
Abstract
Introduction
Genome sequence plays an important role in both basic and applied studies. Gossypium raimondii, the putative contributor of the D subgenome of upland cotton (G. hirsutum), highlights the need to improve the genome quality rapidly and efficiently.
Methods
We performed Hi-C sequencing of G. raimondii and reassembled its genome based on a set of new Hi-C data and previously published scaffolds. We also compared the reassembled genome sequence with the previously published G. raimondii genomes for gene and genome sequence collinearity.
Result
A total of 98.42% of scaffold sequences were clustered successfully, among which 99.72% of the clustered sequences were ordered and 99.92% of the ordered sequences were oriented with high-quality. Further evaluation of results by heat-map and collinearity analysis revealed that the current reassembled genome is significantly improved than the previous one (Nat Genet 44:98–1103, 2012).
Conclusion
This improvement in G. raimondii genome not only provides a better reference to increase study efficiency but also offers a new way to assemble cotton genomes. Furthermore, Hi-C data of G. raimondii may be used for 3D structure research or regulating analysis.
Similar content being viewed by others
Introduction
Over the last decade, next-generation sequencing (NGS) technologies have brought immense improvements in plant genome sequencing throughput and reduced the cost, and many plant genomes have been sequenced using this technology, such as F. vesca (Shulaev et al. 2011), C. cajan (Varshney et al. 2012), G. raimondii (Wang et al. 2012; Paterson et al. 2012), G. arboreum (Li et al. 2014) and G. hirsutum (Li et al. 2015; Zhang et al. 2018). However, de novo assembly of large eukaryotic genomes has remained a great challenge with the NGS platform because of a significant amount of repeat contents. As a result, de novo assembly of chromosome-scale scaffolds has become a major constraint to the completion of a high-quality genome sequence. Compared with traditional methods, like genetic map and physical map, high-throughput chromosome conformation capture (Hi-C) technology (Belton et al. 2012) has assisted in the assembly of long scaffolds to produce chromosome-scale genome assemblies (Lightfoot et al. 2017). The Hi-C technique is based on restriction enzyme cutting sites, which are more evenly distributed and have much higher density than genetic map.
The Hi-C-based proximity-guided assembly was initially developed to study the three-dimensional (3D) conformation of chromosomes of yeast gene expression (Berkum et al. 2010). The working principle based on two regular models with Hi-C data: the first one is that the rate of Hi-C interaction is inversely proportional to the genomic distance between the pairs of loci; the second one is that the rate of Hi-C interaction of pairs of loci within a chromosome is significantly higher than that in different chromosomes (Xie et al. 2015). Based on these two models, Hi-C-based proximity-guided assembly was applied for de novo assembling of human beings, and subsequently for the assembling of mouse and Drosophila genomes which reported good results or improvement (Burton et al. 2013). With the success of testing and verifying this method in Arabidopsis thaliana (Xie et al. 2015), Hi-C based proximity-guide assembly has been reported as an effective and efficient method which subsequently has been used in many other plants.
Besides its commercial value, cotton also serves as a perfect model system for studying cell wall biosynthesis (Zhang et al. 2018), cell elongation (Guo et al. 2017), and polyploidization (Yuan et al. 2016). The Gossypium genus comprises more than 50 species including at least 5 tetraploid species and 45 diploid species. Diploid cotton species are divided into 8 sub-genomes, denoted A-G and K based on chromosome pairing relationships (Wendel 1992). Tetraploid cotton species, such as cultivated G. hirsutum (AD1) and G. barbadense (AD2), had formed by an allopolyploidy event about 1–2 million years ago (Paterson et al. 2012). These tetraploid cotton species share common ancestors with the modern New World species G. raimondii (D5) and the Old World A-genome species G. herbaceum (A1) or G. arboreum (A2). Previously, the genomes of different cotton species were sequenced and assembled including G. raimondii (Wang et al. 2012), G. arboreum (Li et al. 2014), and G. hirsutum (Li et al. 2015), respectively.
Among these cotton species, the genome of G. raimondii has the lowest complexity which has been sequenced and assembled using the next-generation Illumina paired-end sequencing strategy (Wang et al. 2012). Approximately 73% (281 scaffolds) of the assembled sequences were anchored to 13 chromosomes, covering 88.1% of the genome, while only 52.4% (228 scaffolds) of the total sequences were both ordered and oriented. The completeness and accuracy of the previous sequenced and assembled genome of G. raimondii (Wang et al. 2012) were relatively low due to the large number of repeat elements and the small number of genetic markers. In the present study, we conducted a de novo Hi-C sequencing of G. raimondii, and incorporated the new Hi-C data with the existing G. raimondii scaffolds (Wang et al. 2012) to improve the quality of the D-genome.
