receptor with some similar residue involvement to the standard V600E-BRAF inhibitor (vemurafenib). In most of the iterations, CYS532 was the most significant residue associated with vemurafenib-V600E-BRAF (Fig. 9). One conventional-Hbond was formed between the receptor with LYS578 residue. There is the formation of two carbon-Hbond from methoxy group of NI to ASN581 and ASP594 residues. Two Pi-sulfur was observed between the S-atom of the thiazole moiety to PHE583 and the other one between the aromatic benzene ring to CYS532 as shown in Fig. 5. One halogen bond was observed from the Br-atom (new substituent) to CYS532. Pi-Pi Stacked hydrophobic interaction occurred between PHE583 and N1 and that of alkyl was between ALA543 and N1 through the C-atom. The newly introduced substituent on the parent structure of AC1L2OAS increases the binding stability by forming three eight Pi-alkyl hydrophobic interactions between the receptor and N1 as shown in Fig. 5.
N2 binding to V600E-BRAF was presented in Fig. 6. N2 was bound to the active site of the receptor with some similar residue involvement to vemurafenib. Most of the iterations, CYS532 was the most significant residue associated with vemurafenib-V600E-BRAF interaction. Two conventional-Hbond was present, one from LYS483 to the HN- of the thiazole moiety of N2, another one from carbonyl O-atom N2 to SER536. There is one carbon-Hbond from N2 to GLY615 residue. The N2 molecule was found to interact with the receptor through electrostatic (Pi-Sigma) interaction with TYR538. There exist seven pi-alkyl hydrophobic interactions between N2 to the receptor as observed in Fig. 6 and Table 4 respectively. Pi-Pi stacked hydrophobic interaction also exists between TRP531 and PHE583 of the receptor to N2 similar to vemurafenib as shown in Figs. 6 and 9. Three alkyl hydrophobic interaction was present from N2 to LEU618, LEU654 and VAL471 residues respectively.
In Fig. 7, N3 binding to V600E-BRAF was presented. N3 was bound to the active site of the receptor with some similar residue involvement to vemurafenib. Two conventional-Hbond was present from CYS532 to N3 through oxygen atom of the nitro group (new substituent) and the second was between O-atom of methoxy group to LYS578 residue. Carbon-Hbond interaction occurred between N3 to ASN581 and ASP594 residues respectively. Pi-sulfur interaction occurred between S-atom of the thiazole ring to PHE583, another one was observed between benzene ring of N3 and CYS532 residue. There was the formation of one pi-pi stacked interaction between the benzene ring and PHE583 in a similar way to vemurafenib (Fig. 9). There are three important aromatic residues that led to the formation of pi-alkyl hydrophobic interaction from LEU505, LEU514, and THR529 to N3. There are four alkyl hydrophobic interactions, one occurred from PHE468 to the C-atom of N3, the others were from N3 to ILE463, LYS483, and VAL471 respectively.
N4 binding to V600E-BRAF was presented in Fig. 8. N4 was bound to the active site of the receptor with some similar and additional residue involvement as vemurafenib. There was the formation of important conventional-Hbond between N4 and the receptor with SER536. Pi-sulfur interaction occurred between CYS532 of the receptor and N4. There was one Pi-sigma interaction between N4 and TYR538 residue. Stacked pi-pi interaction is the usual form of pi-interaction which occurs between N4 and PHE583 and TRP531 residues similar to vemurafenib (Fig. 9). Some other alkyl hydrophobic interaction was present between N4 to LEU618 and LEU654 residues. Other Pi-alkyl interaction appear between the receptor to N4 with TYR538 (3) and TRP619 (2) residues. Furthermore, it can be observed that there is a formation of additional hydrogen bonds from the newly introduced substituent than the template (Comp.39) used for design and vemurafenib. Much more interaction was found in all the newly designed compounds particularly N4 which shows more similarity to vemurafenib which if synthesize and tested was hoped to be better V600E-BRAF inhibitor with same therapeutic effect as Vemurafenib and more effective.