Methods
Tissue collection and Hi-C sequencing
Plant materials
The seeds of G. raimondii D5–1 were planted in an incubator at constant environmental conditions having 27 °C temperature, 60% relative humidity, 16/8 h light/dark photoperiod, and 100% fluorescent light. When the sixth euphylla came out, these seedlings were transplanted into big pots. Approximately 3 g young leaves from G. raimondii plants were collected and immediately treated with formaldehyde.
Hi-C pipeline
During this study, we have used the same Hi-C pipeline as in Arabidopsis thaliana (Xie et al. 2015). Before starting this experiment, we have tested the integrity of DNA from the formaldehyde-treated tissue, and then the DNA samples were isolated and digested by MboI instead of HindIII because of the shorter recognition site (only four bases of MboI). The resulting sticky ends were filled with nucleotides in which cytosine is biotinylated, and ligated the adjacent blunt ends to a chimeric circle under extremely dilute conditions. Subsequently, DNA was purified and broken into 300–500 base pairs using ultrasonic, pull-down the biotin-labeled DNA and performed the PCR reaction (10 cycles). After DNA purification, the finished Hi-C library was sequenced with an Illumina Hiseq (PE150). A total of 570 412 361 read-pairs were obtained.
Genome assembly based on Hi-C data
Assembling of G. raimondii genome involved three steps. First, valid Hi-C paired-end reads and contact matrix with a resolution of 100 kb were generated by HiC-Pro (Servant et al. 2015). The raw sequence data with low quality, unmapped and invalid mapped paired reads were filtered out by HiC-Pro and contact-matrix based on interaction frequency was created. HiC-Pro results showed that 95.6% of sequences were clean Q30 bases, showing a good quality of sequence data. After filtering out the Hi-C data, 81.95% of uniquely mapped sequences were valid paired-end reads. Thus, the valid paired-end reads (223304666) were used for further genome assembly. At the second step, Errors in scaffolds of the initial draft assembly were identified and corrected following the Aedes aegypti’s de novo assembly procedure (Dudchenko et al. 2017). Briefly, the errors were corrected by identifying the bins where a scaffold’s long-range contact pattern changes abruptly, which was unlikely a correct scaffold. We cut out the error bins as a new scaffold. There are 259 errors within scaffolds. At the third step, the G. raimondii genome was assembled with the Hi-C data by Lachesis (Burton et al. 2013), which contained clustering, ordering, and orienting. Finally, the assembled G. raimondii genome was assessed by heat-map and collinear analysis.
Repeats and gene annotation
Repeat sequences of G. raimondii genome were masked by RepeatMasker (v4.0.8) with a custom library generated from RepeatModeler v2.0.1 (Flynn et al. 2020). Repeat-masked sequences were obtained as a draft genome in gene prediction. de novo prediction, homology-based prediction and transcriptome-based prediction were combined to annotate the draft genome of G. raimondii. GlimmerHMM v3.0.4 (Chrysanthou et al. 2011) and AUGUSTUS v3.3.2 (Stanke et al. 2006) were run for de novo prediction. We used 9 transcriptomes (SRR389181, SRR203240, SRR203250, SRR8878792, SRR8878720, SRR8878562, SRR8878553, SRR8878556, and SRR8878559) of G.raimondii from different organs and growth stages to predict genes. Homologous proteins from Arabidopsis, maize, and rice were input into GenomeThreader (v1.6.1) to train models for homology-based prediction. Sequences of transcriptome samples were aligned to draft genome by HISAT2 v2.1.0 (Kim et al. 2019) and transcripts were assembled by StringTie v1.3.6 (Pertea et al. 2015). All files in general feature format were integrated into a final genome annotation file by EvidenceModeler v1.1.1 (Haas et al. 2008).
The reassembled genome was evaluated in three aspects: completeness, consistency and continuity. We evaluated the continuity according to the length of scaffold N50 calculated by our python script. The completeness was checked by BUSCO (v4.1.4; Simão et al. 2015) and the consistency was evaluated by comparative analysis between our reassembled genome and other published genomes of G. raimondii. Genome alignment was performed by Minimap2 v2.1 (Li, 2018) and dot plots were generated by dotPlotly (https://github.com/tpoorten/dotPlotly/). Orthologous genes of G. raimondii genomes were detected by Orthofinder v2.5.1 (Emms and Kelly, 2015), and the visualization of orthologous gene detection was implemented by jcvi (https://github.com/tanghaibao/jcvi). All parameters of the softwares in this article were set as default.
Results
Assembling results
The genome of G. raimondii was reassembled using Hi-C data. About 98.42% of total sequence length were clustered successfully, among which 99.72% and 97.37% of the clustered sequences were ordered and high-quality ordered, respectively. And approximately 99.92% of the ordered sequences were oriented with high-quality. The statistics of pseudo-chromosome length are shown in Table 1, while the indicator statistics of the initial draft genome and reassembly results are shown in Table 2. From the parameters like scaffolds number, N50 of previously draft genome and reassemble genome, we found that the G. raimondii genome using a Hi-C-based proximity-guided assembly is much better than the reported draft genome (Wang et al. 2012).
Genome assessment results
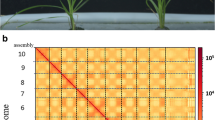
The quality of the reassembled genome can be verified in four aspects. First, BUSCO analysis reported that 97.2% (> 90%) completeness scores for the reassembled genome, indicating that it was complete. Compared with ~ 32 000 genes in the old draft genome (besides scaffolds), 38 975 genes were found in the annotation of the reassembled genome. Second, The heat-map directly proved the validity of the processing methods. Based on the two regular models as we mentioned above, the heat-map of the reassembled results revealed that the diagonal interaction was much higher. The boundaries of each pseudo-chromosome are relatively clear and it is under low background noise that shows good reassembling results (Fig. 1). Third, we compared the draft genome and the reassembled genome with two previous genome assemblies of G. raimondii (Paterson et al. 2012; Udall et al. 2019) (Fig. 2). The reassemble genome had a general agreement in their alignments with two previous genome assemblies than the draft genome. Fourth, we compared homologous genes in the draft genome and reassembled genome with which in two previous genome assemblies of G. raimondii (Paterson et al. 2012; Udall et al. 2019) (Fig. 3). Both the draft genome and reassembled genome showed a good agreement with other two versions, but the sizes of chromosomes (like chromosome 4 and 8) in the reassembled genome were more reliable than the draft genome.

The heat-map of the reassembled result at a 100-kb resolution. Hi-C interactions were detected in the reassembled G.raimondii genome sequences, and most interactions maps of chromosome sequences suggested that the genome sequence was assembled in the correct order. The darker red dot is representing the higher interaction between bins

Genome comparisons between the draft genome and reassembled genome with two previous genome assemblies of G. raimondii. A) Genome alignment between G. raimondii (Wang et al. 2012) and G. raimondii (Paterson et al. 2012). B) Genome alignment between G. raimondii (Wang et al. 2012) and G. raimondii (Udall et al. 2019). C) Genome alignment between G. raimondii (new) and G. raimondii (Paterson et al. 2012). D) Genome alignment between G. raimondii (new) and G. raimondii (Udall et al. 2019)

Discussion
In the present study, we applied de novo Hi-C sequencing of the G. raimondii genome to improve the quality and accuracy of its previously reported draft genome from Wang et al. (2012). Results from the comparative analysis of different parameters between the draft genome (Wang et al. 2012) and the current reassembled genome showed significantly improved quality as compared with the previous one. The reassembled genome is not good enough, because it is based on the next generation sequencing in 2012. The G. raimondii genome (Udall et al. 2019) was assembled to a chromosome level using PacBio long-read technology, but Hi-C and Bionano optical mapping may have a better assembly. But diffirent genome versions can provide different information, like some genes can only be annotated in a certain version of annotation file.
Availability of data and materials
The reassembled genome sequences and annotation files will be upload after received.
Abbreviations
- NGS:
-
Next generation sequencing
- Hi-C:
-
High-throughput chromosome conformation capture
- CAAS:
-
Chinese Academy of Agricultural Science
References
Belton JM, Mccord RP, Gibcus JH, et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58(3):268–76. https://doi.org/10.1016/j.ymeth.2012.05.00.
van Berkum NL, Lieberman-Aiden E, Williams L, et al. Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp. 2010;39:1869. https://doi.org/10.3791/1869.
Burton JN, Adey A, Patwardhan RP, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31(12):1119–25. https://doi.org/10.1038/nbt.2727.