The research revealed that hydrogen bonding is the major force controlling the interactions that exist between the docked compounds and the protein target and also the free binding energy of the compounds increases with the increase in the number of the Hbond [34, 35]. It could be observed that in the conventional hydrogen bonding identified with the designed derivatives, the number of amino acids involved was found to be better compared to vemurafenib as shown in the Figs. 3, 4, 5, 6, 7, 8 and 9 and there are high similarities. This might acquaint the more reliable binding scores of the chosen compounds for V600E-BRAF. Hence, these novel compounds will serve as good inhibitors of V600E-BRAF showing competitive inhibition with vemurafenib as evident from the molecular docking results.
Furthermore, to ensure that the designed derivatives are the viable drugs, the drug-likeness was predicted with vemurafenib as the reference. The SwissADME [36] online tool was used to predict the drug-likeness properties as presented in Table 4. The drug-likeness properties are the main criteria used in screening drug candidates at an initial stage of the drug discovery process. This method can be described as a means to correlate the physicochemical properties of a given compound with the bio-pharmaceutical aspect of it in a human body, particularly its influence in oral bioavailability [37].
The initial thorough analysis of drug-likeness properties was performed by Lipinski’s rule [38], which argues that good absorption or permeation is more likely when: the molecular weight (MW) < 500, the number of hydrogen bond donors (HBDs) < 5 (counting the sum of all NH and OH groups) partition coefficient octanol/water Log P < 5, the number of hydrogen bond acceptors (HBAs) < 10 (counting all N and O atoms). As observed from Table 4, all the designed derivatives meet Lipinski’s rule successfully suggesting that these compounds theoretically have ideal oral bioavailability. These physicochemical parameters are associated with acceptable aqueous solubility and intestinal permeability that are the first steps in oral bioavailability.
Conclusion
In this research, the GFA-MLR modeling tool was used in the construction of a QSAR model and the in-silico screening method was applied to the developed QSAR model which enable the design and prediction of activity (pGI50) of new potentially active compounds on MALME-3M cell line. The accuracy and predictability of the proposed model was illustrated by various criteria, the model is statistically robust both internally (R2 = 0.885, R2adj = 0.868, and Q2cv = 0.842) and externally (R2pred = 0.738). This satisfies the criteria of acceptable QSAR models proposed by different groups. Compound 39 was selected as a template among the data set due to its good pGI50 (9.205) and was utilized to design new potent compounds, thereby enhancing the activity of the parent structure. The activity (pGI50) of new potent compounds were predicted by the built QSAR model as N1 = 9.836, N2 = 12.876, N3 = 10.901, and N4 = 11.263 respectively. Moreover, molecular docking simulation was also applied to investigate the proper binding mode of the designed compounds on V600E-BRAF protein kinase. All the studied ligands were able to inhibit the receptor by totally occupying the active segment in the target. The designed N1, N2, N3, and N4 with free binding energy (FBE) of − 11.7 kcal mol−1, − 12.8 kcal mol−1, − 12.7 kcal mol−1, and − 12.9 kcal mol−1 respectively were found to be more potent than the parent structure of the template (compound 39, FBE = − 7.0 kcal mol−1) and vemurafenib (FBE = − 11.3 kcal mol−1) due to the introduction of the new substituent which has the capability of increasing the overall free binding energy by increasing the number of hydrogen bonds and hydrophobic interactions shown in their complex. Additionally, these molecules have shown good physicochemical properties. Thus, in future studies, there is hope to include the synthesis, in vivo, and in vitro evaluation of these ligands which can establish them as potent V600E-BRAF inhibitors for the treatment melanoma cancer.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- NC:
-
National Cancer Institute
- DFT:
-
Density functional theory
- QSAR:
-
Quantitative structure activity relationship
References
Bhat UG, Zipfel PA, Tyler DS, Gartel AL (2008) Novel anticancer compounds induce apoptosis in melanoma cells. Cell Cycle 7(12):1851–1855
Dubey S, Roulin A (2014) Evolutionary and biomedical consequences of internal melanins. Pigment Cell Melanoma Res 27(3):327–338
Dembitsky V, Kilimnik A (2016) Anti-melanoma agents derived from fungal species. MJ Pharma 1(1):002
Dhillon AS, Hagan S, Rath O, Kolch W (2007) MAP kinase signalling pathways in cancer. Oncogene 26(22):3279
Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467(7315):596
Namba H, Nakashima M, Hayashi T, Hayashida N, Maeda S, Rogounovitch TI, Ohtsuru A, Saenko VA, Kanematsu T, Yamashita S (2003) Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metabol 88(9):4393–4397
Puzanov I, Amaravadi RK, McArthur GA, Flaherty KT, Chapman PB, Sosman JA, Ribas A, Shackleton M, Hwu P, Chmielowski B (2015) Long-term outcome in BRAFV600E melanoma patients treated with vemurafenib: patterns of disease progression and clinical management of limited progression. Eur J Cancer 51(11):1435–1443
Wang L, Berne B, Friesner R (2011) Ligand binding to protein-binding pockets with wet and dry regions. Proc Natl Acad Sci 108(4):1326–1330
Li Z, Jiang J-D, Kong W-J (2014) Berberine up-regulates hepatic low-density lipoprotein receptor through Ras-independent but AMP-activated protein kinase-dependent Raf-1 activation. Biol Pharm Bull 37(11):1766–1775
Wu C-P, Ambudkar SV (2014) The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm Sin B 4(2):105–111
Umar BA, Uzairu A, Shallangwa GA, Sani U (2019) QSAR modeling for the prediction of pGI50 activity of compounds on LOX IMVI cell line and ligand-based design of potent compounds using in silico virtual screening. Netw Mod Anal Health Infor Bioinform 8(1):22
Chtita S, Ghamali M, Larif M, Adad A, Hmammouchi R, Bouachrine M, Lakhlifi T (2013) Prediction of biological activity of imidazo [1, 2-a] pyrazine derivatives by combining DFT and QSAR results. IJIRSET 2(12):7962
Adhikari N, Maiti M (2010) MK tarun, synthesis and SAR of potent inhibitors of the hepatitis C virus NS3/4A protease: exploration of P2 quinazoline substituents. J Bioorg Med Chem Lett 20:20–4021
Aouidate A, Ghaleb A, Ghamali M, Chtita S, Ousaa A, Sbai A, Bouachrine M, Lakhlifi T (2018) Molecular docking and 3D-QSAR studies on 7-azaindole derivatives as inhibitors of Trk a: a strategic design in novel anticancer agents. Lett Drug Design Discov 15(11):1211–1223
Hadaji EG, Bourass M, Ouammou A, Bouachrine M (2017) 3D-QSAR models to predict anti-cancer activity on a series of protein P38 MAP kinase inhibitors. J Taibah Univ Sci 11(3):392–407
Mansouri K, Ringsted T, Ballabio D, Todeschini R, Consonni V (2013) Quantitative structure–activity relationship models for ready biodegradability of chemicals. J Chem Inf Model 53(4):867–878
Larif M, Chtita S, Adad A, Hmamouchi R, Bouachrine M, Lakhlifi T (2013) Predicting biological activity of anticancer molecules 3-ary l-4-hydroxyquinolin-2-(1H)-one by DFT-QSAR models. Int J 3(12):32–42
Abdulfatai U, Uzairu A, Uba S (2017) Quantitative structure-activity relationship and molecular docking studies of a series of quinazolinonyl analogues as inhibitors of gamma amino butyric acid aminotransferase. J Adv Res 8(1):33–43
Parthasarathy K, Praveen C, Saranraj K, Balachandran C, Kumar PS (2016) Synthesis, antimicrobial and cytotoxic evaluation of spirooxindole [pyrano-bis-2H-l-benzopyrans]. Med Chem Res 25(10):2155–2170
Yap CW (2011) PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32(7):1466–1474