Chrysanthou N, Chrysos G, Sotiriades E, Papaefstathiou I. Parallel accelerators for GlimmerHMM bioinformatics algorithm. In: 2011 Design, automation and test in Europe. Grenoble, France, 14–18 March, 2011. https://doi.org/10.1109/DATE.2011.5763024.
Dudchenko O, Batra SS, Omer AD, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356(6333):92–5. https://doi.org/10.1126/science.aal3327.
Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16(157):157. https://doi.org/10.1186/s13059-015-0721-2.
Flynn JM, Hubley R, Goubert C, et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci. 2020;117(17):9451–7. https://doi.org/10.1073/pnas.1921046117.
Guo K, Tu L, He Y, et al. Interaction between calcium and potassium modulates elongation rate in cotton fiber cells. J Exp Bot. 2017;68(18):5161. https://doi.org/10.1093/jxb/erx346.
Haas BJ, Salzberg SL, Wei Z, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9(1):R7. https://doi.org/10.1186/gb-2008-9-1-r7.
Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with hisat2 and hisat-genotype. Nat Biotechnol. 2019;37(8):907–15. https://doi.org/10.1038/s41587-019-0201-4.
Li F, Fan G, Lu C, et al. Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol. 2015;33:524–30. https://doi.org/10.1038/nbt.3208.
Li F, Fan G, Wang K, et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet. 2014;46(6):567–72. https://doi.org/10.1038/ng.2987.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094. https://doi.org/10.1093/bioinformatics/bty191.
Lightfoot DJ, Jarvis DE, Ramaraj T, et al. Single-molecule sequencing and Hi-C-based proximity-guided assembly of amaranth (Amaranthus hypochondriacus) chromosomes provide insights into genome evolution. BMC Biol. 2017;15(1):74. https://doi.org/10.1186/s12915-017-0412-4.
Stanke M, Keller O, Gunduz I, et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34(Web Server issue):W435–9. https://doi.org/10.1093/nar/gkl200.
Paterson AH, Wendel JF, Gundlach H, et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492(7429):423–7. https://doi.org/10.1038/nature11798.
Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. https://doi.org/10.1038/nbt.3122.
Servant N, Varoquaux N, Lajoie BR, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. https://doi.org/10.1186/s13059-015-0831-x.
Shulaev V, Sargent DJ, Crowhurst RN, et al. The genome of woodland strawberry (Fragaria vesca). Nat Genet. 2011;43:109–16. https://doi.org/10.1038/ng.740.
Simão FA, Waterhouse RM, Ioannidis P, et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. https://doi.org/10.1093/bioinformatics/btv351.
Udall JA, Long E, Hanson C, et al. De Novo Genome Sequence Assemblies of Gossypium raimondii and Gossypium turneri[J]. G3: Genes|Genomes|Genetics. 2019;9(10). https://doi.org/10.1534/g3.119.400392.
Varshney RK, Chen W, Li Y, et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotech. 2012;30:83–9. https://doi.org/10.1038/nbt.2022.
Wang K, Wang Z, Li F, et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44(10):1098–103.
Wendel JF. Phylogenics of the cotton genus (Gossypium): character-state weighted parsimony analysis of chloroplast-DNA restriction site data and its systematic and biogeographic implications. Syst Bot. 1992;17(1):115–43. https://doi.org/10.2307/2419069.
Xie T, Zheng JF, Liu S, et al. De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana. Mol Plant. 2015;8(3):489–92. https://doi.org/10.1016/j.molp.2014.12.015.
Yuan D, Tang Z, Wang M, et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci Rep. 2016;5:17662. https://doi.org/10.1038/srep17662.
Zhang J, Huang GQ, Zou D, et al. The cotton (Gossypium hirsutum) NAC transcription factor (FSN1) as a positive regulator participates in controlling secondary cell wall biosynthesis and modification of fibers. New Phytol. 2018;217(2):625–40. https://doi.org/10.1111/nph.14864.
Acknowledgements
This work was supported by CAAS.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Yang QH and Song GL conceived and designed the experiments; all authors performed data analysis and interpretation; Yang QH, Javaria A, Liu S and Song GL wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
YANG, Q., ZUO, D., CHENG, H. et al. Improved Gossypium raimondii genome using a Hi-C-based proximity-guided assembly. J Cotton Res 4, 23 (2021). https://doi.org/10.1186/s42397-021-00096-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42397-021-00096-2