Rajer-Kanduč K, Zupan J, Majcen N (2003) Separation of data on the training and test set for modelling: a case study for modelling of five colour properties of a white pigment. Chemom Intel Lab Syst 65(2):221–229
Kennard RW, Stone LA (1969) Computer aided design of experiments. Technometrics 11(1):137–148
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. Molecular Informatics 22(1):69–77
Leardi R (1996) Genetic algorithms in feature selection. In: Genetic algorithms in molecular modeling. Accademic press, United states, pp 67–86
Eriksson L, Jaworska J, Worth AP, Cronin MT, McDowell RM, Gramatica P (2003) Methods for reliability and uncertainty assessment and for applicability evaluations of classification-and regression-based QSARs. Environ Health Perspect 111(10):1361–1375
Gramatica P, Giani E, Papa E (2007) Statistical external validation and consensus modeling: a QSPR case study for Koc prediction. J Mol Graph Model 25(6):755–766
Jaworska J, Nikolova-Jeliazkova N, Aldenberg T (2005) QSAR applicability domain estimation by projection of the training set descriptor space: a review. ATLA-NOTTINGHAM 33(5):445
Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A (2002) BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 62(23):6997–7000
Choi W-K, El-Gamal MI, Choi HS, Baek D, Oh C-H (2011) New diarylureas and diarylamides containing 1, 3, 4-triarylpyrazole scaffold: synthesis, antiproliferative evaluation against melanoma cell lines, ERK kinase inhibition, and molecular docking studies. Eur J Med Chem 46(12):5754–5762
Hassan AH, Lee K-T, Lee YS (2020) Flavone-based arylamides as potential anticancers: design, synthesis and in vitro cell-based/cell-free evaluations. Eur J Med Chem 187:111965
Wu W, Zhang C, Lin W, Chen Q, Guo X, Qian Y, Zhang L (2015) Quantitative structure-property relationship (QSPR) modeling of drug-loaded polymeric micelles via genetic function approximation. PLoS One 10(3):e0119575
Jalali-Heravi M, Konuze E (2002) Use of quantitative structure property relationships in predicting the Kraft point of anionic surfactants. Electronic J Molecular Design 1:410–417
Chatterjee S, Maity A, Chowdhury S, Islam MA, Muttinini RK, Sen D (2020) In silico analysis and identification of promising hits against 2019 novel coronavirus 3C-like main protease enzyme. J Biomol Struct Dyn 38(18):1–14
Adedirin O, Uzairu A, Shallangwa GA, Abechi SE (2018) Optimization of the anticonvulsant activity of 2-acetamido-N-benzyl-2-(5-methylfuran-2-yl) acetamide using QSAR modeling and molecular docking techniques. Beni-Suef Univ J Basic Appl Sci 7(4):430–440
Umar BA, Uzairu A, Shallangwa GA, Uba S (2019) Rational drug design of potent V600E-BRAF kinase inhibitors through molecular docking simulation. J Eng Exact Sci 5(5):0469–0481
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL (2012) Quantifying the chemical beauty of drugs. Nat Chem 4(2):90
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1-3):3–25
Acknowledgements
The authors sincerely acknowledge the National Cancer Institute for providing the data used in this study and Ahmadu Bello University, Zaria for providing the soft wares used.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
UA designed the study. ABU carried out QSAR analysis, docking simulation, and drafted the manuscript. GAS and SU Participated in designing the study and edited the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Specification of entered descriptors and their mean effect. Table S2. Predicted activities of FBA compounds based on the developed model. Figure S1. The best docking pose of vemurafenib superimposed upon co-crystal structure of ligand to the V600E-BRAF binding pocket (PDB ID: 3OG7)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Umar, A.B., Uzairu, A., Shallangwa, G.A. et al. Ligand-based drug design and molecular docking simulation studies of some novel anticancer compounds on MALME-3M melanoma cell line. Egypt J Med Hum Genet 22, 6 (2021). https://doi.org/10.1186/s43042-020-00126-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-020-00126-9